

Abstract
Solid dispersion systems of telmisartan (a poorly water-soluble antihypertension drug) with biopolymer carrier chitosan have been investigated in this study. The mechanism of solubilization of chitosan for drug has been studied. In addition, the influence of several factors was carefully examined, including the preparation methods, the drug/carrier weight ratios, and the milling time. Drug dissolution and physical characterization of different binary systems were studied by in vitro dissolution test, particle size distribution, Fourier transform infrared spectroscopy, differential scanning calorimetry, powder X-ray diffractometry, and scanning electron microscopy. The results presented that the weak basic property of chitosan appeared as the main driving force for the drug dissolution enhancement. Other effects such as decreased drug crystallinity and size played a positive contributory role. Among the preparation methods, cogrinding was the best method showing strong drug amorphization, reduced particle size, and enhanced dissolution. The drug dissolution markedly improved with increasing the amount of chitosan in solid mixtures. As a result, a significant effect of chitosan increasing telmisartan dissolution has been demonstrated, and cogrinding in a roll ball mill was the best way to prepare solid dispersions, which had high degree of uniformity in drug content and had a practical application in manufacturing.
KEY WORDS: chitosan, cogrinding, dissolution enhancement, solid dispersion, telmisartan
INTRODUCTION
Solid dispersion (SD) is one of the most effective techniques to enhance the dissolution rate of poorly water-soluble drugs to improve drug bioavailability. Generally, SD can be defined as a dispersion of active ingredients in molecular, amorphous, and/or microcrystalline forms into an inert carrier (1–3). The mechanisms whereby SD increases drug dissolution include reduction of the particle size of the drug, change of crystalline drug to amorphous form, formation of soluble complexes, higher dispersion, and better wettability of drug. It is widely believed that often more than one of these phenomena determine the rate and extent of dissolution (4). Among the methods to prepare the formulation of SDs, cogrinding (CG) is environmentally friendly. Unlike other techniques, it does not require toxic solvents (whose removal can be difficult and expensive) and sophisticated equipments, connected with relatively simple processes and a decrease in the total time. On the other hand, this approach is usually effective in increasing the solubility of insoluble drugs (5–8).
Telmisartan (TEL) is an angiotensin-II receptor antagonist, useful in treatment of hypertension diseases, heart diseases, heart strokes, and bladder diseases (Fig. 1). TEL is manufactured and supplied in the free acid form and is characterized by its very poor intestinal solubility, which depended on the pH of the medium as a BCS Class II drug with an aqueous solubility of 0.09 μg/mL and a pKa of 4.45 ± 0.09 (9). Several researches have been reported for the dissolution enhancement, such as incorporating alkalizers into TEL SD systems (9), being loaded with nanoparticles (10,11), or preparation of amorphous form (12).
Fig. 1.
Molecular structures of TEL and CS
Preliminary studies demonstrated that chitosan (CS) was successful in enhancing the dissolution rate of TEL. Moreover, CS exhibits several attractive biological properties, such as nontoxicity, good biocompatibility and biodegradability, wide availability in nature, and low cost (13). Some researches have been reported that CS as carrier increased the dissolution properties and bioavailability of insoluble drugs (14–20). Taking all into account, CS was chosen as a desirable carrier to disperse TEL in SD systems.
The mechanism of solubilization of CS was generally clarified by different explanations. Some authors concluded that the amorphizing effect of CS acted as a primary cause for the enhanced drug dissolution (14–16); whereas, others believed that reduced particle size of the crystal drug (17) or formation of water-soluble complexes (18) played the leading role. In this paper, the reason of solubilization of CS for TEL was carefully investigated. Besides, the influence of several factors was fully examined, such as the preparation methods, the drug/carrier weight ratios, and the milling time. In vitro dissolution test, particle size dispersion, Fourier transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), powder X-ray diffractometry (PXRD), and scanning electron microscopy (SEM) were used for evaluation/characterization of the formulations.
MATERIALS AND METHODS
Materials and Reagents
TEL was kindly supplied by Zhejiang Apeloa Medical Technology Co., Ltd. (Dongyang, Zhejiang). CS was obtained by Zhejiang Aoxing Biotechnology Co., Ltd. (Yuhuan, Zhejiang), which was 90.6% degree of N-deacetylation and the viscosity of 0.5% (w/v) solution in 0.5% (w/v) acetic acid at 20°C was 52 mPa s. Polyethylene glycol (PEG 6000) was purchased from Huiyou Fine Chemical Factory (Haidian, Beijing). Hydroxypropyl-β-cyclodextrin (HP-β-CD) was from Qianhui Fine Chemical Co., Ltd. (Zibo, Shandong). Polyvinylpyrrolidone (PVP k30) and poloxamer 188 were provided by BASF (Ludwigshafen, Germany). Microcrystalline cellulose (MCC PH101) was from Hengxin Chemical Reagents Co., Ltd. (Shanghai). Deionized water was prepared by a Nano-purification system obtained from Thermo, USA. All commercially available solvents and reagents were used without further purification.
Preparation of SDs
TEL-CS SDs in different ratios (w/w = 1:1, 1:3, 1:5, 1:7, and 1:9) were prepared by three methods. Before preparation, CS was sieved through a 200 mesh sieve (75 μm). Physical mixtures (PM) of drug and polymer were obtained by simply rotating in a jar for 10 min. Coevaporation (CE): firstly drug dissolved in a certain volume of ethanol (200 mL/1 g TEL to be added), and then CS was added and stirred for a while. Finally, the systems were evaporated and dried in a vacuum dryer for 2 h at 60°C. CG: using a roll mill (QM-2, Sanxing instruments, Xiangtan) under the following conditions: a polyurethane jar of 500 mL, steel balls diameter of 22 mm, milling time of 1, 2, 4, and 8 h at 700 rpm. All powder products were sieved by 200 mesh sieve and used for subsequent studies. The samples were stored in a desiccator at room temperature.
Solubility Study
An excess of TEL (10 mg) was added to 25 mL of deionized water containing increasing amounts of CS. The resulting suspensions were placed in 37°C shaking table and shaken for 24 h at a speed of 150 rpm. An aliquot of solution was withdrawn and filtered through a 0.45 μm membrane filter. The concentration of TEL was measured using HPLC. Each experiment was performed in triplicate.
Drug Content Uniformity
SDs equivalent to 10 mg of TEL were weighted accurately and suspended in suitable quantity of ethanol. The drug dissolved rapidly in solvent by ultrasonication, and then filtered, diluted to 0.1 mg/mL. The drug concentration was determined by HPLC. Each sample analysis was preformed in triplicate.
Determination of pH in Dissolution Media
The pH of dissolution medium after the dissolution test was measured using a pH meter (Delta 320, Mettler Toledo).
Particle Size Distribution
Particle size distributions of solid systems were analyzed by Mastersizer 2000 (Malvern Instruments Ltd., UK). All samples were measured in a dry mode from 0.02 to 2,000 μm.
Fourier Transform Infrared Spectroscopy
Infrared spectra of the samples were obtained using a Niclolet Avatar 370 instrument (Thermo Niclolet Corporation, USA). All samples were mixed with KBr for compressing into a thin tablet. The wave number of FTIR spectra ranged from 400 to 4,000 cm−1 with a resolution of 2 cm−1.
Differential Scanning Calorimetry
Thermal analysis was investigated using a Mettler Q100 apparatus (TA Corporation, USA) equipped with a differential scanning calorimeter. About 5∼7 mg sample was weighed and placed in an aluminum pan with lid and crimp sealed. Samples were heated from 50°C to 300°C at the rate of 10°C/min, using nitrogen as a purge gas at 50 mL/min.
Powder X-ray Diffractometry
The powder X-ray diffraction patterns of the samples were recorded with an X’ Pert PRO diffractometer (PANalytical, Holland) using Cu-K radiation at a voltage of 40 kV and a current of 30 mA. The samples were scanned in increments of 0.034° from 3° to 45° (diffraction angle 2θ).
Scanning Electron Microscopy
The morphology of samples was characterized by a Hitachi S-4700 SEM (Hitachi, Japan), operated at 15 kV. Each sample was blown onto adhesive carbon tape on aluminum stub followed by sputter coating of gold.
HPLC Analysis (10)
TEL was analyzed by the Agilent HPLC system (1200 series), a UV–vis tunable absorbance detector, and with a reverse phase column (150 × 4.5 mm, Zorbax SB-C18). The mobile phase consisted of a 80:20 (in percent, v/v) mixture of methanol and 10 mM potassium dihydrogen phosphate solution; the flow rate was 1.0 mL/min; the detection wavelength was 296 nm; the injection volume was 50 μL; and the running time was 6 min. The solvent system was filtered using a 0.45 μM membrane filter (Xingya, Shanghai) before running the HPLC analysis.
In Vitro Dissolution Test
Dissolution testing was performed using a HTY-EU802 rotating paddle apparatus (Tailin Bioengineering Equipments Co., Ltd., Hangzhou). The dissolution medium (900 mL deionized water) was maintained at 37 ± 0.5°C and stirred at 50 rpm. The SD powder equivalent to 40 mg TEL was added to deionized water. Aliquots of 5 mL dissolution medium were withdrawn at 2, 5, 10, 20, 30, 40, and 60 min and replaced with an equal volume of the same medium. All samples were filtered through a 0.45 μm membrane filter. The amounts of TEL were determined by HPLC. Each experiment was performed in triplicate.
RESULTS AND DISCUSSION
Mechanism of Solubilization of CS for TEL
In preliminary studies, several excipients were used for dispersing the drug TEL by CG method for 30 min at the ratio of 1:3 (drug/polymer, w/w). These included PEG 6000, PVP k30, poloxamer 188, HP-β-CD, MCC, and CS. Except for TEL-CS system, no significant drug solubility enhancement of these binary systems was observed (Table I). Since the molecular structure of TEL is bulky, there is a great steric hindrance between HP-β-CD and TEL. Hence, it was difficult to form inclusion complex (21). It was assumed that there was also no interaction between PEG 6000/PVP k30/poloxamer 188/MCC and TEL.
Table I.
Solubility of TEL in SDs Prepared by Different Excipients
| Excipients | Solubility (μg/mL) |
|---|---|
| PEG 6000 | 0.31 ± 0.04 |
| PVP k30 | 0.25 ± 0.02 |
| Poloxamer 188 | 0.22 ± 0.02 |
| HP-β-CD | 0.25 ± 0.05 |
| CS | 3.48 ± 0.13 |
| MCC | 0.22 ± 0.03 |
TEL telmisartan, SD solid dispersion, PEG 6000 polyethylene glycol, PVP k30 polyvinylpyrrolidone, HP-β-CD hydroxypropyl-β-cyclodextrin, CS chitosan, MCC microcrystalline cellulose
There were various explanations to illustrate the mechanism of solubilization of CS, including the amorphizing effect toward the drug, reduction of the particle size of the drug, and formation of water soluble complexes with the drug. More details of CS in the SDs were investigated next in order to elucidate the mechanism of solubilization of CS for TEL.
First of all, in order to get insight into the nature of a possible interaction in solution between TEL and CS, the solubility of TEL in different drug/carrier weight ratios of PM was preformed. In addition, the pH values of the dissolution media (water) were measured for understanding whether pH changes in solution was a mechanism for enhancing drug solubility. As shown in Fig. 2, the drug solubility markedly improved with an increasing amount of CS in solid mixtures and a nearly linear dependence was found. The concentration of drug increased to 12.4 μg/mL at the ratio of 1:9 (drug/polymer, w/w); whereas, the aqueous solubility of TEL was 0.09 μg/mL. Meanwhile, the pH value of the solution gradually became higher with increasing CS content, and the solution was in a weak basic condition (Table II). These results are probably consistent with the presence of electrostatic interactions between TEL and CS due to the anionic nature of the drug and the strong positive charge of the polymer at pH values of <6.3 (13).
Fig. 2.
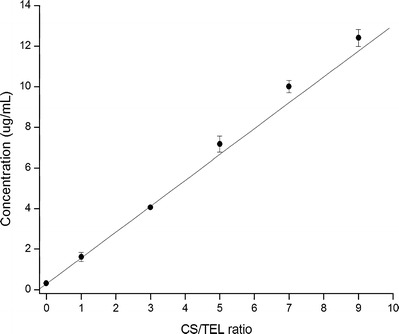
Solubility of TEL in various drug/carrier weight ratios of PM in deionized water at 37°C
Table II.
Solution pH after the Dissolution of PM (TEL, 10 mg) at Varying Weight Ratio in Water (25 mL)
| TEL/CS (w/w) | pH |
|---|---|
| Water | 5.50 ± 0.10 |
| 1:1 | 6.71 ± 0.11 |
| 1:3 | 7.05 ± 0.05 |
| 1:5 | 7.36 ± 0.06 |
| 1:7 | 7.65 ± 0.05 |
| 1:9 | 7.73 ± 0.03 |
TEL telmisartan, PM physical mixtures, CS chitosan
After grinding pure TEL for 30 min, the intensity of the characteristic peaks (6.7° and 15.0°) in PXRD patterns of drug noticeably became less relating to drug amorphousness (Fig. 3); nevertheless, no difference between original drug and the drug after grinding in the dissolution properties, indicating that change of crystalline drug to amorphous form had no influence on drug solubilization.
Fig. 3.

PXRD patterns of TEL and TEL (milling for 30 min). The amount of both samples used for PXRD was equal (90 mg)
With the purpose to further attest that drug amorphousness was not a dominant force to enhance dissolution, MCC was employed for comparison with CS, just because the monomer molecular structure of MCC is same to CS, except that in C-2 position of glucose, a hydroxyl group and an amino group presented in MCC and CS, respectively. The PXRD patterns of TEL, CS, MCC, and CG products (drug/carrier, 1:3; milling time, 30 min) are given in Fig. 4. The intensity of the characteristic peak (6.7°) in TEL-MCC system is less than TEL-CS, indicating the extent of amorphizing of MCC to drug better than CS. Besides, the average particle size of TEL-CS system is 57.1 μm, bigger than 37.0 μm of TEL-MCC shown in Fig. 5. Although the drug amorphousness and the particle size of TEL-MCC system are better than TEL-CS, these changes have no effect to improve the dissolution of drug.
Fig. 4.
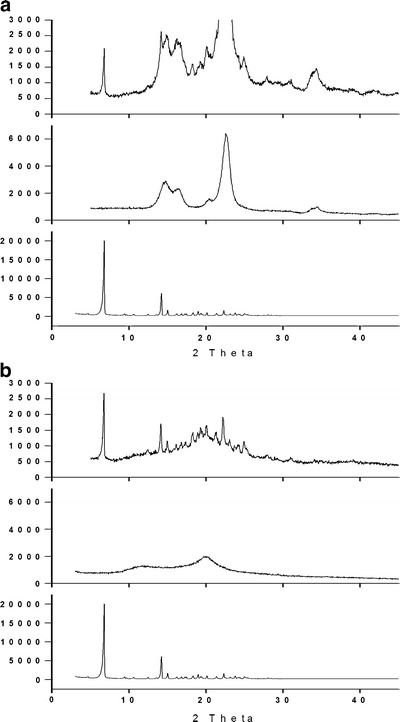
a PXRD patterns of TEL, MCC, and CG product (drug/carrier, 1:3); b PXRD patterns of TEL, CS, and CG product (drug/carrier, 1:3). The amount of both samples TEL-CS SD and TEL-MCC SD used for PXRD was equal
Fig. 5.
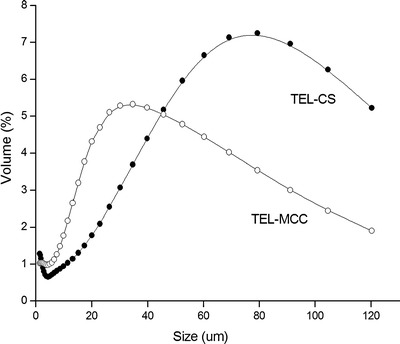
Particle size distribution of TEL-CS SD and TEL-MCC SD
The interaction of drug and carrier can lead to changes in bonding between functional groups that can be detected by FTIR spectroscopy (14,22). The FTIR spectra of TEL, CS, PM, and CG are shown in Fig. 6. The characteristic peak of TEL was a carbonyl absorption band at 1,697.55 cm−1 assigned to the carboxyl group, whereas the spectrum of CS showed a broad band at high wave number and a band at 1,601.36 cm−1 belonging to stretching vibration in hydroxyl groups and amino group, respectively. The spectrum of PM was equivalent to the addition spectrum of TEL and CS indicating there was no interaction between polymer and drug. The spectrum of CG had no difference from that of PM in the field of the main TEL absorption bands, and particularly, the absorption band of carbonyl appeared almost unchanged, which suggested the absence of intermolecular hydrogen bonding via carbonyl group of TEL and hydroxyl groups of CS.
Fig. 6.

FTIR spectra of TEL, CS, PM, and CG at drug/polymer (1:1, w/w) SDs
By analyzing all the results above, it was concluded that the drug solubilization was mainly attributed to the weak basic feature of CS as carriers. Besides, drug amorphousness and reduced particle size played a positive contributory role, which was demonstrated in next studies about effect of factors.
Effect of Factors
Methods
The methods used to disperse TEL within CS could be a factor affecting the drug dissolution rate. The common methods (physical mixing, CE, and CG) were employed for preparing TEL-CS SDs. Dissolution profiles of PM, CE and CG SDs at 1:5 (w/w) of drug/polymer are clearly seen in Fig. 7. After 1 h dissolution test, the concentration of drug in solution of CG product was close to seven times than PM, whereas CE was near three times than PM. The pH values of the solution of PM, CE, and CG samples were 6.05, 6.11, and 6.21, separately.
Fig. 7.
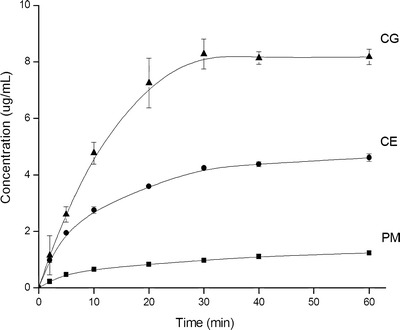
Dissolution profiles of PM, CE, and CG at 1:5 (w/w) of drug/polymer
The crystallinity of TEL in the products of PM, CE, and CG SDs was compared with that of pure TEL by DSC and PXRD. The DSC thermograms of samples are shown in Fig. 8a. The DSC curve of pure TEL exhibited a single sharp endothermal peak at 271.5°C, which corresponding to intrinsic melting point. Under same amount of pure TEL of samples of PM, CE, and CG, the intense of endothermal peak decreased, as follows: PM < CE < CG. The PXRD patterns of TEL, PM, CE, and CG are given in Fig. 8b. The reduced intense of the characteristic peaks of drug was ordered from PM < CE < CG. The results of DSC and PXRD were consistent with the partial amorphous effect of drug during SD preparation.
Fig. 8.

a DSC curves of PM, CE, and CG at 1:5 (w/w) of drug/polymer; b PXRD patterns of PM, CE, and CG at 1:5 (w/w) of drug/polymer
PM were produced by simply mixing drug with carrier together, and the drug remained in the original form. CE products were a consequence of the closer contact between the components and the better dispersion of the drug into the hydrophilic carrier through the coprecipitation technique. In addition, the reduced drug crystallinity appeared in the coevaporated products. The best performance presented by CG mixtures could be attributed to the very intimate physical contact between drug and carrier, reduction of the particle size and change of crystalline drug to amorphous form brought about by the mechanical treatment.
The morphology of TEL-CS systems prepared by the different methods was investigated by means of SEM analysis (Fig. 9). TEL particles appeared as needle crystals with smooth surfaces of homogeneous morphology, whereas CS consisted of amorphous particles of rather irregular size and shape. The needle crystals of TEL were clearly spread over the surface of carrier particles in the drug-polymer PM. The morphology of drug appeared as partial amorphous form in the coevaporated product, in accordance to the results obtained from DSC and PXRD studies. The needle shape of crystal drug disappeared in the CG product of the same composition. At the same time, it was not clear to differentiate the carrier CS.
Fig. 9.

SEM photographs of TEL (a), CS (b), PM (c), coevaporated product (d), and CG product (e) at 1:5 (w/w) of drug/polymer
To summarize, CG as the best method could cause drug amorphization and decrease particle size, which promoted electrostatic interactions between TEL and CS in solution and enhanced drug dissolution.
The uniformity of the drug content of SD systems is a requirement for SD technique applied in industry, resulting in uniform drug bioavailability in vivo. To investigate the drug content uniformity, we separated the drug from the solid mixtures and detected drug concentration. The results showed that coefficient of variation of samples was less than 0.9%, indicating TEL-CS SDs prepared by CG with high degree of uniformity in drug content.
Ratios of Drug/Carrier
Different drug/polymer weight ratios of SDs were produced by CG method for 60 min. The effects of ratios of drug/polymer on drug dissolution are presented in Fig. 10a. The solution pH after dissolution test of SDs was available in Table III. The drug dissolution of TEL-CS CG products markedly improved compared with that of TEL alone. The higher the polymer content, the higher the solution pH value and the faster was the dissolution. The drug concentration reached the highest value of nearly 14 μg/mL at the ratio of 1:9 (drug/polymer, w/w). Different from other drug/polymer ratios dissolution profiles, the concentration of drug was lower at the ratio of 1:1. This was maybe caused by drug aggregation at high drug content. After 30 min, there was almost 0% dissolved of the pure drug, compared with nearly 21% and 34% for 1:5 and 1:9 TEL/CS ratio, respectively. It was clearly evident that a nearly linear relationship was found between percents dissolved after 30 min and the polymer content (Fig. 10b).
Fig. 10.

a Dissolution profiles of TEL, different drug/polymer ratio CG products; b percent dissolved of different drug/polymer ratios after 30 min
Table III.
Solution pH after the Dissolution of CG (TEL, 40 mg) at Varying Weight Ratio in Water (900 mL)
| TEL/CS (w/w) | pH |
|---|---|
| 1:1 | 5.80 ± 0.11 |
| 1:3 | 6.00 ± 0.05 |
| 1:5 | 6.21 ± 0.03 |
| 1:7 | 6.33 ± 0.10 |
| 1:9 | 6.56 ± 0.12 |
TEL telmisartan, CG cogrinding, CS chitosan
As shown in Fig. 11a, the DSC curve of TEL was typical of a crystalline anhydrous substance. In contrast to drug, CS was a type of amorphous hydrated compounds, showing a broad endothermal effect varying from 50∼180°C associated with dehydration. Under equal amount of pure TEL of SDs, the intensity of the endothermal peak slightly decreased by increasing the amount of CS.
Fig. 11.
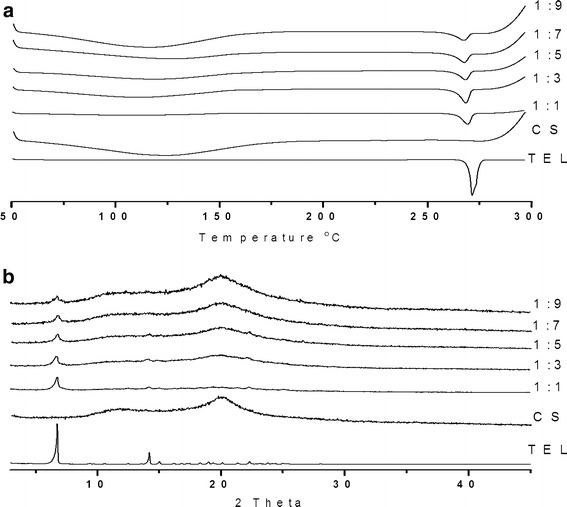
a DSC curves of TEL, CS, and different drug/polymer ratios CG products; b PXRD patterns of TEL, CS, and different drug/polymer ratios CG products. Samples equivalent to equal amount of TEL used for DSC and PXRD
The PXRD patterns of TEL, CS, and different drug/polymer ratio SDs are given in Fig. 11b. The PXRD pattern of TEL had two characteristic peaks of intensity at 6.7° and 14.0°, and CS showed a broad peak at 10∼15°, indicating close to amorphousness. The characteristic peak (6.7°) of PXRD patterns also slightly decreased by increasing the content of CS, similar with the change of DSC curves, indicating the amount of CS had no significant impact on drug crystallinity.
Milling Time
The milling time which is also an important factor was investigated. The effects of the milling time on drug dissolution rate are shown in Fig. 12. It was clearly evident that the drug concentration progressively improved with increasing milling time and reached the highest values at 8 h, which was consistent with the change of the pH value of the solution (Table IV). The highest concentration of drug reached to 27.5 μg/mL.
Fig. 12.

Dissolution profiles of different milling time at the ratio of 1:5 (drug/polymer, w/w)
Table IV.
Solution pH after the Dissolution of CG (TEL/CS, 1:5 (w/w); TEL, 40 mg) in Water (900 mL)
| Time (h) | pH |
|---|---|
| 1 | 6.21 ± 0.03 |
| 2 | 6.40 ± 0.11 |
| 4 | 6.63 ± 0.01 |
| 8 | 6.72 ± 0.07 |
TEL telmisartan, CG cogrinding, CS chitosan
The PXRD diffraction patterns of different milling time are given in Fig. 13. Each sample for PXRD analysis had same weight of pure drug. It can be seen that the characteristic peak of intensity at 6.7° was gradually decreasing by extending the milling time. When the milling time reached 4 h, the characteristic peak disappeared, indicating drug complete amorphousness. The particle size distributions of different milling time are shown in Fig. 14. The longer milling time, the smaller the particle size. When the milling time was increased, the crystal drug turned amorphous and the particle size of solid systems also decreased, which benefitted drug dissolution because of less lattice energy, thinner dissolution layer and more surface areas.
Fig. 13.

PXRD patterns of different milling time at the ratio of 1:5 (drug/polymer, w/w)
Fig. 14.

Particle size distributions of different milling time at the ratio of 1:5 (drug/polymer, w/w)
In addition, the drug dissolution profiles of CG powders appeared unchanged after 3 months of storage in a desiccator at room temperature. Since CS as an excipient can be used for direct compression, the CG products were able to give directly compressed tablets, containing the proper dosage of the drug. It took less than 5 s to disintegrate, with dissolution behavior was practically similar to the starting CG powder.
CONCLUSIONS
In this present work, CS can favorably enhance dissolution of TEL, which was fundamentally resulted from the weak basic property of CS. Drug amorphousness and reduced particle size are helpful for drug dissolution rate. The most effective method was the CG technique, which acted the best dissolution performance. Moreover, high degree of uniformity in drug proportion can be acquired by this way. Therefore, CG technique has the potential for industrial application, avoiding the requirement of adding organic solvents.
ACKNOWLEDGMENTS
We are grateful to the Natural Science Foundation of China (No. 21006097, 21076191, and 21176222), the Science and Technology Program of Zhejiang Province (No. 2009C14001), and the Program of International S&T Cooperation (No. 2008DFR40280) for financial support.
REFERENCES
- 1.Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 2.Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poorly water soluble drugs. Drug Discov Today. 2007;12(23/24):1068–1074. doi: 10.1016/j.drudis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Bikiaris DN. Solid dispersions, part I: recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin Drug Deliv. 2011;8(11):1501–1519. doi: 10.1517/17425247.2011.618181. [DOI] [PubMed] [Google Scholar]
- 4.Craig DQM. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int J Pharm. 2002;231:131–144. doi: 10.1016/S0378-5173(01)00891-2. [DOI] [PubMed] [Google Scholar]
- 5.Barzegar-Jalali M, Valizadeh H, Shadbad MS, Adibkia K, Mohammadi G, Farahani A, et al. Cogrinding as an approach to enhance dissolution rate of a poorly water-soluble drug (gliclazide) Powder Technol. 2010;197:150–158. doi: 10.1016/j.powtec.2009.09.008. [DOI] [Google Scholar]
- 6.Colombo I, Grassi G, Grass M. Drug mechanochemical activation. J Pharm Sci. 2009;98(11):3961–3986. doi: 10.1002/jps.21733. [DOI] [PubMed] [Google Scholar]
- 7.Dushkin AV. Mechanochemical synthesis of organic compounds and rapidly-soluble materials. High-energy ball milling: mechanochemical processing of nanopowders. Cambridge, UK: Woodhead Publishing Limited; ; 2010. pp. 224–247. [Google Scholar]
- 8.Corti G, Capasso G, Maestrelli F, Cirri M, Mura P. Physical–chemical characterization of binary systems of metformin hydrochloride with triacetyl-β-cyclodextrin. J Pharm Biomed Anal. 2007;45:480–486. doi: 10.1016/j.jpba.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 9.Tran PHL, Tran HTT, Lee BJ. Modulation of microenvironmental pH and crystallinity of ionizable telmisartan using alkalizers in solid dispersions for controlled release. J Control Release. 2008;129:59–65. doi: 10.1016/j.jconrel.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Zhi Z, Jiang T, Zhang J, Wang Z, Wang S. Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J Control Release. 2010;145:257–263. doi: 10.1016/j.jconrel.2010.04.029. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Jiang T, Zhang Q, Wang S. Inclusion of telmisartan in mesocellular foam nanoparticles: drug loading and release property. Eur J Pharm Biopharm. 2010;76:17–23. doi: 10.1016/j.ejpb.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Koilkonda P, Lekkala AR, Mala C, Golla K, Ganji SR, Konda RB. Amorphous telmisartan. US Patent 2006/0111417 A1.
- 13.Ravi Kumar MNV, Muzzarelli RAA, Muzzarelli C, Sashiwa H, Domb AJ. Chitosan chemistry and pharmaceutical perspectives. Chem Rev. 2004;104:6017–6084. doi: 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- 14.Mura P, Zerrouk N, Mennini N, Maestrelli F, Chemtob C. Development and characterization of naproxen–chitosan solid systems with improved drug dissolution properties. Eur J Pharm Sci. 2003;19:67–75. doi: 10.1016/S0928-0987(03)00068-X. [DOI] [PubMed] [Google Scholar]
- 15.Kumar SGV, Mishra DN. Preparation, characterization and in vitro dissolution studies of solid systems of valdecoxib with chitosan. Chem Pharm Bull. 2006;54(8):1102–1106. doi: 10.1248/cpb.54.1102. [DOI] [PubMed] [Google Scholar]
- 16.Portero A, Remuñán-López C, Vila-Jato JL. Effect of chitosan and chitosan glutamate enhancing the dissolution properties of the poorly water soluble drug nifedipine. Int J Pharm. 1998;175:75–84. doi: 10.1016/S0378-5173(98)00245-2. [DOI] [Google Scholar]
- 17.Sawayanagi Y, Nambu N, Nagai T. Dissolution properties and bioavailability of phenytoin from ground mixtures with chitin or chitosan. Chem Pharm Bull. 1983;31:2062–2068. doi: 10.1248/cpb.31.2064. [DOI] [PubMed] [Google Scholar]
- 18.Sheng Y, Zheng F, Zhong F. The effect of chitosan on dissolution properties of griseofulvin. J China Pharm Univ. 1993;24(6):376–379. [Google Scholar]
- 19.Maestrelli F, Cirri M, Mennini N, Zerrouk N, Mura P. Improvement of oxaprozin solubility and permeability by the combined use of cyclodextrin, chitosan, and bile components. Eur J Pharm Biopharm. 2011;78:385–393. doi: 10.1016/j.ejpb.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 20.Corti G, Maestrelli F, Cirri M, Mura P, Zerrouk N. Dissolution and permeation properties of naproxen from solid-state systems with chitosan. Drug Deliv. 2008;15:303–312. doi: 10.1080/10717540802006955. [DOI] [PubMed] [Google Scholar]
- 21.Brewster ME, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007;59:645–666. doi: 10.1016/j.addr.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 22.Lina S, Hsua C, Sheub M. Curve-fitting FTIR studies of loratadine/hydroxypropyl-β-cyclodextrin inclusion complex induced by co-grinding process. J Pharm Biomed Anal. 2010;53:799–803. doi: 10.1016/j.jpba.2010.06.010. [DOI] [PubMed] [Google Scholar]