

Abstract
Polymers are extensively used in the pharmaceutical and medical field because of their unique and phenomenal properties that they display. They are capable of demonstrating drug delivery properties that are smart and novel, such properties that are not achievable by employing the conventional excipients. Appropriately, polymeric refabrication remains at the forefront of process technology development in an endeavor to produce more useful pharmaceutical and medical products because of the multitudes of smart properties that can be attained through the alteration of polymers. Small alterations to a polymer by either addition, subtraction, self-reaction, or cross reaction with other entities have the capability of generating polymers with properties that are at the level to enable the creation of novel pharmaceutical and medical products. Properties such as stimuli-responsiveness, site targeting, and chronotherapeutics are no longer figures of imaginations but have become a reality through utilizing processes of polymer refabrication. This article has sought to review the different techniques that have been employed in polymeric refabrication to produce superior products in the pharmaceutical and medical disciplines. Techniques such as grafting, blending, interpenetrating polymers networks, and synthesis of polymer complexes will be viewed from a pharmaceutical and medical perspective along with their synthetic process required to attain these products. In addition to this, each process will be evaluated according to its salient features, impeding features, and the role they play in improving current medical devices and procedures.
KEY WORDS: alteration, blending, drugs, grafting, interpenetrating polymer networks, medicine, pharmaceutical, polymer complexes, polymer modification
INTRODUCTION
The demand for safer and more efficient products for pharmaceutical and medical applications has increased over the years. Consequently, their exploration has become a fundamental area of research with great importance being placed on exploring novel systems for the enhanced delivery of drugs and for improved medical applicators. Among these, polymeric systems are of particular interest for a wide expanse of medical applications such as drug delivery as they predominantly demonstrate enhanced pharmacokinetic and pharmacodynamic parameters over conventional drug delivery systems (1,2). In addition, polymeric medical systems/devices comprehensively demonstrate improved biocompatibility, strength, stability, and biodegradability over their respected conventional systems (1,2). A few of the captivating properties of polymeric systems for pharmaceutical and medical applications are extraordinarily elevated stability, variable controllable solubility, superior structural design, lowered immunogenicity, enhanced biocompatibility and cytocompatibility, three-dimensional geometry, antigenicity, and often specific tissue/cell targeting (2,3).
Currently, the biomedical market has numerous drug delivery systems and medical devices synthesized from polymeric sources (4). These polymeric systems and devices are therapeutically more advantageous as well as superior in other aspects such as application, durability, and efficiency. Despite having all these smart systems, there is still a developing demand for novel or altered polymers that can accommodate for enhanced and newer parameters. These novel polymers will enable the gratification of a comprehensive array of medical applications. As a result, polymer refabrication remains an active area of process technology development in an endeavor to produce more beneficial pharmaceutical and medical products.
Furthermore, each polymeric systems has strengths, but they also have limitations, and by altering these polymers, it is possible to embellish the polymers strengths and to integument its limitations. Polymeric alteration can take place in a great number of ways whether reversible or irreversible most of which are by mixing, chemically altering, or exposure to external factors such as electromagnetic radiation, gamma radiation, or enzymes. Discovering and implementing various approaches to manipulate the architectural and functional operation and performance of polymeric materials is an imperative exigency for the delineation of advanced polymeric materials (5,6). These approaches permit for the fastidious and precise modification of polymeric properties, parameters, functions, and performance (5,6). By conscientiously selecting the appropriate constituent(s) for modification and by selecting the optimum process for the constituent’s alteration whether reversibly or irreversibly, the physical prosperities can be altered, enhanced, removed, or created (5,7). These changes satiate a more comprehensive variety of applications in the pharmaceutical and medical disciplines for modification (5,7). Together with the above, if the most appropriate technique is selected, the altered product can be attained while still maintaining a significant amount of cost-effectiveness.
Another important factor that is become increasingly prominent is the emphasis on the employment of natural polymers as they are acquired from renewable sources consequently decreasing the environmental concerns. However, to derive expedient behavior from natural polymers for their utilization in the pharmaceutical and medical disciplines, some alteration process are initially required before they can synergistically function with active ingredients (8). In addition, the hybridization of natural polymers with synthetic polymers has displayed phenomenal significance in the pharmaceutical and medical disciplines achieved by interfusing natural polymers with synthetic polymers (9). To achieve this, the polymers in question need to undergo processes that allow for the conglomeration of natural and synthetic polymers (9). Alteration techniques such as grafting or interpenetrating polymer networks increase the compatibility between the different classes of polymers (9).
In addition, it has been observed that immediate and/or sustained release drug delivery systems are not always convenient because of the extraordinary complexities of certain disease states (10). In such cases, it is preferred to use a method of treatment that employs an externally modulated drug release system (10). These smart delivery systems are formally known as stimuli-responsive delivery systems and are regulated by the application or change in certain parameters such as temperature, pH, or body metabolites demonstrating an enhanced prognosis (10). To attain such a delivery system, some technique of polymeric alteration needs to be performed to actualize a drug delivery system with such ingenuity.
Accordingly, this review undertakes to discuss the practicality of polymeric refabrication in the advancement of more beneficial products within the pharmaceutical and medical disciplines. It aims to discuss how the various techniques of modification of polymers is performed to engineer polymers that exhibit a desired property such as stimuli-sensitivity, enhanced strength, increased biocompatibility, and altered properties on the whole. In addition, the different techniques employed for polymer processing are also concisely described to enable the reader to gain an understanding of how these processes can enable one to achieve a desired outcome in the disciplines of pharmaceutics and medicine. Simultaneously, this review seeks to provide a concise overview on the application of polymeric refabrication techniques. Applications such as tissue engineering scaffolds, drug release vehicles, and specific examples of how the polymer modifications have been translated into devices used in animal or human studies.
METHODOLOGICAL APPROACHES AND CATEGORIZATION OF POLYMERIC REFABRICATION TECHNIQUES
There are many different methods by which polymers are being altered at a molecular level to provide altered pharmacochemical and pharmacodynamic parameters; the major techniques are defined in Table I. Expedient properties can be formulated by the appropriate selection of constituents and an acceptable method of alteration and processing therefore permitting a comprehensive array of application.
Table I.
Major Types of Polymeric Refabrication Techniques and Their Respective Definitions
| Technique | Definition | Product |
|---|---|---|
| Grafting | Grafting is process where a parent polymer is used as a backbone onto which branches of a second polymer are attached at different points along the parent polymer backbone consequently altering the surface properties of the parent polymer (11,12) | Formation of a polymer with altered surface properties while still maintaining the bulk properties of the parent polymer (11,12) |
| Blending | This technique is a relatively simplistic process as it only encompasses the mechanical mixing of two or more polymers to form a mixture that displays different characteristics to each separate constituent (13–16) | Formation of a polymer mixture devoid of permanent bonds that exhibits unique properties to the individual constituents (14,17) |
| Interpenetrating polymer networks | An IPN is an amalgamation of two or more polymers that coordinates into a network devoid of covalent bonds and where at least one polymer is polymerized and/or cross-linked within the microenvironment of the other polymer(s) (18–20) | Formation of a product much similar to a blend, however, IPNs contain cross-links appropriately inferring a greater stability to them (9,18,21) |
| Polymer complexes | In general it is the synthesis of a complex between polymers with opposite characteristics such as ionic charges, stereo-conformation, or charge transfers (22–28) | Formation of a polymer complex that has different properties to either complexation agent (22–28) |
Polymer Grafting
This is a process whereby the external or surface layer of a polymer is altered while it retains its bulk properties as shown in Fig. 1 (12). This type of polymer modification is being pursued in pharmaceutical laboratories throughout the world because of many applications which would be impacted by the ability to only tailor surface properties of a material. It huge impact is due to the surface being the first entity to makes contact with the body and hence dictates the initial biological response to the foreign body (11,12,30–32). It is this biological surface reaction that mediates the performance of a delivery device; for example, a contact lens or eye delivery system will damage the cornea of the eye if the device is not wettable with tears. Likewise, the ability of an immune adsorbent is dependent on the affinity of the adsorbent to preferentially bind to pathogenic moieties over binding to proteins and body fluids which will block the binding sites consequently decreasing its efficacy (12,30–32). Many biomaterials are not clinically pertinent, and an effective way of formulating them to become clinically effective is by altering the surface properties. One such way is by grafting appropriate moieties onto the polymer surface to alter, enhance, or remove properties of the bulk polymer (30,32). In addition, the grafting technique provides a stable platform against desorption and establishes long-term chemical stability because of its covalent nature in contrast to surface coating modifications which are unstable over time and do not prevent desorption (32). Some of the surface characteristics being successfully manipulated by grafting include chemical resistance, wettability, biocompatibility, and dyeability allowing for their utilization in countless pharmaceutical areas (29). Grafting desired functionalities onto the surface of materials to render them capable of binding enzymes, proteins, and similar species has yielded products useful for site-specific targeted drug delivery systems (33). There are different types of polymeric grafting processes in place, and the main techniques are summarized in Table II.
Fig. 1.

Illustration of the general grafting technique of polymer re-fabrication to alter surface properties of polymers while retaining their bulk properties; monomer is irreversibly added to the polymer backbone (adapted from (29))
Table II.
Summary of the Different Types of Polymeric Grafting Methods, Outlining the Core Processes, Salient Features, and Impeding Features of Each Major Technique
| Technique | Sub-techniques | Core process | Salient features | Impeding features |
|---|---|---|---|---|
| Chemical grafting | Free radical/ionic | Generation of radicals/ions with chemicals (34) | Alters surface properties and not bulk properties (12) | Synthesis of homopolymer as a by-product difficult to extract |
| Living polymerization | Propagating moiety takes no part in chain transfer/termination allowing > graft control (35) | Grafting is controlled, synthesizing uniform, narrow M w distribution grafts (4,36) | By-products less pronounced (37–39), use of harsh chemicals | |
| Photo-initiated grafting | Generation of radicals by direct irradiation via UV/microwaves (40,41) | Direct radical generation eliminates need of initiators and washout steps (42,43) | Irradiation is non-penetrative and only allows surface modification (42,43) | |
| Enzymatic grafting | Generation of radicals by enzymes (44) | High specificity allows the synthesis of pure products without homopolymers (45,46) | Enzymes require narrow range of pH, temp and conc. to work optimally (45,46) | |
| Radiation grafting | Free radical/ionic | Generation of radicals/ions by gamma radiation (11,47) | Direct radical/ion generation eliminates the need of initiators and washout steps (48) | Serious degradation and/or decomposition of the polymer can occur (49–51) |
Chemical Grafting
Chemical grafting is one of the most widely used methods of grafting and is probably in the forefront of manufacturing processes being utilized (52). Grafting by chemical means can advance in two distinct pathways, namely free radical generation and/or ionic generation methods (32,53,54). In chemical means of grafting, a great importance is placed on the initiatory chemical as it is this entity that establishes the principle pathway by which the grafting process will eventually occur (40,41,53). The grafting process of a polymer can be accomplished under different conditions of the monomer: The parent polymer can be treated with an aqueous solution, vapor phase, or plasma phase of the monomer (31,41,53). However, the major limitation confronted and the explanation for the privation of its extensive employment in industrial graft copolymerization, expressly with the free radical-based systems, is the concurrent formation of homopolymer (11,38,55,56). Homopolymer is inherently unattached polymer having the equivalent chemical structure as that of the grafted chains grown from the parent polymer where there is actual covalent bonding between the parent polymer backbone and the monomer (11,38,52,55). The leading incitement of homopolymer synthesis is the generation of nonspecific macroradicals, isomerization of intermediate radicals, and chain transfers of monomer from growing grafted chain ends (56). In addition, not only is homopolymer an extrinsic expenditure of monomer, but the purification of the grafted polymer from the parent polymer/homopolymer is notably an intricate process as their properties are appreciably similar. This similarity makes their separation a substantially arduous process that requires the employment of solvents that dissolve only one or the other and consequently also introducing complications in the characterizing of the graft derivative (11,38,52,55).
Free Radical Grafting via a Chemical Approach
In this process, free radicals are generated from the initiator chemicals. These free radicals are intermediary molecules or molecular fragments having at least a single unpaired electron which provides an appreciable measure of reactivity to the molecule capable of triggering a reaction (34). The reaction that eventuates is the transfer of this high energy radical to a substrate polymer that can subsequently react with monomer composites to effect a modification on the polymer surface presenting a grafted product (32). There are two ways in which radicals can be formed, directly on the initiator chemical or indirectly through redox reaction generating active species other than the initiator chemical such as reactive oxygen (57). The grafting process through direct radical formation is summarized in Table III, and a reaction scheme depicting the mechanism by which the initiator chemical, di-tert-butyl peroxide, is employed to activate the grafting process is demonstrated in Fig. 2.
Table III.
A Summary of the Grafting Process Presenting the Possible Mechanisms of the Initiation, Propagation, and Termination Processes [Adapted from (58)]
| Initiator steps | |
| Initiator chemicals (I2) are decomposed to generate radicals (R˙) | |
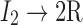
|
(Eq. 1) |
| Radicals can transfer electrons to monomer (M) to synthesize monomer radicals (M˙) | |
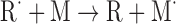
|
(Eq. 2) |
| Propagation steps | |
| Addition of monomer to radical | |
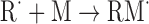
|
(Eq. 3) |
| Increasing the graft size | |
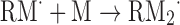
|
(Eq. 4) |
| Or reaction with a polymer molecule for a single monomer graft | |
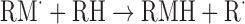
|
(Eq. 5) |
| The new R˙ can continue to propagate as according to Eq. (3) | |
| Intramolecular hydrogen abstraction | |
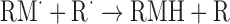
|
(Eq. 6) |
| Termination steps | |
| Loss of main propagation species | |
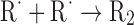
|
(Eq. 7) |
| Loss of radicals | |
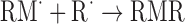
|
(Eq. 8) |
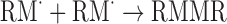
|
(Eq. 9) |
Fig. 2.

Reaction scheme showing the generation of radicals from the initiator chemical, di-tert-butyl peroxide by action of heat. The radical formation is according to Table III: Eq. 1, and the grafting process can proceed as according to Table III (29)
Common initiators employed for the generation of free radicals and with considerable success include ammonium persulfate, potassium persulfate, ceric ammonium nitrate, dibenzoyl peroxide, and azobisisobutyronitrile at temperatures up to 80°C (54,59). As with any manufacturing technique, lower temperatures are often preferred as it provides a cost-effective method of production considering heating is an extremely expensive procedure. In addition, lower temperatures also decrease the incidence of homopolymer synthesis because at elevated temperatures, radical–radical reintegration is intensified as according to Table III: Eq. 7, as compared to lower temperatures where radical flux presents at a much higher rate (52,60). However, certain reactions require temperatures above 200°C as these polymers need to attain a molten state for a feasible reaction to transpire (58). For example, maleic anhydride is grafted to polyethylene by employing di-tert-butyl peroxide as the initiator, but the reaction can only be performed directly at 200°C when the polymer is in a molten state (40,53,59). Despite the attempt to refrain from the use of high temperatures because of rising production costs, the higher temperatures can influence the rate constants of the initiation and propagation reactions. In addition, higher temperatures can dictate that the depropagation is the dominating factor thus ensuring that grafts are relatively short and that homopolymerization of the monomer is not a significant side reaction subsequently producing a higher yield of uniform product (59).
Although several measures can be appropriated to prevent side reactions, the practicality is that many side reactions can eventuate thus appreciably limiting the use of the radical initiated grafting technique (40,41). During the initiation process, the R˙ radicals that are synthesized can spontaneously undergo isomerization such as intramolecular hydrogen abstraction prior to monomer addition according to Table III: Eq. 6 (41,59). This isomerization changes the position at which the monomer attaches to the parent polymer thus synthesizing a byproduct that adulterates the expected product. Consequently, the purity of the product is decreased and could render it unsuitable for human consumption and ultimately preventing its use in pharmaceutical and medical applications (59). In addition to the above limitation, this conventional technique of free-radical polymerization requires a continuous flow of initiatory chemicals. That is to say, a continuous initiation is necessary for chain growth, and for the termination of the grafting process to transpire, either radical coupling or disproportionation reactions should eventuate (40,55). Concertedly these two reactions incite unreactive (“dead”) polymers, accordingly developing time invariant degrees of polymerization that emanate into expansive molecular weight distributions (56,59).
Ionic Grafting via a Chemical Approach
This process is very similar to the free radical method of grafting; however, the initiator chemicals do not form free radicals to initiate the grafting process, but instead they generate ionic (cationic or anionic) centers that initiate the grafting process (52). Initiator chemicals such as tertiary butyl phosphazene (t-BuP4) have been employed in the anionic modification of polyethylene oxide by generating a cationic center on the initiator molecule by reaction with acid as depicted in Fig. 3 (52).
Fig. 3.
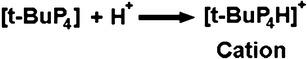
Illustration of the formation of a cationic centre on tertiary butyl phosphazene required for the ignition of the ionic grafting process (adapted from (52))
Living Polymerization Grafting via a Chemical Approach
There are various different categories of “living” polymerization techniques. These processes are also known as controlled systems because transfer and termination reactions are more controlled as compared to conventional radical polymerization systems thus producing more uniform grafted polymers with a narrow molecular weight distribution (6,11,36). This control is achieved because in living polymerization, chain growth is achieved by an active propagation moiety that plays no role in chain transfer or termination reactions (11,35). In contrast, conventional radical polymerization has numerous reactions transpiring simultaneously, specifically, initiation, propagation, termination, and chain transfer (11,35) One unique characteristic and distinguishing trait of a living system is that the active propagation moiety maintains a consistent concentration for the infinite duration of the polymerization process (11,35,53). Once 100% monomer conversion is achieved, any addition of supplementary monomer to the system results in a continuation of the polymerization process again until 100% monomer conversion is achieved (11,35,53). This results in a linear relationship being obtained when the molecular weight of the grafted polymer is plotted against the concentration of monomer conversion that has transpired (11,35,36,53).
In pharmaceutics, the living polymerization technique has been utilized in the formation of poly(N-isopropylacrylamide), a thermoresponsive polymer that alters its structure and density of its cross-links according to the environmental temperature consequently altering its solubility at different temperatures that it is subjected to (39). The conventional polymerization technique employed peroxide initiators that produced a size variant product in contrast with the living system which produces a more consistent size distribution appropriately producing a more sensitive temperature-dependent product (39). The living process employs azo-initiators and dithioesters as chain transfer agents (39). There are many advantages of implementing a living system for radical-based grafting. Specific macroradical formation can be effectuated at pertinent sites along the parent polymer backbone allowing for an enhanced controlled propagation of the polymer chain resulting in uniform graft products (11,39,56). In addition, when the monomer-to-initiator ratio is low, chain transfer is attenuated at the growing chain ends resulting in a narrower molecular weight distribution consequently generating a polymer with superior uniform properties (36,56). The above features also restrain the synthesis of homopolymerization because the two main sources of homopolymer in the radical grafting technique are nonspecific macroradical generation, isomerization, and chain transfer of growing grafted chain ends (36,39). However, this is not to say that this technique is a conclusive deterrent to homopolymer synthesis and in application homopolymer is often found as a byproduct of these systems consequently contaminating and decreasing the product yield (36,39).
Photo-initiated Grafting
The photo-initiated grafting technique is comparable to the free radical grafting technique with the exception that free radicals are generated by the employment of electromagnetic radiation such as ultraviolet light or microwaves. It is a rather simplistic and effortless process requiring only a limited number of steps to accomplish a graft product as depicted in Fig. 4 where ethyl cellulose is affected by electromagnetic radiation to generate a free radical capable of reacting with acrylonitrile (31,42,43,61). The photo-initiated grafting process can advance via two different approaches, either by employment or devoid of a photosensitizer depending on the properties of the polymer in question (36,43,62,63). The mechanism is a relatively simplistic process where a macromolecule/polymer or a chromophore/sensitizer absorbs light energy and acquires an excited state/high energy state (43,61). This high energy state then progresses to cause dissociation in the molecule thereby generating free radicals and initiating the grafting process (43,61). This process can occur directly on the polymer itself if it is photolabile (31,43,61). However, if the absorption of light does not lead to the direct synthesis of free radicals on the polymer through bond cleavage, then the process can be advocated by adding photosensitizes such as benzophenone, benzoin ethyl ether, or dyes such as acrylated dye as previously specified (31,43,63). Photosensitizers are capable of absorbing photo-energy and subsequently imparting that energy onto the polymer to effectuate the grafting process (43,63). This photo-initiated technique to attain a graft product is often favorable as it simply requires brief irradiation times and the equipment required is relatively economical (31,36). There are many types of photo sources all of which are encompassed in the electromagnetic spectra; however, many of the sources do not provide enough energy to cause free radical generation or the number of bounds affected is limited, as such the two main sources employed are ultraviolet and microwave.
Fig. 4.

Photoinitiated grafting producing radicals on the backbone of polyethylene without the employment of sensitizer and consequent addition of acrylonitrile monomers to produce a graft (adapted from (29))
Ultraviolet Induced Photo-initiated Grafting
The most promising form of photo-initiated grafting is by the employment of ultraviolet radiation (62). This is a surface modification technique that is employed to attach monomers onto a polymeric surface with the intention of constructing an entity that has capabilities unlike the parent polymer. The surface is changed to allow for surface wettability, increased absorption of dyes, and/or increased adhesion properties (43,63,64). The inert parent polymer surface is activated by exposure to ultraviolet radiation generating free radicals and enabling the reaction with monomer consequently functionalizing the surface of the polymer (64). In general, there are two main methods by which ultraviolet modification of polymer surfaces can be accomplishing: (a) irradiation of the polymer in the immediate presence of a monomer containing solution and (b) pre-irradiation method. The first technique is rather self-explanatory where the reaction takes place upon irradiation. In the second technique, the parent polymer is irradiated individually to generate reactive radical sites or peroxides on the surface of the polymer. This is then exposed to a monomer solution in the absence of irradiation where the generated reactive sites on the parent polymer have the capacity to eventualize a reaction with the monomer solution. This technique of polymer modification was exploited for the creation of an altered styrene–butadiene–styrene (SBS) triblock copolymer membrane in an attempt to enhance the hydrophilicity of a membrane (64,65). The SBS membrane was synthesized by solvent casting and then exposed to ultraviolet radiation after which the polymer was treated with monomer to initialize the actual grafting process (64,65). Fourier transform infrared performed during the study displayed that the photo-initiated grafting technique by employment of ultraviolet radiation was successful, and this was confirmed by the contact angle measurement which had decreased after the membrane was exposed to irradiation and the monomer solution (65). As previously mentioned, this is a rather easy process as there is no complex steps required to effectuate the chemical reaction, and the process is devoid of harsh chemical treatments (64,65).
Microwave Induced Photo-initiated Grafting
Another technique that is popular within the photo-initiated grafting techniques is the employment of microwave radiation. This technique is comparable to the ultraviolet technique as it also includes the radiation of a sample with electromagnetic radiation, but differs in the electromagnetic radiation source which in this case is microwaves. The same steps and conditions are required as with any electromagnetic radiation technique. However, as with any radical-based grafting system, the same complications regarding homopolymerization and isomerization apply, and to complicate homopolymerization further, the grafting process only occurs at the point of incidence of photons (42). Areas where the incident radiation was absent, monomer cannot attach to the polymer resulting in further synthesis of homopolymer (42). In addition, photo-systems do not have a great penetration depth; therefore, perplexities are encountered when considerable magnitudes of polymer need to be grafted corresponding to industrial amounts (31,42,43,61).
Enzymatic Grafting
This technique of grafting is relatively recent compared to other technique and it employs an enzyme to initiates the grafting process (66). The need to develop this method originated from the increase in safety and environmental considerations as the chemical means of grafting extensively utilizes highly reactive reagents under harsh conditions which are environmentally hazardous (8,44). In addition, the products require additional steps of product purification to remove these hazardous chemicals ensuring it is safe for use in humans (8,44). The application of enzymes is conceivably an environmentally cleaner process and offers a better product due to the known selectivity of enzymes that leads to the formation of an almost pure modified polymer product (5,8,45). The use of enzymes leads to the acquisition of a more efficient product for employment in the pharmaceutical and medical disciplines (5,8,45). This selectivity also offers the potentiality for improved control and an economically viable technique of grafting functionalities as there are no requirements for wasteful protection/deprotection procedures (44,54,66–68)
One captivating class of enzymes that has been employed in grafting of biopolymers is the oxidative enzymes such as peroxidases or laccases (44). These enzymes possess the ability to generate radicals on polymer backbones which can then react with monomer to achieve the grafting process in a domino type reaction (44,69). Domino reactions present when sequential processes occur. The first process is the formation or breaking of bonds followed by a subsequent process which occurs at the functionalities generated in the preceding step. Consequently, these two steps together circumvent the need for exuberant purification processes which would have had to occur after each synthetic step, resulting in an economical process (69). One example of enzymatic initiated grafting is with the use of tyrosinase, a peroxidase enzyme (8,46,70,71). Tyrosinase is capable of converting phenol via a two-step mechanism into o-quinone, a reactive species that is freely diffusible and can actualize a non-enzymatic reaction with the nucleophilic amino groups of chitosan (8,46,67,70,71). This modified chitosan can then be incorporated into a drug delivery system that will exhibit altered physicochemical and physicomechanical properties to the parent chitosan such as making chitosan soluble in both acidic and basic media where the parent polymer is only soluble in acidic media as shown in Fig. 5 (54,67). This allows for the creation of immediate release drug delivery systems with the enzymatic modified polymer. Since the enzyme is required for modification to take place, the generation of o-quinone is the rate determining step as it relies on the activity of the enzyme (8,72,73). Consequently, all elements that promote their synthesis will complement the grafting process and increase the rate at which grafting is achieved (8,72,73).
Fig. 5.

Solubility behavior of chitosan and chlorogenic acid-modified chitosan. Chlorogenic acid-modified chitosan was prepared using the conditions described in the text, and this modified chitosan was observed to be soluble under acidic and basic conditions. At neutral pH, the chlorogenic acid-modified chitosan is insoluble, and the precipitate was allowed to settle for 0.5 h before taking the photograph. Unmodified chitosan is soluble at low pH, while large precipitates were observed when the pH was adjusted to 7. When the pH of a dissolved chitosan solution was rapidly adjusted from acidic to basic conditions (pH 4.9), a gel is formed (reprinted with permission from (67) © 1999 John Wiley & Sons, Inc.)
This concept of adopting enzymes is astonishingly captivating for various reasons. Firstly, tyrosinase has an extensive substrate range, and it can oxidize a diverse number of low molecular weight natural and synthetic phenols as well as high molecular weight oligomers and phenolic containing polymers and even cause the cross-linking of these reagents (67,70,71). Secondly, the enzyme only requires simple co-substrate requirements employing molecular oxygen as the only oxidant (70). Thirdly, the enzyme tyrosinase has the capability of converting primarily unreactive phenolic substrates into reactive o-quinones that are capable of executing a non-enzymatic reaction resulting in the emanation of the grafting process (44,68,70). Finally, by using this “domino” effect reaction mechanism, the steric complications conventionally experienced when enzymes are employed in the catalysis of high molecular weight non-physiological substrates (e.g., for synthesizing and functionalizing polymers) are eliminated (66,69,73). This is largely a result of the generation of reactive o-quinones being reliant on the chemical that eventuates the o-quinone rather than the polymer being grafted (66,69,73).
Despite the many attractions provided by the enzymatic modification technique, it has some debilitating limitations considering the narrow pH, temperature, and concentration ranges for enzyme activity (45,46,66,74). For optimum enzymatic activity to transpire a chemical reaction, the microenvironment must be in a liquid state at the optimum temperature and pH for the particular enzyme being employed (45,46,66,74). These variable ranges are very narrow and even minute changes in pH and temperature can significantly incite the reaction yield significantly (8,46,74). To further augment the complication, the acquisition of a high yield of the grafted polymer requires a pH that not only accommodates the enzymes variable range but in addition it must compliment the monomers and the polymer to be grafted as many moieties are affected by pH (8,45,66). The pH often changes the molecules charge, reactivity, and possible configuration introducing complications (8,45,66). Thus, an appropriate solvent must be selected that not only dissolves and work synergistically with the substrates but additionally must maintain enzyme activity/stability by displaying the optimum pH required for enzymatic action (45,66,70). Besides the above limitations, the most debilitating limitation of enzymatic refabrication is the characterization of the products synthesized as it is difficult to control the reactions of o-quinones and without proper control, grafts can be produced with a wide weight distribution range (67,72,73).
Radiation Grafting
High-energy γ-radiation causes changes to polymeric structures as well as other macromolecules causing the induction of radicals, cations, and free electrons much like the photo-initiated technique of grafting (11,47). These high energy sites/radicals on a polymer backbone can then react with monomer to synthesize a grafted chain polymer in the same manner as explained before (11,47,48). This process is extremely resourceful as it eliminates the requirement for initiator chemicals thereby excluding further contamination, has a greater penetration depth than photo-initiated means, and only requires polymer, monomer, and occasionally a suitable solvent (11,47,70). Radiation grafting can progress along two pathways, free radical or ionic production much like chemical means of grafting except that the intermediates mentioned are generated via excitation actuated by the radiation in a dry, semi-dry, or wet process (47,49). This radiation technique has been employed in medicine by inducing the cross-linking of polyethylene oxide to effectuate heat shrinkable properties in the polymer (11). This shrinkage is employed in the connection of blood vessels in a sutureless method and also to create a pH-sensitive device from polyethylene oxide for ovarian cancer targeted delivery systems (11).
Free Radical Grafting via a Radiation Approach
The irradiation of macromolecules can cause homolytic fission generating free radicals on the polymer itself (48). This is the only grafting process where an initiator is not imperative (48). However, the medium in which this technique is carried out is important (48,49). If irradiation is carried out in air, then peroxides maybe formed on the polymer due to oxygen in the atmosphere (49). The lifetime of the free radical depends on the backbone nature of the polymer. Grafting is feasible in three different ways, namely (a) pre-irradiation, (b) peroxidation, and (c) mutual irradiation approach. In the pre-irradiation approach, the parent polymer is irradiated in a vacuum or under an inert atmosphere such as nitrogen to generate free radicals (48,49,75). The irradiated polymer is then amalgamated with the monomer as a liquid, solution in solvent, or gas state to initiate grafting (48,49,75). In the peroxidation approach, the parent polymer is irradiated in the presence of air or oxygen to form stable hydroperoxides or diperoxides (49,50). These oxides are then amalgamated with monomer at elevated temperatures at which the peroxides are capable of decomposing to form radicals required to initiate the grafting process (49,50). The final approach is mutual irradiation where both the parent polymer and the monomer are simultaneously subject to radiation to form free radicals subsequent causing annexation of the two to produce a graft (75). The limitation of this method is in the actual process. Radicals are generated by electron abstraction; naturally radical generation is considerably unselective and is constrained along the path of the incident radiation beam; it follows that radical generation is desultory (11,49,51). In addition, radiation is very deleterious and its employment to certain polymers can result in serious degradation and decomposition of the polymer (11,49,51). However, despite having no selectivity, there are no synthetic steps to be performed but like previous methods of radical grafting there remains the hazard of homopolymers formation and isomerization (11).
Ionic Grating via a Radiation Approach
Ionic grafting can proceed in two different ways, namely cationic and anionic. The polymer is irradiated to form the polymeric ion and then reacted with monomer to form the grafted polymer (49,51). The potential of radiation grafting over other methods is high reaction rates; thus, minute radiation times can produce a significant amount of grafting (48,49,75).
Co-blending in Polymer Refabrication
The polymer blending process is another technique that has been extensively employed in polymeric refabrication to improve properties of polymers (16,76,77). Beneficial properties can be acquired by incorporating the correct polymers into the blend providing an economically favorable method over synthesizing a new polymeric resin because it is simply the mechanically mixing of two or more polymers together as shown in Fig. 6 (13–16,76).
Fig. 6.
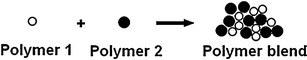
Illustration of a mechanical blend constituted of two miscible polymers with different chemical structures (adapted from (29))
Polyethylene oxide (PEO) is a low toxic synthetic uncharged polymer that is mucoadhesive because of its hydrophilicity and high viscosity; therefore, it has been comprehensively employed in drug delivery systems (37,77,78). However, pure PEO films have unsatisfactory mechanical and physical characteristics, and their extreme water solubility greatly delimitates their employment in sustained pharmaceutical products (14,39,77). To improve PEO’s use in drug delivery systems, chitosan has been blended together with it to synthesize films that express the capacity to be employed in sustained release delivery systems (16,37,77,78). The chitosan component of the blend decreases the water solubility of the films by limiting water permeability into the film; in addition, chitosan also deliberates an antimicrobial effect and increases the flexibility of polyethylene oxide therefore making it even more useful in the pharmaceutical and medical disciplines (16,37,77,78).
There are many different types of polymer blends available, but they may be classified into two general classes as either miscible or immiscible blends, depending on the interactional behavior of the polymers that constitute the blend (16,21,39). Miscible polymer blends are comparable to homopolymers or random copolymers as they have a degree of altered properties with the absence of covalent or permanent bonds (14,17). Immiscible polymer systems are frequently identified by multiple glass transition temperatures owing to the distinct separation between the constituents (14,17). The properties of immiscible blends are primarily controlled by morphology, which is consequently dependent on the thermodynamic, rheological, deformation, and thermal history properties (15,17). To increase the miscibility of polymers in a blend, the use of a compatibilizer such as clays is employed, which is an additive that physically reduces interfacial tension between the polymers subsequently increasing the interactional forces between the constituents (14,17,79).
The major limitation of polymer blends is that it demonstrates a multiphase nature and this variation in structure infers that flow responses could possibly be complex (14,17,39,79). Consequently, the performance of processing and fabrication of blends depends on the knowledge of not only the parameters controlling the homogeneity of the polymer system (such as molecular weights, their size distribution and thermal and stress–strain profiles) but also on morphology and its evolution (17,80). Theoretical and experimental studies indicate that complete determination of morphology must include viscosity and elasticity, interfacial tension, composition, and the thermal profile to provide minimal structural defects such as phase separation (15,39,76,80). This seems easy enough as most data have previously been collected; however, most data have been collected on infinitely diluted Newtonian fluids, and blending is invariably done at high concentration of viscoelastic liquids (79–81). In such systems, the shape of the droplets is not only influenced by the dissipative, viscous forces, but also by the pressure distribution around the droplet arising from the elasticity (79–81). Therefore, the characteristics of drop deformation could be quite contradistinctive from those in Newtonian systems appropriately introducing complications in understanding the process dynamics (79–81).
Newer blends are using preprocessed constituents to provide a final result with unique characteristics to those of the constituents. One such are being explored is by combining nano-composites into a blends (13). Such system can form exceptional characteristics as they will exaggerate properties originating from the polymer blend in addition to presenting unique characteristics of nano-composites (13). It is worth mentioning that from a simple blend, the generation of products that fall under other systems becomes a rather easy task. By simply introducing a cross-linking agent into a blend, it is possible to form an interpenetrating polymer network as explained in the following section. In addition by creating a system that causes the blend constituents to behave oppositely in some regard and form interpolymeric bonds, it is possible to form a polymer complex. Some opposite behaviors that are possible to create are the charge on each polymer, or maybe the stereochemistry of the constituents; in certain cases, the complex formation can be spontaneous and in other cases it may need to be advocated by some external influence, as will be elaborated in a later section.
Interpenetrating Networks in Polymer Refabrication
An interpenetrating polymer network (IPN) is an amalgamation of two or more polymers that coordinates into a network devoid of covalent bonds and where at least one polymer is polymerized and/or cross-linked within the microenvironment of the other polymer(s) preferably synthesizing permanent mesh formations as depicted in Fig. 7 (18–20). The principle use of IPNs is to deliberate novel characteristics to one of the constituents while preserving the imperative characteristics of the other constituent (7,20). There are numerous categories of IPNs such as semi-IPNs, sequential IPNs, simultaneous IPNs, and gradient IPNs to mention only a few; however, IPNs will be discussed as a single extensive category herein, and for understanding purposes, a few definitions of the main types of IPNs are displayed in Table IV.
Fig. 7.

a Depicts a semi-interpenetrating polymer network where only one polymer is cross-linked in the presence of a linear polymer and b depicts an interpenetrating polymer network where both polymers are cross-linked (adapted from (18))
Table IV.
Definitions of Four Types of IPNs, Where Full-IPNs and Semi-IPNs Are the Two Main Characterizations Under Which Most Other Types of IPNs Can Fall Under (82)
| IPN type | Definition |
|---|---|
| Full-IPN | A polymer combination consisting of 2 or more networks which are at minimal partially interlaced at a molecular level but absent of covalent bonds and cannot be separated unless chemical bonds are broken. A mixture of 2 or more preformed polymer networks is not an IPN. |
| Semi-IPN | A polymer combination comprising of 1 or more networks and 1 or more linear or branched polymer(s) where the linear component is locked into the network component. |
| Sequential IPN | A polymer combination synthesized by polymerizing an initial mixture of monomer with a cross-linking agent to form a network. This network is then swollen with a second combination of monomer and cross-linking agent and polymerized to form an IPN. |
| Simultaneous IPN | An IPN formed by polymerizing 2 different monomers and cross-linking agents in 1 step. The 2 constituents must polymerize without interfering with each other. |
IPN interpenetrating polymer network
Certain IPNs may have a similar end product, but the method of synthesis differs thereby characterizing them differently such as sequential IPNs and simultaneous IPNs where their respective syntheses. Although the end products are the same, the difference in the synthesis method gives rise to subtle differences such as swellability. Simultaneous IPNs are capable of swelling to a greater extent as compared to sequential IPNs since the later lacks the intermediate swelling step which causes a less relaxed network to be formed. In addition, the differing methods allows for certain procedures to be accomplished in some methods and not others. For example, IPNs synthesized via the simultaneous reaction is straightforward as it only requires one step and permits injection molding of the product while the sequential method requires a two-step reaction and necessitates either compression molding or the formation of IPN beads (82). The materials produced by the two methods are not exactly the same either. Due to the intermediate swelling step in the sequential process, the first network formed usually has an extended chain conformation. The networks in simultaneous IPNs (SINs) usually have relaxed conformations. One consequence of this difference is that sequential IPNs swell less than SINs.
IPNs are related to polymer blends given that they are both combinations of two or more polymers devoid of permanent covalent bonds but they differ in composition where the former is usually fashioned from linear polymers, while the latter contains cross-links within each polymer (7,18,21). This difference in composition infers superior mechanical strength and thermal stability to IPNs due to the enhanced interpenetration of the two component phases as assessed to the analogous mechanical blend which is culpable of separating easily (21,83). This difference is because of the weak interactional behaviors that are present in blends as explained previously (21,83). There are different ways of achieving the same output
One use of IPNs in the medical discipline is to fashion more beneficial hydrogels (2,78,83). Hydrogels are exceptionally biocompatible, have a high water content, and are 3D in nature appropriately allowing for the construction of a material that can potentially form scaffolds for tissue engineering because of its similarities with natural tissue and as such it could be employed as artificial ligaments and cartilage where needed (2,84,85). However, one comprehensive limitation of many hydrogels is that they are mechanically delicate consequently demarcating their employment in biological and biomedical applications as they cannot withstand long periods of time and loose shape with use (2,20,84). To preempt this limitation, a hydrogel composite is synthesized by fabricating an IPN conceivably increasing the practicability to simulate the properties of a natural extracellular matrix consequently making it more applicable for employment (2,84,86). One such case is with the use of a unique class of hydrogels consisting of biologically cross-linkable hyaluronic acid (HA) and semi-interpenetrating collagen constituents (2,84,86). These constituents react synergistically to produce a biologic with enhanced mechanical properties that has the capacity to encourage cell adhesion and proliferation and when micro-engineered has the ability to arrange into architectural scaffolds to form artificial replacements as represented in Fig. 8 (84,86).
Fig. 8.
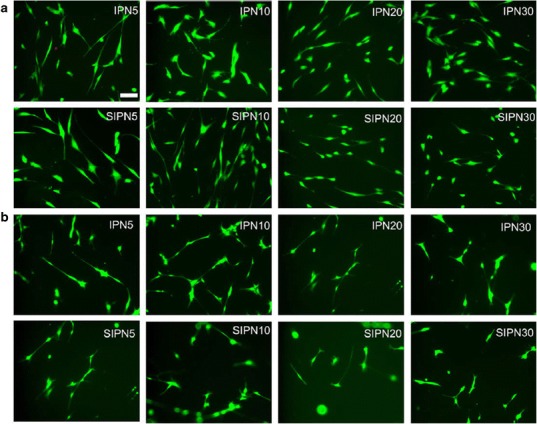
a Live/dead assay results of fibroblasts seeded on IPN and SIPN hydrogels after 24 h. b Live/dead assay results of Schwann cells seeded on IPN and SIPN hydrogels after 24 h. Live cells were stained in green by calcein and dead cells were stained red by ethidium (reprinted with permission from (86) © 2009 Acta Materialia Inc. Published by Elsevier Ltd.)
Despite IPNs unique use, there are limitations in attaining an IPN. IPN structures are determined by the chemical attributes of the constituents and by the properties such as the density of cross-linking and the polymerization kinetics of individual networks (7,19,86). This structure directly imparts different physicomechanical properties to the ultimate product, and accordingly, the elaborate knowledge of all reactions and side reactions that transpire during synthesis should be evaluated to synthesize a practical product devoid of defects (7,86). This is because an operational system is considerably complex with multiple reagents, some of which may be highly reactive and some superfluous subsidiary reactions may result which can actively cripple or distort the progression of the preferred network formation (19,86). Ultimately this complexity increases the possibilities for the formation of toxic byproducts that are useless in the pharmaceutical and medical disciplines (19,86).
Complex Synthesis in Polymer Refabrication
Complex synthesis is another process utilized in polymeric science to create and enhance the properties of parent polymers. Polymer complexes can be classified into four general classes: polyelectrolyte complexes (PEC), hydrogen-bonding complexes (HBC), stereocomplexes, and charged-transfer complexes (CTC).
Polyelectrolyte Complexes
When a polycation solution is mixed with a polyanion solution, a complex is generally synthesized as a precipitate because the oppositely charged polymers are attracted to each other and this affinity culminates in the two components attaching together as depicted in Fig. 9 (26,88). This interaction of ionic polymers and the properties synthesized from this collaboration is contingent on the anionic-to-cationic charge ratio of the polymers, the ionic strength, the degree of neutralization, and the number of valence ions in the electrolyte solution (26,88). Consequently, for a strong polyelectrolyte complex to be synthesized, the polyanions and polycations must contain strong acids and bases for the polyions to achieve their fully ionized forms. The converse is true where weak polyelectrolyte complexes are synthesized in the environment of both weak acids and bases (87,88).
Fig. 9.
Showing the self-formation of a polyelectrolyte complex between oppositely charged polyanions and polycations (adapted from (87))
PEC may be prepared from different types of polymers; however, when synthesized from natural polymers, such as the polysaccharides, the complexes have the superiority of being biocompatible in that they are non-toxic and bioabsorbable (88,158). Within the discipline of pharmaceutics PEC are being used to increase the shelf life of α-amylase containing formulations by entrapping the enzyme in a biodegradable PEC (89). This entrapment enhances the enzymes stability by preventing possible interference from other excipients (89,158). The PEC is assembled from chitosan and alginate with the electrostatic attraction evolving from the cationic amino groups of chitosan and the anionic carboxyl groups of the alginate (158,89).
A more sophisticated PEC was synthesized from chitosan/pectin/polyacrylamide generating a tri-polymeric ionic complex (TPIC). This TPIC formed a matrix that was capable of controlling the release of a hydrophilic drug that was trapped with the complex providing a sustained release formulation as shown in Fig. 10. The complex was also capable of site specificity where drug release was hindered within the acidic environment of the stomach and rather be released within the alkali environment of the intestine (90). The impediment of PECs is that they are generally stable at neutral pH only and are liable at acidic and basic conditions thus decreasing their use in humans as the pH conditions of the gastrointestinal tract are acidic in the stomach (88). This instability is caused by the dissociation of the complex bonds due to either the protonation in acidic conditions or deprotonation in basic conditions of the ionic centers (88).
Fig. 10.
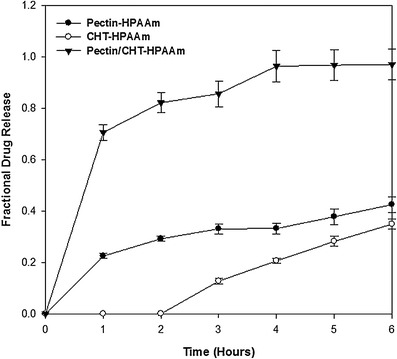
Illustration of PEC formation between the cationic centre of chitosan and the anionic center of alginate as depicted by the arrow in the illustration (reprinted with permission from (90) © 2011 American Association of Pharmaceutical Scientists)
Hydrogen-Bonding Complexes
Non-covalent interactions have been recognized to participate in modeling the secondary and tertiary structures of natural macromolecules such as polypeptides, polynucleotides, and polysaccharides (91,92) This perception has been capitalized in the molecular self-assembly of synthetic polymers with particular interest in hydrogen bonding to design new polymers of exceptional sophistication (91–94). Such polymers that are capable of self-assembling into contemporaneously integrated complexes considering multiple hydrogen bonds possess strength, orientation, and specificity (91–94). As the number of hydrogen bonds increases in a given complex, the magnitude of favorable properties could possibly increase proportionally (93,95). One such example is the polymer network formed from the polymerization of acrylic acid in aqueous sodium hydroxide with acrylamide which occurs via hydrogen bonding (10). This bond is temperature dependent therefore providing a thermosensitive polymer that is externally regulated; on exposure to heat, the polymer expands thus providing pores in the surface for the release of drug (10,93,94). Hydrogen bonds can be established between comparable molecules to form a homomeric/homodimer hydrogen bonding synthon and also between different molecules to form a heteromeric/heterodimer hydrogen bonding synthon as shown in Fig. 11a so long as the architectural requirements below are satisfied (93). For the success of synthesizing a HBCs, the following architectural requirements are essential: (a) a polymerizable group must be approximal to the hydrogen bonding receptor site, (b) the synthesized complex must be soluble in low polarity solvents, (c) directed hydrogen bonding interactions must be viable, and (d) the binding site must be easily synthesized (92–94). 2-Acrylamidopyridine satisfies these requisites as shown in Fig. 11b.
Fig. 11.

Schematic depiction of a homomeric hydrogen bonding synthon and heteromeric hydrogen bonding synthon, b 2-acrylamidopyridine illustrating the architectural requirements for hydrogen bonding complexes, c self-association of 2-acrylamidopyridine in solution by a double hydrogen bonding system, d a centrosymmetric dimer of 3-N-methyl-6-tridecyluraci, and e one of three other possible dimer geometries of 3-N-methyl-6-tridecyluracil where there are two additional repulsive interactions (depicted by double arrows) (adapted from (92,96))
Double Hydrogen Bonded Complexes
The double hydrogen bond system is the simplest HBC that can be synthesized (92,93,96). 2-Acrylamidopyridine is one such molecule that forms a double hydrogen bond. It satisfies the requisites of forming a hydrogen bond and is able to self-associating in solution by forming a pair of hydrogen bonds as shown in Fig. 11c (92,96).
Triple Hydrogen Bonded Complexes
The triple hydrogen bonding system exhibits stronger networks over the double hydrogen bonding systems, correspondingly offering more possibilities for the formation of new polymers (90,93,95). However, the greater the number of possible hydrogen bonds that can be formed, the greater the influence on complex stability since this relies on the arrangement of donor and acceptor groups (90,95). In the case where two donor or two acceptor groups are situated adjacent to one another (even if on different molecules), there is approximately 7 kJ/mol causing secondary electrostatic repulsion (92–95). This interaction causes a destabilizing effect consequently effectuating variances in the dimerization strength and subsequently variances in the complexation strength of triple HBCs (92–95). A worse situation arises when the groups accountable for the secondary interaction do not participate in hydrogen bonding. For instance, in the case of two spectator oxygens, the destabilizing effect is estimated to be approximately 11 kJ/mol generating an unstable complex (92,93). This concept of dimers being synthesized with variances in complexation strengths predicated on the existence or exemption of secondary repulsive electrostatic interactions and the interactional set up of the hydrogen bonds can be seen in Fig. 11d (92,97).
In the centrosymmetric dimer of 3-N-methyl-6-tridecyluracil, there are two attractive forces between acceptor–acceptor and donor–donor groups. However, the other three possible dimer geometries offer two additional repulsive secondary interactions due to the spectator oxygens as depicted by double arrows in Fig. 11e (92,93). Despite the many attractions of HBCs, there are some major drawbacks. As already explained, the molecules to be employed in the complex need to meet certain requirements, and to create advantageous properties for drug delivery, it requires multiple hydrogen bonded complexes which results in the synthesis of unstable dimmers (92,93,97). Furthermore, although hydrogen bond design fundamentals have been substantiated to show specificity most reactions are not entirely understood and often lack orientation leading to micro-phase-separated structures or gelation due to the network formation (96).
Stereocomplexes
Stereocomplexation is a network formation process that is possible between isotactic (cis-) and syndiotactic (trans-) configured polymers (i.e., enantiomeric polymers) having similar or different chemical structures synthesizing a composite with variant properties to the individual initiator polymers (22,23). This occurs when the affinity between two polymers of contrasting stereochemistry is greater than the affinity between polymers with the same stereochemistry consequently synthesizing a stereoselective network between the different polymers via van der Waal forces (22,23). A characteristic example of stereocomplexation transpires between isotactic and syndiotactic poly(methyl methacrylate) known as homo-polymerization since it transpires between congruent polymers (22). As such, complexation between polymers with variant chemical structures is known as hetero-stereocomplexation. In pharmaceutics, this technique has been exploited to form tissue scaffolds. The ultimate tissue scaffolding structure should be elastic and resistant to creep to prohibit long-term deformations that usually occur as well as degrading harmlessly with no adverse effects (24,98). High molecular weight poly-1,3-trimethylene carbonate (poly (TMC)) is an amorphous polymer that degrades in vivo by surface erosion without the liberation of acidic compounds making it suitable for cell cultures and tissue engineering. Unfortunately, the creep resistance of poly(TMC) is passably sparse (98). Stereocomplexation of poly(l-lactide) and poly(d-lactide) transpires consequentially upon solvent casting of mixed solutions comprising of equivalent amounts of polymers (23,24). This solvent cast film of the stereocomplex poly(ST-TMC-ST) showed good mechanical properties and excellent creep resistance (98).
Charge Transfer Complexes
A CTC is an electrostatic association of two or more polymers, in which a partial measure of electrostatic charge is transferred between one molecular entity to the other (25,27,28). This transfer could possibly form an ionic bond or an electron resonance bond in which the existing electrostatic interactions accommodates for the formation of a stabilizing force for complex establishment (25,27,28). The contributing polymer from which the charge is transferred is known as the electron donor and the receiving polymer is known as the electron acceptor and the amalgamation of these two entities conceives the complex (25,27,28). In the pharmaceutical discipline, CTCs have been synthesized between the electron donor 1,10-phenanthroline in combination with the acceptor, p-nitrophenol in equal concentrations (28). This association enables the activation of antifungal and antibacterial activity against many strains which is absent in the individual constituents of the complex (28). CTCs have certain debilitating stability limitations. Firstly, the bond that is formed between CTC constituents is created by an electrostatic transition into a high energy electron state and is superlatively characterized as a weak electron resonance which is not ideally stable as compared to covalent bonds, and therefore, there lies a great possibility for polymer complex separation (28). Secondly, the stability is also compromised owing to the nature of the entities; since the entities are charged, there are repulsive forces that exist between similar charges effectuating an electrostatic destabilizing consequence, the repercussion of which emanates into complex separation (27).
Evaluation of Polymer Techniques, Their Uses, and Future Development
Each technique that has been presented here has its own advantages and disadvantages regarding cost and implementation. Particular advantages and disadvantages of each process have been described in text; for example, under the section chemical grating, the advantage “Grafting desired functionalities onto the surface of materials to render them capable of binding enzymes, proteins, and similar species has yielded products useful for site-specific targeted drug delivery systems” (33) was mentioned in addition to a disadvantage “the major limitation confronted and the explanation for the privation of its extensive employment in industrial graft copolymerization, expressly with the free radical-based systems, is the concurrent formation of homopolymer” (11,38,55,56). Since each technique is unique and different in its own regard, it is a superfluous effort to carry out comparative studies of the different techniques because each technique is suited and fitted to particular situation. For example, the chemical means of grafting is a process where it is possible to accurately predict the outcome of a reaction by knowing the reagents that will be used and how the reaction will affect the physicochemical properties of the reagents when combined. In contrast, the process of blending is a much cheaper and quicker process that could possibly offer the same changes to the physicochemical properties, but it is near impossible to predict the outcome without extensive analysis making it very difficult to make needed/wanted changes to the product.
Another example is enzymatic grafting which is capable of creating a very pure product with no requirements of purification steps, and in the medical and pharmaceutical fields, purity of products are extremely important. This would seem the best method then; however, if the reagents used are unaffected by enzymes due to enzyme specificity, the method becomes useless.
In industry, the main concern is cost-effectiveness. This would mean that the photochemical means of grafting would be the best-suited process as it does not require the use of process chemicals, but only the parent polymer and monomer to be used. However, when working with reagents that are photolabile, such a process is rendered useless as it will degrade or adversely affect the product. As already mentioned, each technique is unique and there is no one single particular technique that is exceptionally advantageous or disadvantageous comparative to the other. Rather each technique is important in its own regard and depending on the wants and needs of the researcher; the appropriate most suitable technique should be selected in order to achieve the goals of the research. Future recommendations are to combine the different methods to create polymers that are very specific in their properties, for example, grafting hydrogen bonding moieties onto a polymer to render it capable of forming hydrogen bonds, or the use of interpenetrating polymer networks that are charged and able to complex together forming a 3D structure. The next section explains where some of the techniques have been utilized for the formation of specific medical and pharmaceutical applications.
Polymer Refabrication Applied to Drug Delivery Matrices and Tissue Engineering
Perspicacious contrivance of biocompatible polymeric delivery vehicles has initiated the amelioration of drug delivery systems such as controlled release systems that are capable of delivering small molecules, lipids, peptides, and/or proteins, both systemically and site specific targeting (99). During the formulation of such systems, both synthetic and natural polymers have been applied to various compositions based on their unique properties that each exhibits, accommodating for a platform of desired functions (99). In addition, the advancement in synthetic processes of polymers with specific chemical architecture has increased the collection of biocompatible materials that can be employed. Extraction and purification processes have also facilitated the employment of a multitudinous amount of natural polysaccharides that can be utilized for drug delivery and tissue engineered scaffolds (99). Different methods of modifications have been employed for the delivery of drugs such as polymer–drug conjugation for micellar delivery systems, nano- and micro-particles for local injection or systemic release, drug-loaded hydrated hydrogel networks to epitomize the physicochemical properties of soft tissue environments for tissue regeneration, and dehydrated hydrogels for rigid scaffolds for hard tissue engineering (99). Once the suitable polymers and process are selected, a felicitous design must be generated to synthesize a polymer with the required architectural composition to satisfy the requisites of it applications.
Tissue-Engineered Scaffolds
Biomaterial scaffolds are increasingly being pushed in the forefront of research of tissue engineering as these systems are capable of interacting with living systems in a specifically designed manner thereby amplifying prognosis (100). The ultimate scaffold should be designed in such a manner that it encourages the natural wound healing and regeneration mechanisms of the body by proximately emulating the structure and biological activity of the extracellular matrix present (100–102). For such a scaffold to exist, certain criteria need to be met: (a) the architecture of the biomaterial must harmonize with the body’s natural wound healing processes, (b) the scaffold should have selectivity associated with it as the natural system does, and (c) the scaffold interface should mediate bimolecular entities similar to those found in a wound. Simultaneously with these characteristics, the potential scaffold materials should be biocompatible, non-immunogenic, have controlled biodegradability, and able to manipulate into three dimensional structures (100–102). Figure 12 depicts the process of using polymers to form a formidable tissue scaffold.
Fig. 12.

Summarization of the synthesis of a tissue scaffold for transplant and use in a patient. Also depicted is the three main entities required for such a scaffold to be established (adapted from (103))
Several researchers have fashioned and assessed in vivo various scaffolds composed of different polysaccharides, proteins, and synthetic molecules as reviewed elsewhere. The scaffolds synthesized had variant magnitudes of accomplishment, but the majority of developed scaffolds did not meet all the above criteria. To overcome the imperfections of these various scaffolds that were synthesized, modification processes of polymers were employed. One of the principal candidates was naturally derived HA. Hyaluronic acid is a glycosaminoglycan of natural origin and satisfies many of the above criteria for scaffold formation (84). Additionally, HA also possess the capacity to support the wound healing mechanisms as it is naturally angiogenic when it disintegrates (84,101,102). HA reacts with the body mechanisms in such a way that it initially stimulates inflammation, which is integral to wound healing, but then subsequently regulates inflammation in the later phases consequently decreasing inflammation and allowing for repair matrix stabilization (101,102). Additionally, it has been found that the environment engulfing the migrating and proliferating fetal cells is substantially supplemented with HA and by employing exogenous HA supplementation it promotes faster and more extensive regeneration in adult injuries by proximally imitating the embryonic environment (101,102). However, in its innate structure, HA has no place in tissue scaffolds. By application of photopolymerization to effectuate the cross-linking of HA to form an IPN (as described in “Radiation Grafting” section 5 above), it is possible to synthesize a material that significantly acquires the potential to perform as an ideal tissue engineered scaffold. Table V below summarizes the collaborated function of HA with the bodies repair mechanisms.
Table V.
The Collaboration of Hyaluronic Acid with the Body’s Natural Wound Repair Mechanisms
| Wound healing phase | Collaborated function of hyaluronic acid |
|---|---|
| Inflammation | Macrophage and neutrophil activation |
| Pro-inflammatory cytokine synthesis | |
| Matrix stabilization by inhibiting inflammatory proteinases | |
| Granulation | Cellular differentiation, proliferation, and migration |
| Angiogenesis | |
| Remodeling | Reduced scar tissue formation |
Stimuli Responsiveness from Polymer Refabrication
There has been an improved perceptive on the theories of disease that have indicated the existence of disease variations due to diurnal rhythms. This has been taken into consideration when synthesizing drug delivery systems as they need to augment the symptomatic constraints of the disease (104). These deliberations have directed the delivery of drugs in the direction of idealized drug delivery, where the desired quantity of active ingredient is released at a desired site at the desired time for the most effective treatment (104). This significantly decreases the side effects associated with drugs as the drug is only available to the body at a time and place it is required therefore unable to cause deleterious effects elsewhere. It has been found that drug can be released under specific conditions of an environment such as pH, temperature changes, pathological sites, and certain spaces when a disease manifests (104). These stimuli-sensitive drugs, also known as “smart” drugs, are polymers having the acquired capabilities of drastically changing their physicochemical properties in response to minute changes in the environment, pH and temperature demonstrating exceptional results. The pH in a humans changes along the length of gastrointestinal tract, as well as showing variation in other areas such as tumor sites. By using chemically ionizable moieties which respond to either acids or bases, it is possible to delivery drugs at specific times and areas. Likewise, there may exist areas in the body where the temperature is slightly different to normal physiological pH.
pH-Responsive Drug Delivery Devices
Stimuli-responsive drug delivery systems are synthesized by the employment of the grafting (as described under “Polymer Grafting” section) special moieties onto a parent polymer (105). The most widely applied grafting method for the synthesis of these polymers is the living polymerization technique as it has greater graft product specificity (as described under the “Living Polymerization Grafting via a Chemical Approach” section). It is known that the pH of cancerous cells is slightly more acidic than the normal cells of the human body; using this knowledge, acid degradable delivery systems were created to specifically target cancerous cells and deliver antineoplastics at that site (105,106). Liposomes were utilized as antineoplastics carriers due to their biocompatibility, biodegradability, and their capacity to carry both hydrophilic and hydrophobic drugs (105). The use of doxorubicin is widely used in this regard as a base onto which acid labile polymers are incorporated (105). The polymer plays two roles in such a system; initially, it stabilizes the liposomes structure during blood circulation after which it disarticulates the integrity of the liposomal membrane integrity when encountered by an acidic environment depictive of cancerous cells. Accordingly, this establishes a selective drug delivery release at a specific site, namely a cancerous site (105). The process is shown in Fig. 13.
Fig. 13.

Depiction of the entrapment of drug in a micelle system that is disrupted by a higher pH as found around cancer cells
An efficacious pH-responsive block copolymer was synthesized from poly (sodium styrene sulfonate) as the parent with sodium 4-vinylbenzoic acid (VBA) added as a graft moiety (105). The block copolymer works by protonating the carboxylic acid of VBA at low pHs thereby rendering the poly(VBA) block hydrophobic (105). This pH change induces micellization that is fully reversible when the pH is increased thereby trapping the drugs inside the micelles until a higher pH is encountered at a particular site; the deformation of the micelles allows for the release of drug (105).
Thermoresponsive Drug Delivery Devices
There exist various different types of thermoresponsive systems that work by having a temperature-related property called a critical solution temperature (107). Systems that display a homogenous solution above a certain solution and a heterogeneous solution below that temperature have an upper critical solution temperature (107). The opposite system has a lower critical solution temperature (LCST) where a homogenous solution is displayed below a certain temperature and above that temperature the heterogeneous solution is displayed (107). The later represents the polymers with the greatest potential for application. N-Alkyl acrylamides have shown thermoresponsive properties with N-isopropylacrylamide showing very promising results due to it having a LCST of 32°C (107). This type of delivery system has proven to be a good point of research for anti-cancer drugs as these drugs are highly deleterious being cytotoxic and the exposure to non-cancer cells in the body causes a sequence of undesirable effects such as myelosuppression, mucositis, and alopecia which can now be suppressed by site targeting (105,106). Various concepts have been utilized in the synthesis of tissue engineering to develop temperature sensitive scaffolds. Once such scaffold was synthesized from poly(NIPAAm-co-acrylic acid) gel which was applied as an extracellular matrix for pancreatic islets in biohybrid pancreas (107). In addition, the NIPAAm was cross-linked to form a gel that was temperature sensitive and allowed the release of salicylic acid and bovine serum albumin when temperature conditions were met at 40°C (107).
Dual Stimuli-Responsive Drug Delivery Devices
These are systems that respond to changes in both temperature and pH by the incorporation of both ionizable and hydrophobic functional groups (107). This combinatory effect is achieved by using methods of copolymerization of two different monomers, each responding to a different stimulus (107). This combinatory effect can be used to delivery drug with greater specificity at sites that are susceptible to changes in both stimuli. Elastin-like polymers have shown the greatest significance in this area showing a modulated temperature and pH sensitivity to the surrounding environment of the polymer (107). The ELPs have also been created to respond to photo-initiation such as the azobenzenes and spiropyrans (107).
Chronotherapeutism from Polymer Refabrication
It is not perpetual that sustained release therapy is the best therapy for any disease state. It has been recognized that circadian rhythms performs an important role in certain disease states as these disease states have chronological aspects related to them (108). One such example is asthma which has shown to become formidable in the early hours of morning (2–8 am) when most patients are asleep (108). It is favorable for patients to take medicate before they sleep rather than waking up at midnight to take a dose in order to relive these early morning symptoms. In addition to this time complication, theophylline, used for asthma has an expansive inter-individual variation and a narrow therapeutic window with a short half-life of 7–8 h only (109). To overcome these patient-related complications, chronotherapy is applied to the dosage form which initially releases a burst of a drug when it is initially administered, and thereafter, there is a lag phase after which drug is released again at a later stage. This pattern was achieved by using effervescent tablets of theophylline that were coated in a polymeric blend (as described in “Enzymatic Grafting” section above) of Eudragit RS 100 blended with 10% hydroxypropyl methylcellulose (HPMC) and then second coated with enteric coating, cellulose acetate phthalate (110). The enteric coating resisted the acidic environment of the stomach, and the second coat delayed the release of drug by approximately 2 h then allowing for the release of theophylline (110). Alternately the system is put into an insoluble capsule with the entities placed in a well-designed order as shown in Fig. 14.
Fig. 14.

Depiction of a chronotherapeutic capsule used for the treatment of asthma (adapted from (111))
The above device works by synthesizing and insoluble capsule body from polypropylene and creating an erodible plug made from a blend of lactose and HPMC, compressed and placed in front of the drug to be release (111). When the drug is taken, the gelatin capsule cover dissolves easily thereby exposing the erodible plug. Depending on the thickness of the plug at any particular compression, force will determine the length of time required for drug release (111). If an initial release is required, some drug can be placed between the gelatin cap and the erodible tablet.
CONCLUSIONS
There exist many techniques for the refabrication of polymers that have been employed in industry and research to synthesize polymers that meets specific needs required by the employer. From site specificity, to stimuli-responsive, enhanced biocompatibility, and chronotherapeutics, a vast majority of desirable properties can be attained through the use of one or more of these polymeric alteration techniques. These techniques can and have been manipulated and employed in the disciplines of pharmaceutics and medicine to enhance the prognostic outcomes of diseases and illnesses. However, despite the many techniques that are available for polymer refabrication, there is still a need to either develop these techniques or expand their employment to more polymers or to create novel techniques to allow for a greater variety of modified polymers that can be employed in more novel systems in pharmaceutics and medicine.
REFERENCES
- 1.Kaparissedes C, Alexandridou S, Kotti K, Chaitidou S. Recent advances in novel drug delivery systems. J Nanotechnol. 2006; 2.
- 2.Pergal MV, Antic VV, Tovilovic G, Nestorov J, Vasiljevic-Radovic, Djonlagic J. In vitro biocompatibility evaluation of novel urethane-siloxane co-polymers based on poly(ε-caprolactone)-block-co-poly(dimethylsiloxane)-block-poly(ε-caprolactone) J Biomater Sci Polym Ed. 2012;23:1629–1657. doi: 10.1163/092050611X589338. [DOI] [PubMed] [Google Scholar]
- 3.Pasut G, Veronese FM. Polymer–drug conjugation, recent achievements and general strategies. Prog Polym Sci. 2007;32:933–961. doi: 10.1016/j.progpolymsci.2007.05.008. [DOI] [Google Scholar]
- 4.Subhash D, Mandal C, Mandal M. Current status and future prospects of new drug delivery systems. Pharma Times. 2010;42:13–16. [Google Scholar]
- 5.Kim P-H, Kim SW. Polymer-based delivery of glucagon-like peptide-1 for the treatment of diabetes. ISRN Endocrinol. 2012 doi: 10.5402/2012/340632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saito T, Mather BD, Costanzo PJ, Beyer FL, Long TE. Influence of site-specific sulfonation on acrylic graft copolymer morphology. Macromolecules. 2008;41:3503–3512. doi: 10.1021/ma800178d. [DOI] [Google Scholar]
- 7.Kamal M, Dwivedi A. Photo polymerized interpenetrating polymer network of poly(antimony acrylate) and poly(arsenic acrylate): synthesis and characterization. J Macromol Sci Pure Appl Chem. 2008;45:548–554. doi: 10.1080/10601320802100648. [DOI] [Google Scholar]
- 8.Sampaio S, Taddei P, Monti P, Buchert J, Freddi G. Enzymatic grafting of chitosan onto Bombyx mori silk fibroin: kinetic and IR vibrational studies. J Biotechnol. 2005;116:21–33. doi: 10.1016/j.jbiotec.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Kaith BS, Jindal R, Jana AK, Maiti M. Characterization and evaluation of methyl methacrylate-acetylated Saccharum spontaneuml L. graft copolymers prepared under microwave. Carbohydr Polym. 2009;78:987–996. doi: 10.1016/j.carbpol.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 10.Qiu Y, Park K. Environmentally-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev. 2012 doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 11.Jenkins DW, Hudson SM. Review of vinyl copolymerization featuring recent advances toward controlled radical-based reactions and illustrated with chitin/chitosan trunk polymers. Chem Revolut. 2001;101:3245–3274. doi: 10.1021/cr000257f. [DOI] [PubMed] [Google Scholar]
- 12.Roach P, Eglin D, Rohde K. Modern biomaterials: a review—bulk properties and implications of surface modifications. J Mater Sci Mater Med. 2007;18:1263–1277. doi: 10.1007/s10856-006-0064-3. [DOI] [PubMed] [Google Scholar]
- 13.Yan W, Qin S, Guo J, Zhang M, He M, Yu J. Morphology and mechanical properties of acrylonitrile-butadiene-styrene (ABS)/polyamide 6 (PA6) nanocomposites prepared via melt mixing. J Macromol Sci B Phys. 2012;51:70–82. doi: 10.1080/00222348.2011.562113. [DOI] [Google Scholar]
- 14.Fang Z, Harrats C, Moussaif N, Groeninckx G. Location of a nanoclay at the interface in an immiscible poly(ε-caprolactone)/poly(ethylene oxide) blend and its effect on the compatibility of the components. J Appl Polym Sci. 2007;106:3125–3135. doi: 10.1002/app.26331. [DOI] [Google Scholar]
- 15.Kuo S-W, Cheng R-S. DNA-like interactions enhance the miscibility of supramolecular polymer blends. Polymer. 2009;50:177–188. doi: 10.1016/j.polymer.2008.10.047. [DOI] [Google Scholar]
- 16.Ludwiczak S, Mucha M. Modeling of water sorption isotherms of chitosan blends. Carbohydr Polym. 2010;79:34–39. doi: 10.1016/j.carbpol.2009.07.014. [DOI] [Google Scholar]
- 17.Si M, Araki T, Ade H, Kilcoyne ALD, Fisher R, Sokolov JC, et al. Compatibilizing bulk polymer blends by using organoclays. Macromolecules. 2006;39:4793–4801. doi: 10.1021/ma060125+. [DOI] [Google Scholar]
- 18.Sperling LH, Mishra V. The current status of interpenetrating polymer networks. Polym Adv Technol. 1996;7:197–208. doi: 10.1002/(SICI)1099-1581(199604)7:4<197::AID-PAT514>3.0.CO;2-4. [DOI] [Google Scholar]
- 19.Jayasuriya MM, Hourston DJ. The effect of composition and the level of crosslinking of the poly(methylmethacrylate) phase on the properties of natural rubber-poly(methylmethacrylate semi-2 interpenetrating polymer networks. J Appl Polym Sci. 2012;124:3558–3564. doi: 10.1002/app.35107. [DOI] [Google Scholar]
- 20.Myung D, Waters D, Wiseman M, Duhamel P, Noolandi J, Ta CN, et al. Progress in the development of interpenetrating polymer network hydrogels. Polym Adv Technol. 2008;19:647–657. doi: 10.1002/pat.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johns J, Nakason C. Novel interpenetrating polymer networks based on natural rubber/poly(vinyl alcohol) Polym Plast Technol Eng. 2012;51:1046–1053. doi: 10.1080/03602559.2012.689053. [DOI] [Google Scholar]
- 22.Kumaki J, Kawauchi T, Okoshi K, Kusanagi H, Yashima E. Supramolecular helical structure of the stereocomplex composed of complementary isotactic and syndiotactic poly(methyl methacrylate)s as revealed by atomic force microscopy. Angew Chem. 2007;119:5444–5447. doi: 10.1002/ange.200700455. [DOI] [PubMed] [Google Scholar]
- 23.Tsuji H, Bouapao L. Stereocomplex formation between poly(L-lactic acid) and poly(D-lactic acid) with disproportionately low and high molecular weights from the melt. Polym Int. 2012;61:442–450. doi: 10.1002/pi.3219. [DOI] [Google Scholar]
- 24.Kim SH, Park O, Nederberg F, Topuria T, Krupp LE, Kim H, et al. Application of block-copolymers supramolecular assembly for the fabrication of complex TiO2 nanostructures. Small. 2008;4:2162–2165. doi: 10.1002/smll.200801115. [DOI] [PubMed] [Google Scholar]
- 25.Wegner D, Yamachika R, Wang Y, Brar VW, Bartlett BM, Long JR, et al. Single-molecule charge transfer and bonding at an organic/inorganic interface: tetracyanoethylene on noble metals. Nano Lett. 2008;8:131–135. doi: 10.1021/nl072217y. [DOI] [PubMed] [Google Scholar]
- 26.Shaovsky A, Varga I, Makuška R, Claesson PM. Formation and stability of water-soluble, molecular polyelectrolyte complexes: effects of charge density, mixing ratio, and polyelectrolyte concentration. Am Chem Soc. 2009;25:6113–6121. doi: 10.1021/la804189w. [DOI] [PubMed] [Google Scholar]
- 27.Stein T, Kronik L, Baer R. Reliable prediction of charge transfer excitations in molecular complexes using time-dependant density functional theory. J Am Chem Soc. 2009;131:2818–2820. doi: 10.1021/ja8087482. [DOI] [PubMed] [Google Scholar]
- 28.Khan IM, Ahmad A. Synthesis, spectral investigations, antimicrobial activity and DNA-binding studies of novel charge transfer complex of 1,10-phenanthroline as an electron donor with π-acceptor p-nitrophenol. J Mol Struct. 2010;977:189–196. doi: 10.1016/j.molstruc.2010.05.031. [DOI] [Google Scholar]
- 29.Bhattacharya, Misra BN. Grafting: a versatile means to modify polymers techniques, factors and applications. Programmed Polym Sci. 2007;29:767–814. doi: 10.1016/j.progpolymsci.2004.05.002. [DOI] [Google Scholar]
- 30.Li M-Z, Li J-H, Shao X-S, Miao J, Wang J-B, Zhang Q-Q, et al. Grafting zwitterionic brush on the surface of PVDF membrane using physisorbed free radical grafting technique. J Membr Sci. 2012;405–406:141–148. doi: 10.1016/j.memsci.2012.02.062. [DOI] [Google Scholar]
- 31.Kato K, Uchida E, Kang E, Uyama Y, Ikada Y. Polymer surface with graft chains. Programmed Polym Sci. 2002;28:209–259. doi: 10.1016/S0079-6700(02)00032-1. [DOI] [Google Scholar]
- 32.McGinty KM, Brittain WJ. Hydrophilic modification of poly(vinyl chloride) film and tubing using physisorbed free radical grafting technique. Polymer. 2008;49:4350–4357. doi: 10.1016/j.polymer.2008.07.063. [DOI] [Google Scholar]
- 33.Siegwart DJ, Oh JK, Matjaszewski K. ATRP in the design of functional materials for biomedical applications. Prog Polym Sci. 2012;37:18–37. doi: 10.1016/j.progpolymsci.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krivoguz YM, Guliyev AM, Pesetskii SS. Free radical grafting of trans-ethylene-1,2-dicarboxilic acid onto molten ethylene-vinyl acetate copolymer. Appl Polym Sci. 2012 [Google Scholar]
- 35.Moad G, Rizzardo E, Thang SH. Radical addition–fragmentation chemistry in polymer synthesis. Polymer. 2008;49:1079–1131. doi: 10.1016/j.polymer.2007.11.020. [DOI] [Google Scholar]
- 36.Durmaz YY, Kumbaraci V, Demirel AL, Talinli N, Yagci Y. Graft copolymers by the combination of ATRP and photochemical acylation process by using benzodioxinones. Macromolecules. 2009;42:3743–3749. doi: 10.1021/ma900360s. [DOI] [Google Scholar]
- 37.Zhang N, Huber S, Schulz A, Luxenhofer R, Jordan R. Cylindrical molecular brushes of poly(2-oxazoline)s from 2-isopropenyl-2-oxazoline. Macromolecules. 2009;42:2215–2221. doi: 10.1021/ma802627y. [DOI] [Google Scholar]
- 38.Zhao J, Mountrichas G, Zhang G, Pispas S. Thermoresponsive core-shell brush copolymers with poly(propylene oxide)-block-poly(ethylene oxide) side chains via a “grafting from” technique. Macromolecules. 2010;43:1771–1777. doi: 10.1021/ma902590w. [DOI] [Google Scholar]
- 39.Aoshima S, Kanaoka S. Synthesis of stimuli-responsive polymers by living polymerization: poly(N-isopropylacrylamide) and poly(vinyl ether)s. Adv Polym Sci. 2008;210:169–208. doi: 10.1007/12_2007_120. [DOI] [Google Scholar]
- 40.Huang L-P, Zhou X-P, Cui W, Xie X-L, Tong S-Y. Maleic anhydride-grafted linear low-density polyethylene with low gel content. Polym Eng Sci. 2009;49:673–679. doi: 10.1002/pen.21285. [DOI] [Google Scholar]
- 41.Vicente G, Aguado J, Serrano DP, Sánchez N. HDPE chemical recycling promoted by phenol solvent. J Anal Appl Pyrolysis. 2009;85:366–371. doi: 10.1016/j.jaap.2008.10.007. [DOI] [Google Scholar]
- 42.Na C-K, Park H-J. Preparation of acrylic acid grafted polypropylene nonwoven fabric by photoinduced graft polymerization with preabsorption of monomer solution. J Appl Polym Sci. 2009;114:387–397. doi: 10.1002/app.30364. [DOI] [Google Scholar]
- 43.Wang X, Colavita PE, Streifer JA, Butler JE, Hamers RJ. Photochemical grafting of alkenes onto carbon surfaces: identifying the roles of electrons and holes. J Phys Chem C. 2010;114:4067–4074. doi: 10.1021/jp911264n. [DOI] [Google Scholar]
- 44.Aracri E, Fillat A, Colom JF, Gutierrez A, del Rio JC, Martinez AT, et al. Enzymatic grafting of simple phenols on flax and sisal pulp fibres using laccases. Bioresour Technol. 2010;101:8211–8216. doi: 10.1016/j.biortech.2010.05.080. [DOI] [PubMed] [Google Scholar]
- 45.Hanefeld U, Gardossi L, Magner E. Understanding enzyme immobilisation. Chem Soc Rev. 2009;38:453–468. doi: 10.1039/b711564b. [DOI] [PubMed] [Google Scholar]
- 46.Hossain KMG, Gonzalez MD, Lozano GR, Tzanov T. Multifunctional modification of wool using an enzymatic process in aqueous-organic media. J Biotechnol. 2009;141:58–63. doi: 10.1016/j.jbiotec.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 47.Lv X, Song W, Ti Y, Qu L, Zhao Z, Zheng H. Gamma radiation-induced grafting of acrylamide and dimethyl diallyl ammonium chloride onto starch. Carbohydr Polym. 2012 doi: 10.1016/j.carbpol.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 48.Qiu J, Wang Z, Li H, Xu L, Peng J, Zhai M, et al. Adsorption of Cr(VI) using silica-based adsorbent prepared by radiation-induced grafting. J Hazard Mater. 2009;166:270–276. doi: 10.1016/j.jhazmat.2008.11.053. [DOI] [PubMed] [Google Scholar]
- 49.Gürsel SA, Gubler L, Gupta B, Scherer GG. Radiation grafted membranes. Adv Polym Sci. 2008;215:157–217. [Google Scholar]
- 50.Goel NK, Rao MS, Kumar V, Bhardwaj YK, Chaudhari CV, Dubey KA, et al. Synthesis of antibacterial cotton fabric by radiation-induced grafting of [2-(methacryloyloxy)ethyl]trimethylammonium chloride (MAETC) onto cotton. Radiat Phys Chem. 2009;78:399–406. doi: 10.1016/j.radphyschem.2009.03.011. [DOI] [Google Scholar]
- 51.Liu R, Saunders BR. Thermoresponsive surfaces prepared using adsorption of a cationic graft copolymer: a versatile method for triggered particle capture. J Colloid Interface Sci. 2009;338:40–47. doi: 10.1016/j.jcis.2009.05.073. [DOI] [PubMed] [Google Scholar]
- 52.Zhao J, Mountrichas G, Zhang G, Pispas S. Amphiphilic polystyrene-b-poly(p-hydroxystyrene-g-ethylene oxide block-graft copolymers via a combination of conventional and metal-free anionic polymerization. Macromolecules. 2009;42:8661–8668. doi: 10.1021/ma9016604. [DOI] [Google Scholar]
- 53.Zhao J, Li J, Feng Y, Yin J. A novel approach to synthesis of functional CPVC and CPE or graft copolymers-in situ chlorination graft. Polym Adv Technol. 2007;18:822–828. doi: 10.1002/pat.941. [DOI] [Google Scholar]
- 54.Alves NM, Mano JF. Chitosan derivatives obtained by chemical modifications for biomedical and environmental applications. Int J Biol Macromol. 2008;43:401–414. doi: 10.1016/j.ijbiomac.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Scherf U, Gutacker A, Koenen N. All-conjugated block copolymers. Acc Chem Res. 2008;41:1086–1097. doi: 10.1021/ar7002539. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J-F, Yang D-Z, Xu F, Zhang Z-P, Yin R-X, Nie J. Electrospun core–shell structure nanofibres from homogenous solution of poly(ethylene oxide)/chitosan. Macromolecules. 2009;42:5278–5284. doi: 10.1021/ma900657y. [DOI] [Google Scholar]
- 57.Price M, Reiners JJ, Santiago AM, Kessel D. Monitoring singlet oxygen and hydroxyl radical formation with fluorescent probes during photodynamic therapy. Photochem Photobiol. 2009;85:1177–1181. doi: 10.1111/j.1751-1097.2009.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verbeek CJR, Hanipah SH. Grafting Itaconic anhydride onto polyethylene using extrusion. J Appl Polym Sci. 2010;116:3118–3126. doi: 10.1002/app.31901. [DOI] [Google Scholar]
- 59.Russell KE. Free radical graft polymerization and copolymerization at higher temperatures. Prog Polym Sci. 2001;27:1007–1038. doi: 10.1016/S0079-6700(02)00007-2. [DOI] [Google Scholar]
- 60.Thurecht KJ, Gooden PN, Goel S, Tuck C, Licence P, Irvine DJ. Free-radical polymerization in ionic liquids: the case for a protected radical. Macromolecules. 2008;41:2814–2820. doi: 10.1021/ma7026403. [DOI] [Google Scholar]
- 61.Colavita PE, Sun B, Wang X, Hamers RJ. Influence of surface termination and electronic structure on the photochemical grafting of alkenes to carbon surfaces. J Phys Chem C. 2009;113:1526–1535. doi: 10.1021/jp805933h. [DOI] [Google Scholar]
- 62.Deng J, Wang L, Liu L, Yang W. Developments and new applications of UV-induced surface graft polymerizations. Prog Polym Sci. 2009;34:156–193. doi: 10.1016/j.progpolymsci.2008.06.002. [DOI] [Google Scholar]
- 63.Krekova J, Lacher NA, Svec F. Highly efficient enzyme reactors containing trypsin and endoproteinase LysC immobilized on porous polymer monolith coupled to MS suitable for analysis of antibodies. Anal Chem. 2009;81:2004–2012. doi: 10.1021/ac8026564. [DOI] [PubMed] [Google Scholar]
- 64.Zhang P, Henthorn DB. Synthesis of PEGylated single wall carbon nanotubes by a photoinitiated graft from polymerization. AICHE J. 2010;56:1610–1615. [Google Scholar]
- 65.Wu G, Xie Y, Ou E, Zhang L, Xiong Y, Xu W. Preparation, characterization, and properties of sodium montmorillonite clay/poly(styrene–butadiene–styrene) containing quaternary ammonium cations and photoinitiator nanocomposites via ultraviolet exposure. J Appl Polym Sci. 2010;118:1675–1682. [Google Scholar]
- 66.Sheldon RA. Enzyme immobilization: the quest for optimum performance. Adv Synth Catal. 2007;349:1289–1307. doi: 10.1002/adsc.200700082. [DOI] [Google Scholar]
- 67.Kumar G, Smith PJ, Payne GF. Enzymatic grafting of a natural product onto chitosan to confer water solubility under basic conditions. Biotechnol Bioeng. 1998;63:154–165. doi: 10.1002/(SICI)1097-0290(19990420)63:2<154::AID-BIT4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 68.Aljawish A, Chevalot I, Piffaut B, Rondeau-Mouro C, Giradin M, Jasniewski J, et al. Functionalization of chitosan by laccase-catalyzed oxidation of ferulic acid and ethyl ferulate under heterogeneous reaction conditions. Carbohydr Polym. 2012;87:537–544. doi: 10.1016/j.carbpol.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 69.Muller HG, Waldmann H. An enzyme-initiated domino hydroxylation–oxidation-carbo-Diels–Alder reaction cascade. Tetrahedron Lett. 1996;37:3833–3836. doi: 10.1016/0040-4039(96)00727-7. [DOI] [Google Scholar]
- 70.Shah B, Chen A. Novel electrochemical approach for the monitoring of biodegradation of phenolic pollutants and determination of enzyme activity. Electrochem Commun. 2012;25:79–82. doi: 10.1016/j.elecom.2012.09.009. [DOI] [Google Scholar]
- 71.Fatarella E, Ciabatti I, Cortez J. Activation of polymeric materials towards enzymatic postgrafting and cross-linking. Enzyme Microb Technol. 2012;51:252–257. doi: 10.1016/j.enzmictec.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 72.Sugimoto H, Nakamura S, Ohwada T. Retro-Diels–Alder reaction of 4H-1,2, benzoxazines to generate o-quinone methides: involvement of highly polarized transition states. J Org Chem. 2007;72:10088–10095. doi: 10.1021/jo702246w. [DOI] [PubMed] [Google Scholar]
- 73.Zhao A, Sadik OA. Comparative analysis of quercetin oxidation by electrochemical, enzymatic, autoxidation, and free radical generation techniques: a mechanistic study. J Agric Food Chem. 2008;56:12081–12091. doi: 10.1021/jf802413v. [DOI] [PubMed] [Google Scholar]
- 74.Mateo C, Palomo JM, Fernandez-Lorente G, Guisan JM, Fernandez-Lafuente R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb Technol. 2007;40:1451–1463. doi: 10.1016/j.enzmictec.2007.01.018. [DOI] [Google Scholar]
- 75.Li S, Gao K. The study on methyl methacrylate graft-copolymerization composite separator prepared by pre-irradiation method for Li-ion batteries. Surf Coat Technol. 2010;204:2822–2828. doi: 10.1016/j.surfcoat.2010.02.056. [DOI] [Google Scholar]
- 76.Smith J, Zhang W, Sougrat R, Zhao K, Li R, Cha D, et al. Solution-processed small molecule-polymer blend organic thin-film transistors with hole mobility greater than 5 cm2/Vs. Adv Mater. 2012;24:2441–2446. doi: 10.1002/adma.201200088. [DOI] [PubMed] [Google Scholar]
- 77.Zivanovic S, Li J, Davidson PM, Kit K. Physical, mechanical, and antibacterial properties of chitosan/PEO blend films. Biomacromolecules. 2007;8:1505–1510. doi: 10.1021/bm061140p. [DOI] [PubMed] [Google Scholar]
- 78.Oh KT, Bronich TK, Kabanov VA, Kabanov AV. Block polyelectrolyte networks from poly(acrylic acid) and poly(ethylene oxide): sorption and release of cytochrome C. Biomacromolecules. 2007;8:490–497. doi: 10.1021/bm060599g. [DOI] [PubMed] [Google Scholar]
- 79.Elias L, Fenouillot F, Majeste JC, Cassagnau P. Morphology and rheology of immiscible polymer blends filled with silica nanoparticles. Polymer. 2007;48:6029–6040. doi: 10.1016/j.polymer.2007.07.061. [DOI] [Google Scholar]
- 80.Ravati S, Favis BD. Morphological states for a ternary polymer blend demonstrating complete wetting. Polymer 2010;51:3669. doi:10.1016/j.polymer.2010.07.014.
- 81.Harvie DJE, Davidson MR, Cooper-White JJ, Rudman M. A parametric study of droplet deformation through a microfluidic contraction: shear thinning liquids. Int J Multiphase Flow. 2007;33:545–556. doi: 10.1016/j.ijmultiphaseflow.2006.12.002. [DOI] [Google Scholar]
- 82.Sperling LH. An overview of interpenetrating networks. In: Salamone JC, editor. 5 polymeric materials encyclopedia. Boca Raton: CRC; 1996. [Google Scholar]
- 83.Jayasuriya MM, Hourston DJ. The effect of composition on the dynamic and mechanical properties of natural rubber–poly(methylmethacrylate) blends. J Appl Polym Sci. 2009;112:3217–3224. doi: 10.1002/app.29737. [DOI] [Google Scholar]
- 84.Brigham MD, Bick A, Lo E, Bendali A, Burdick JA, Khademhosseini A. Mechanically robust and bioadhesive collagen and photocrosslinkable hyaluronic acid semi-interpenetrating networks. Tissue Eng. 2009;15:1646–1653. doi: 10.1089/ten.tea.2008.0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Su J, Wall ST, Healy KE, Wildsoet CF. Scleral reinforcement through host tissue integration with biomimetic enzymatically degradable semi-interpenetrating polymer network. Tissue Eng. 2010;16:905–916. doi: 10.1089/ten.tea.2009.0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suri S, Schmidt CE. Photopatterned collagen-hyaluronic acid interpenetrating polymer network hydrogels. Acta Biomater. 2009;5:2385–2397. doi: 10.1016/j.actbio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 87.Dakhara SL, Anajwala CC. Polyelectrolyte complex: a pharmaceutical review. Syst Rev Pharm. 2010;1:121–127. doi: 10.4103/0975-8453.75046. [DOI] [Google Scholar]
- 88.Kim SJ, Yoon SG, Lee KB, Park YD, Kim SI. Electrical sensitivity of a polyelectrolyte complex composed of chitosan/hyaluronic acid. Solid State Ionics. 2003;164:199–204. doi: 10.1016/j.ssi.2003.08.005. [DOI] [Google Scholar]
- 89.Sankalia MG, Mashru RC, Sankalia JM, Sutariya VB. Reversed chitosan-alginate polyelectrolyte complex for stability improved alpha-amylase: optimization and physicochemical characterization. Eur J Pharm Biopharm. 2007;65:215–232. doi: 10.1016/j.ejpb.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 90.Bawa P, Pillay V, Choonara YE, du Toit LC, Ndesendo VMK, Kumar P. A composite polyelectrolyte matrix for controlled oral drug delivery. AAPS PharmSciTech. 2011;12:227–238. doi: 10.1208/s12249-010-9576-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Llanes-Pallas A, Matena M, Jung T, Prato M, Stöhr M, Bonifazi D. Trimodular engineering of linear supramolecular miniatures on Ag(111) surfaces controlled by complementary triple hydrogen bonds. Angew Chem. 2008;120:7840–7844. doi: 10.1002/ange.200802325. [DOI] [PubMed] [Google Scholar]
- 92.Sherrington DC, Taskinen KA. Self-assembly in synthetic macromolecular systems via multiple hydrogen bonding interactions. Chem Soc Rev. 2001;30:83–93. doi: 10.1039/b008033k. [DOI] [Google Scholar]
- 93.Wilson AJ. Non-covalent polymer assembly using arrays of hydrogen-bonds. Soft Matter. 2007;3:409–425. doi: 10.1039/b612566b. [DOI] [PubMed] [Google Scholar]
- 94.Kitagawa S, Uemura K. Dynamic porous properties of coordination polymers inspired by hydrogen bonds. Chem Soc Rev. 2005;34:109–119. doi: 10.1039/b313997m. [DOI] [PubMed] [Google Scholar]
- 95.Blight BA, Camara-Campos A, Djurdjevic S, Kaller M, Leigh DA, McMillan FM, et al. AAA-DDD triple hydrogen bond complexes. J Am Chem Soc. 2009;131:14116–14122. doi: 10.1021/ja906061v. [DOI] [PubMed] [Google Scholar]
- 96.Steinke JHG, Dunkin IR, Sherrington DC. A simple carboxylic acid receptor: 2-acrylamido pyridine. TrAC Trends Anal Chem. 1999;18:159–164. doi: 10.1016/S0165-9936(98)00093-4. [DOI] [Google Scholar]
- 97.Castro TG, Araujo CMU, Braga CF, Silvia LS, Pereira AM, Lopes KC, et al. Theoretical calculations of the substituent effect on molecular properties of the R_C ≡ N…H_F hydrogen-bonded complexes with R = NH2, CH3O, CH3, OH, SH, H, Cl, F, CF3, CN and NO2. Vib Spectrosc. 2009;49:133–141. doi: 10.1016/j.vibspec.2008.06.005. [DOI] [Google Scholar]
- 98.van Leeuwen AC, Bos RRM, Grijpma DW. Composite materials based on poly(trimethylene carbonate) and -tricalcium phosphate for orbital floor and wall construction. J Biomed Mater Res B Appl Biomater. 2012;100B:1610–1620. doi: 10.1002/jbm.b.32729. [DOI] [PubMed] [Google Scholar]
- 99.Baldwin AD, Kiick KL. Polysaccharide-modified synthetic polymeric biomaterials. Biopolymers. 2010;94:128–140. doi: 10.1002/bip.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Humbert P, Mikosinski J, Benchikhi H, Allaert F-A. Efficacy and safety of a gauze pad containing hyaluronic acid in treatment of leg ulcers of venous or mixed origin: a double-blind, randomized, controlled trial. Int Wound J. 2012 doi: 10.1111/j.1742-481X.2012.00957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Voigt J, Driver VR. Hyaluronic acid derivates and their healing effect on burns, epithelial surgical wounds, and chronic wounds: a systematic review and meta-analysis of randomized controlled trials. Wound Repair Regen. 2012;20:317–331. doi: 10.1111/j.1524-475X.2012.00777.x. [DOI] [PubMed] [Google Scholar]
- 102.Chen WY, Abatangelo G. Functions of hyaluronan in wound repair. Wound Repair Regen. 1999;7:79–89. doi: 10.1046/j.1524-475X.1999.00079.x. [DOI] [PubMed] [Google Scholar]
- 103.Khademhosseini A, Langer R, Borenstein J, Vacanti JP. Microscale technologies for tissue engineering and biology. PNAS. 2005;103:2480–2487. doi: 10.1073/pnas.0507681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.He C, Zhuang X, Tang Z, Tian H, Chen X. Stimuli-sensitive synthetic polypeptide-based materials for drug and gene delivery. Adv Healthc Mater. 2012;1:48–78. doi: 10.1002/adhm.201100008. [DOI] [PubMed] [Google Scholar]
- 105.York AW, Kirkland SE, McCormick CL. Advances in the synthesis of amphiphilic block copolymers via RAFT polymerization: stimuli-responsive drug and gene delivery. Adv Drug Deliv Rev. 2007;60:1018–1036. doi: 10.1016/j.addr.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 106.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2012 doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 107.Aguilar MR, Elvira C, Gallardo A, Vazquez B, Roman JS. Smart polymers and their applications as biomaterials. Biomaterials. 2007; 3.
- 108.Litinski M, Scheer FAJL, Shea SA. Influence of the circadian system in disease severity. Sleep Med Clin. 2009;4:143–163. doi: 10.1016/j.jsmc.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee TA, Schumock GT, Bartle B, Pickard AS. Mortality risk in patients receiving drug regimens with theophylline for chronic obstructive pulmonary disease. Pharmacotherapy. 2009;29:1039–1053. doi: 10.1592/phco.29.9.1039. [DOI] [PubMed] [Google Scholar]
- 110.Vijaya KB, Nagaraj B, Agaiah B, Rambhau D. Design and in vitro evaluation of chrono modulated theophylline tablets. S J Pharm Sci. 2008;1:25–28. [Google Scholar]
- 111.Ross AC, Macrae RJ, Walther M, Stevens HNE. Chronopharmaceutical drug delivery from a pulsatile capsule device based on programmable erosion. J Pharm Pharmacol. 2000;52:903–909. doi: 10.1211/0022357001774787. [DOI] [PubMed] [Google Scholar]