

Abstract
Propranolol (PPL) imprinted microspheres (MIP) were successfully prepared via oil/water polymerization using a methyl methacrylate (MMA) monomer, PLL template, and divinylbenzene (DVB) cross-linker and favorably incorporated in a Eudragit-RS100 nanofiber membrane. A non-PPL imprinted polymer (NIP), without a template, was used as a control. The morphology and particle size of the beads were investigated using scanning electron microscopy. The results revealed that both MIP and NIP had a spherical shape with a micron size of approximately 50–100 μm depending on the amounts of DVB and PPL used. NIP2 (MMA/DVB, 75:2.5) and MIP8 (PPL/MMA/DVB, 0.8:75:2.5) were selected for reloading of PPL, and the result indicated that increasing the ratio of PPL to polymer beads resulted in increase PPL reloading (>80%). A total of 10–50% NIP2 or MIP8 was incorporated into a 40% (w/v) Eudragit-RS100 fiber membrane using an electrospinning technique. PPL could be bound to the 50% MIP8 composite fiber membrane with a higher extent and at a higher rate than the control (NIP2). Furthermore, the MIP8 composite fiber membrane showed higher selectivity to PPL than the other β-blockers (atenolol, metoprolol, and timolol). Thus, the MIP8 composite fiber membrane can be further developed for various applications in pharmaceutical and other affinity separation fields.
Key words: membrane, molecularly imprinted, propranolol, selective molecular imprinting
INTRODUCTION
In recent years, the affinity membrane separations have received more attention as a novel technology for separation and purification. To heighten selectivity in the filtration process, affinity membranes have been developed to isolate molecules based on differences in physicochemical features rather than molecular size (1,2). Owing to the high productivity of membranes and the inherent selectivity of chromatography resins, affinity membrane chromatography is now an attractive and competitive method for purifying proteins or other biomolecules from biological fluids (1–3). The fabrication of molecularly imprinted polymers (MIPs) has been rapidly growing. The molecular imprinting technique permits the specific recognition of macromolecules using templates (4,5). In molecular imprinting, the complex formed between the template molecule and the functional monomer is fixed via polymerization in the presence of an excess amount of a cross-linking monomer. After removal of the template, MIPs can rebind the original template very specifically (6). MIPs possess several advantages including low cost, ease of preparation, stability during storage, retention of activity over repeated operations, high mechanical strength, good thermal and chemical stability, and applicability in aggressive media (7,8). Thus, MIPs have been widely applied in an increasing number of applications including use as sensors (9–13), in chromatographic separation (14–16), as artificial antibodies (17,18), in binding assays (19), and for drug separation (20,21). In the molecular imprinting processes, the selection of the template and cross-linkers are important factors affecting the binding affinity and specificity of the imprinted polymer. Yoshimatsu et al. have demonstrated that imprinted microspheres selective for propranolol (PPL) could be synthesized using the precipitation polymerization method. When the ratio of two different cross-linkers (divinylbenzene (DVB) and trimethylolpropane trimethacrylate (TRIM)) was altered, the resulting microspheres displayed different binding affinity and specificity for PPL and different sizes. Use of the DVB cross-linker resulted in superior binding affinity and specificity for PPL compared to use of the TRIM cross-linker (22).
Electrospinning is a simple and versatile technique that can be used to fabricate ultrafine fibers with diameters ranging from submicrons to nanometers. The fibers generated using this method exhibit excellent attributes, such as very large surface to volume ratios and a high porosities with a small pore size (23). Because of these attractive properties, electrospun nanofibers have been used in biomedical sciences, filtration, optical sensors, and affinity membranes (24–26)
Previous studies have demonstrated that preformed MIP particles can be incorporated into membranes (22) and due to a high productivity of membranes, a high surface-to-volume ratio, and high porosities with a small pore size which could be increase separation efficiency and the inherent selectivity of MIP. Thus, the objective of this study was to develop a novel material for affinity membrane separation using electrospun fiber containing PPL-selective MIP. First, PPL-selective MIP was synthesized using o/w emulsion polymerization. PPL, methyl methacrylate (MMA), benzoyl peroxide (BP), and DVB were used as the molecular template, monomer, initiator, and cross-linker, respectively. A non-molecular imprinted polymer (NIP) prepared using the same approach as the MIP but without the template was used as a control to determine the selectivity of the resultant MIP. The morphologies and particle sizes of the resulting MIP and NIP were determined using scanning electron microscopy (SEM). The percentage of PPL loaded into the obtained PPL-selective imprinted polymer beads compared with NIP was investigated. The selectivity for the PPL of imprinted polymer beads was also investigated by comparing the binding ability to other β-blockers including atenolol (ATE), metoprolol tartrate (MET), and timolol maleate (TIM). Finally, the PPL-selective MIP was incorporated into an electrospun fiber and was characterized to create a PPL-selective MIP membrane that can be applied in various fields.
MATERIALS AND METHODS
Materials
Poly(ethyl acrylate-co-methyl methacrylate-co-trimethylammonioethyl methacrylate chloride) (Eudragit-RS100) was purchased from Rohm GmbH, co., Germany. Polyvinyl alcohol (PVA; MW 85,000–124,000, 87–89% hydrolyzed), MMA 99%, BP 75%, DVB 80%, ATE, MET, PPL hydrochloride, TIM, and N,N dimethylformamide (DMF, ≥99.8%) were purchased from Sigma Chemical Co., USA. All other reagents and solvents were commercially available and were of analytical grade.
Synthesis of the PPL-Selective Imprinted Polymer Beads
PPL-imprinted polymer beads were synthesized by o/w emulsion polymerization. The formulations contained MMA (75 ml), BP (3 g), and various amount of PPL template and DVB as shown in Table I. The PPL in free base form was dissolved in the oil phase containing MMA, DVB, and BP. The oil phase was gradually added to the aqueous phase, a 500-ml 0.5% (w/v) PVA solution, at 85°C with fixed stirring (400 rpm). The solution was maintained under these conditions until polymerization was complete (4 h). The bead was then rinsed several times with deionized water and methanol. The template was extracted from the polymer microspheres by repeatedly rinsing with a 1:9:9 (v/v/v) acetic acid, dichloromethane, and methanol mixture until no template could be detected from the washing solvent by UV–visible spectrophotometry at a wavelength of 290 nm (NanoVue™, GE Healthcare, UK). The NIPs were prepared using the same method as the MIP but without the addition of the PPL template during polymerization. The percentage yields of NIP and MIP beads were calculated using Eq. 1.
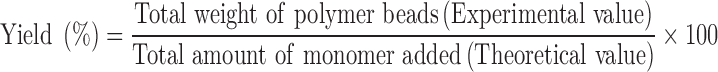 |
1 |
Table I.
Formulations and Percentage Yield of PPL-Imprinted and Non-PPL Imprinted Polymer Beads
| Formula | PPL template (g) | MMA (ml) | DVB (g) | BP (g) | % yield |
|---|---|---|---|---|---|
| NIP1 | 0 | 75 | 2 | 3 | 53.2 |
| NIP2 | 0 | 75 | 2.5 | 3 | 48.3 |
| NIP3 | 0 | 75 | 3 | 3 | 42.6 |
| MIP1 | 0.4 | 75 | 2 | 3 | 31.4 |
| MIP2 | 0.4 | 75 | 2.5 | 3 | 26.5 |
| MIP3 | 0.4 | 75 | 3 | 3 | 44.4 |
| MIP4 | 0.6 | 75 | 2 | 3 | 18.6 |
| MIP5 | 0.6 | 75 | 2.5 | 3 | 18.2 |
| MIP6 | 0.6 | 75 | 3 | 3 | 25.0 |
| MIP7 | 0.8 | 75 | 2 | 3 | 8.4 |
| MIP8 | 0.8 | 75 | 2.5 | 3 | 6.4 |
| MIP9 | 0.8 | 75 | 3 | 3 | 12.5 |
NIP non-molecular imprinted polymers, MIP molecularly imprinted polymers, PPL propranolol, MMA methyl methacrylate, DVB divinylbenzene, BP benzoyl peroxide
Alkali Hydrolytic Reaction of Polymer Beads
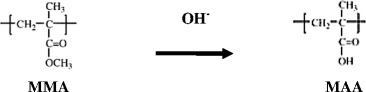
The purpose of this reaction is to remove the methyl groups of MIP to generate carboxylic acid functional groups which are used for binding with propranolol. Briefly, 1 g of MIP or NIP was hydrolyzed in a mixture of 4 g of NaOH and 1.8 ml of deionized water in 80 g of isopropyl alcohol at 85°C for 12 h. The morphologies and particle sizes of the obtained MIP and NIP beads were evaluated by SEM. The chemical structures were characterized using Fourier transform infrared (FTIR) spectrophotometry.
Binding Ability of Polymer Beads to PPL
After the polymerization step, the polymer beads were rinsed with a mixture of acetic acid, methanol, and dichloromethane with a 1:9:9 (v/v/v) ratio. Dichloromethane was added to swell the beads, and acetic acid and methanol were added to remove any un-reacted monomers and template molecules. Then, the methyl groups of the polymer beads were removed by alkali hydrolysis to generate the carboxylic acid functional groups as described above. The ability of the PPL-imprinted polymer beads to selectively bind to PPL was determined by the amount of bound drug from the aqueous solution. In this process, the weight ratio of PPL/MIP or NIP was varied at 1:0.5, 1:1, and 1:2. The mixture was stirred at 200 rpm at 30°C for 48 h. The incubated dispersions were sampled every 2 h after centrifugation at 3,000 rpm for 5 min. The concentration of PPL in the supernatant was determined by UV–visible spectrophotometry at a wavelength of 290 nm (NanoVue™, GE Healthcare, UK). The amount of bound drug was calculated from the difference in the concentrations before and after incubation. All of the experiments were run in triplicate (n = 3). To compare the binding ability, other β-blockers (ATE, MET, and TIM) were similarly prepared using a weight ratio of β-blocker (ATE, MET, or TIM)/MIP or NIP of 1:1.
Preparation Eudragit-RS100 Fiber Membrane
The homogeneous electrospun solutions were prepared by dissolving Eudragit-RS100 in DMF/ethanol (67:33) at the concentrations varied from 30% to 60% (w/v). Before electrospinning process, shear viscosity and conductivity of each solution was measured using a Brookfield DV-III programmable viscometer and an ECtestr11+ conductivity, respectively at 25°C. The Eudragit-RS100 solutions were contained in a glass syringe with a plane tipped stainless steel needle. The electrospinning process was conducted at 25°C with the fixed applied voltage, the distance between a tip and a collector, and the feeding rate at 15 kV, 15 cm, and 0.5 ml/h, respectively. The electrospun nanofibers were collected on aluminum foil that was covering the rotating collector. The electrospinning setup is shown in Fig. 1.
Fig. 1.
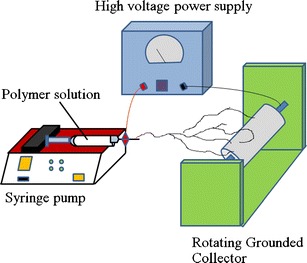
Electrospinning setup scheme
Preparation of MIPs and NIPs Composite Eudragit-RS100 Fiber Membrane
An Eudragit-RS100 solution at a concentration of 40% (w/v) was selected for the preparation of Eudragit-RS100 fibers containing 10–50% (w/w) MIP and NIP. The size of the MIP or NIP beads was reduced using a ball mill (speed 400 rpm). The beads were then added into the 40% (w/v) Eudragit-RS100 in DMF/ethanol (67:33) solution and the mixture was stirred. The obtained solutions underwent the electrospinning process described above, and the obtained fibers were then dissolved in ethanol to liberate the embedded MIP beads. The MIP was then weighed and the % loading efficiency was calculated according to Eq. 2.
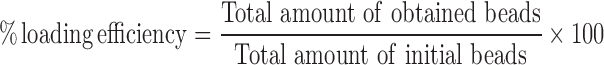 |
2 |
Characterization of Fiber Membranes
The morphology and diameter of the nanofiber mats and NIPs and MIPs composite Eudragit-RS100 fiber membranes were determined using a SEM (Camscan Mx2000, England). In this process, a small section of the electrospun fiber mats was sputtered with a thin layer of gold prior to SEM observation. The average diameter of the fibers was determined by using image analysis software (JMicroVision V.1.2.7, Switzerland). The chemical structure of the fibers was characterized using a FTIR spectrophotometer (Nicolet 4700, USA). The fiber samples were ground and pressed into KBr plates prior to the FTIR analysis with a wave number range of 500–4,000 cm−1.
Binding Affinity of β-Blockers to NIP and MIP Composite Eudragit-RS100 Fibers
The electrospun fiber mats containing polymer beads were cut into circular plates with diameters of 1.5 cm and then pulled away from the aluminum plate substrate. A mixture of the β-blocker solution (PPL, ATE, MET, or TIM) was incubated with the electrospun fiber containing MIPs and NIPs at a weight ratios of 1:1. The mixture was agitated at 200 rpm for 2 h at 30°C. The incubated dispersions were withdrawn after 3, 5, 10, 20, 40, 60, and 120 min. The amount of bound drug was calculated from the difference in the concentrations before and after incubation. All of the experiments were run in triplicate (n = 3). The concentration of each β-blocker in the supernatant was determined using high-performance liquid chromatography (HPLC) (Agilent Technology, USA). A Thermo (USA) C18 column (4.6 mm × 250 mm, 5 μm particle sizes) was used. The elution was performed with a solvent system containing acetonitrile and phosphate buffer (pH 7.4), and 0.2% (w/v) triethylamine at a flow rate of 0.8 ml/min at ambient temperature. The detection wavelength of the diode array detector was set at 225, 227, 290, and 294 nm for ATE, MET, PPL, and TIM, respectively. The content of each drug was determined using its calibration curve with respect to the dilution factor.
Statistical Analysis
All of the experimental measurements were performed in triplicate. Resulted values were expressed as mean value ± standard deviation. The different percentage loading between NIPs and MIPs were analyzed using the independent samples t test. Statistical significance of differences in percentage loading of all polymer bead formulations was examined using one-way analysis of variance followed by least significant difference post hoc test. The significant level was set at p < 0.05.
RESULTS AND DISCUSSION
MIP Bead Synthesis and Characterization
In molecular imprinting processes, the selection of the template and cross-linker are important factors that affect the binding affinity and specificity of the imprinted polymer. Previous studies have demonstrated that imprinted microspheres selective for PPL can be synthesized using the precipitation polymerization method. Yoshimatsu et al. studied an effect of the cross-linker ratio (DVB and TRIM) on the properties of microspheres including their binding affinity, specificity, and size. The results indicated that use of the DVB cross-linker resulted in a higher binding affinity and specificity for PPL than the use of TRIM because of the additional π–π interaction with the aromatic moiety of (S)-propranolol in acetonitrile during the imprinting reaction with DVB (27). Therefore, the cross-linker (DVB) was selected for use in this work.
As shown in Table I, the formulations were prepared by the oil in water (o/w) emulsion polymerization method using MMA as the monomer, BP as the initiator, and different amounts of DVB and PPL as the cross-linker and imprinted template, respectively. The ratio of the monomer (MMA), DVB, and PPL affected the particle sizes and % yields of the obtained MIP and NIP. The percentage yield of MIP beads slightly increased with an increase in the amount of DVB (2–3 g), whereas the % yield of MIP beads decreased with an increase in the amount of PPL. The morphologies and particle sizes of the dried beads were determined using SEM, and the results are provided in Table II. The SEM images of the NIP revealed spherical and smooth surface without evidence of collapsed particles, whereas those of the MIP revealed spheres with small pores on the surface. The particle size of the beads was approximately 50–100 μm depending on the amount of DVB and PPL used. The particle size of the NIP decreased with an increasing amount of DVB. An increase in cross-linker content resulted in stronger binding interaction between the cross-linker and monomer during the polymerization process, which later resulted in a decrease in the bead size. As indicated in Table II, with an increasing the amounts of PPL, smaller particles were formed in all systems suggesting that the template compound has an important influence on the particle growth during the precipitation polymerization. A previous study reported that in the absence of a template, MAA can form hydrogen-bonded dimers in the non-imprinted system. In the imprinted system, there is an additional molecular interaction between MAA and propranolol, which might affect the growth of the cross-linked polymer nuclei, resulting in smaller polymer beads (27).
Table II.
SEM Images of Polymer Beads Obtaining PPL as a Template, MMA as a Functional Monomer, DVB as Cross-linker, and BP as an Initiator (at Magnification of ×100)

PPL propranolol, MMA methyl methacrylate, DVB divinylbenzene, BP benzoyl peroxide
The FTIR spectra of the NIP2 and MIP8 microspheres are presented in Fig. 2. The NIPs spectra exhibited a sharp intense peak at 1,732 cm−1, which is attributed to C=O stretching vibrations. The broad peak ranging from 1,300 to 1,000 cm−1 is attributed to C–O (ester bond) stretching vibrations. The broad peak from 3,100 to 2,900 cm−1 is attributed to the presence of C–H stretching vibrations. The FTIR spectrum of the MIP8 microspheres (Fig. 2b) exhibits the same pattern of peaks as that of the NIP2 microspheres except for the peaks at 799 cm−1, which are assigned to the substituted naphthalene of PPL. Because no change was observed in the FT-IR spectra in the area corresponding to the carbonyl group and C–O of ester bond and a change was observed in the area assigned to naphthalene of PPL, PPL is believed to have been embedded into the MIP beads without bonding.
Fig. 2.
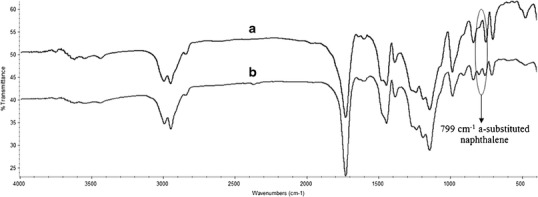
FTIR spectra of a NIP2 and b MIP8 beads
Because of PPL could not be bound in the drug solution during the drug loading process, therefore, the methyl groups of MIP were removed by alkali hydrolysis reaction to generate carboxylic acid functional groups. To evaluate the success of the alkali hydrolytic reaction of polymer beads, IR spectra of the obtained polymer beads were collected. The spectra of the non-hydrolyzed MIP8 and MIP8 microspheres are presented in Fig. 3. The FTIR spectra of the hydrolyzed MIP8 microspheres (Fig. 3b) exhibit all of the peaks that were observed for the non-hydrolyzed MIP8 microspheres (Fig. 3a); in addition, a broad and strong peak associated with O–H stretching of the carboxyl group is observed between 3,000 and 3,500 cm−1 in the spectrum of the hydrolyzed MIP8 microspheres.
Fig. 3.
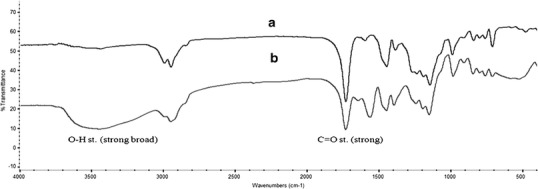
FTIR spectra of a non-hydrolyzed and b hydrolyzed MIP8 beads
Binding Ability of Polymer Beads to PPL
NIP3 and MIP9 were selected for this study. After the polymerization step, the template was rinsed from the polymer beads, and then the methyl groups of the polymer beads were removed via alkali hydrolysis to generate the carboxylic acid functional groups. The polymer beads were reloaded with PPL to evaluate the binding of PLL from the aqueous solution. In this reloading process, the weight ratios of PPL/MIP9 (1:0.5, 1:1, 1:2) or PPL/NIP3 (1:0.5, 1:1, 1:2), as a control, were varied. The results are presented in Fig. 4. The percentages of drug reloading at 48 h with the NIP3 formulation were 20%, 60%, and 100% for PPL to bead ratio of 1:0.5, 1:1, and 1:2, respectively (Fig. 4a). However, the percentages of drug reloading at 48 h for MIP9 increased more rapidly than that of NIP3 with values of 40%, 80%, and 100% for PPL to bead ratios of 1:0.5, 1:1, and 1:2, respectively (Fig. 4b). The percent loading of PPL increased with an increased the weight ratio of PLL/MIP. The equilibrium time of PPL loading to MIP was shorter than that with NIP, demonstrating a faster rate of binding. In addition, the percent drug loading of MIP was greater than that of NIP. This result was most likely due to the incorporation of PPL in the bead synthesis process which created the molecularly specific spaces. At the1:1 ratio, the PPL could be reloaded in the MIP9 at a higher extent and a faster rate than in NIP3. Therefore, the ratio of the drug to polymer bead of 1:1 was selected for further experiments.
Fig. 4.
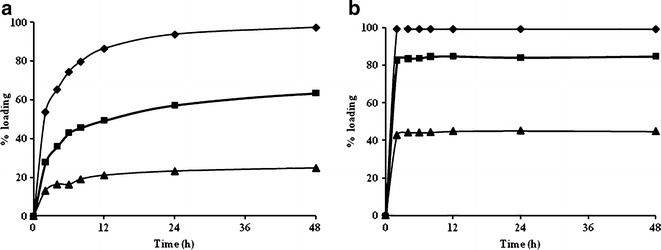
The percentage loading of PPL at drug-to-polymer beads ratios of triangle 1:0.5, square 1:1, and diamond 1:2 a NIP and b MIP
Figure 5 demonstrates the effect of the cross-linker (DVB) content on the percentage of PPL reloading for a drug to polymer bead ratio of 1:1. Figure 5b shows the percentage of PPL reloading in the polymer bead formulations MIP7 (DVB 2 g), MIP8 (DVB 2.5 g), and MIP9 (DVB 3 g) for a drug-to-polymer bead ratio of 1:1. The results demonstrate that the DVB cross-linker content affects the percentage of PPL reloading in the beads. The percentage of PPL reloading increased to a maximum when the DVB content was increased from 2 to 2.5 g, then decreased with a further increase in DVB content (3 g). This result might be due to the condensation of the polymer beads, which decreased drug penetration into the beads. The percentage of PPL reloading in the polymer bead formulations NIP1 (DVB 2 g), NIP2 (DVB 2.5 g), and NIP3 (DVB 3 g) for a drug to polymer bead ratio of 1:1 (Fig. 5a) exhibited the same pattern for the percentages of PPL reloading as that for the polymer bead formulations MIP7-9. However, the PPL could be loaded in all of the MIPs at a higher extent and a faster rate than in the NIPs. Therefore, NIP2 (MMA/DVB, 75:2.5) and MIP8 (PPL/MMA/DVB, 0.8:75:2.5) were selected for further experiments.
Fig. 5.
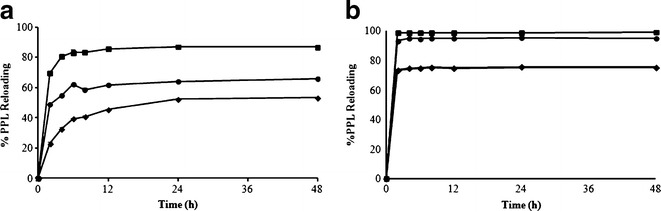
The percentage PPL reloading in a NIP; circle NIP1 (DVB 2 g), square NIP2 (DVB 2.5 g), diamond NIP3 (DVB 3 g) and b MIP; circle MIP7 (DVB 2 g), square MIP8 (DVB 2.5 g), diamond MIP9 (DVB 3 g) at a drug-to-polymer bead ratio of 1:1
Binding Ability of Polymer Beads to β-Blocker Drugs
In this study, the MIP8 and NIP2 formulas were used to study the effect of percentage of the β-blocker drug reloading. Comparing the binding ability, other β-blocker drugs (ATE, MET, and TIM) were similarly prepared using the weight ratio of β-blocker drug: MIP8 or NIP2 of 1:1. The mixture of the β-blocker drug solutions (PPL, ATE, MET, or TIM) was incubated with MIP8 and NIP2 at the weight ratio of 1:1. The concentration of each drug in the supernatant was determined using HPLC. The percentage reloading of the β-blocker drugs in NIP2 and MIP8 is indicated in Fig. 6. Comparing the binding ability to other β-blocker drugs (ATE, MET, and TIM), PPL exhibited the highest percentage drug reloading in MIP8 (>80%). In addition, the percentage drug loading of other β-blocker drugs in MIP8 was similar to that in NIP2 (40–60%). Thus, these MIP8 were selective for PPL.
Fig. 6.
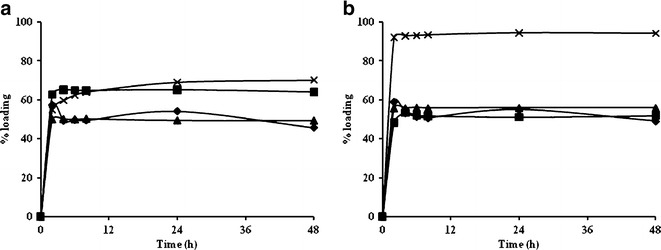
The percentage loading of β-blockers at a drug-to-polymer bead ratio of (1:1) a NIP2 and b MIP8; multiplication symbol propranolol, square metoprolol, triangle timolol, and diamond atenolol
Characterization of Fiber Membrane Containing MIP
Because it is water insoluble and exhibits pH independent solubility, Eudragit-RS100 was selected for the preparation of fibers containing NIP2 and MIP8. The MIP8 particles were reduced in size using ball mill and sieved through a 400 mesh. An irregular, rough morphology with diameters of 20.4 ± 6.5 μm was obtained. In this study, the MIP8 content was varied from 10 to 50% (w/w) with a 40% (w/v) Eudragit-RS100 in DMF/EtOH (33:67) solution before being spun into fibers. The Eudragit-RS100 solution containing MIP8 (>50%, w/w) could not be electrospun because of an excess amount of MIP, which clogged at capillary tip during the electrospinning process. Thereafter, the obtained fibers were dissolved in ethanol to liberate the embedded MIP beads. The % loading efficiency was increased by increasing the number of initially added polymer beads (Table III). SEM images confirmed that smooth, bead-free uniform fibers were observed in the blank fiber mats (Fig. 7a, b), whereas bead-on-string fibers were observed in the 50% (w/w) MIP8 fiber mats (Fig. 7c, d). Therefore, the 50% (w/w) MIP was selected for initial addition into the Eudragit-RS100 solution to produce affinity fiber mats.
Table III.
The Bead Content After the Electrospinning Process for an Initially Added 10–50% (w/w) of MIP8 Beads in a 40% (w/v) Eudragit-RS100 Solution
| Initial polymer beads (%, w/w) | Bead content (%) |
|---|---|
| 10 | 67.00 |
| 20 | 72.73 |
| 30 | 84.22 |
| 40 | 87.21 |
| 50 | 89.55 |
Fig. 7.

SEM images of Eudragit-RS100 fibers at magnifications of a ×100 and b ×350 and of 50% (w/w) MIP8 Eudragit-RS100 fibers at magnifications of c ×100 and d ×350
β-Blocker Binding Affinity of the MIP Fiber Mats
The selectivity for PPL of the fiber membrane containing MIP beads was investigated by determining its binding ability compared with other β-blocker drugs (ATE, MET, and TIM). A testing solution containing 0.01 M of each drug (PLL, ATE, MET, or TIM) was prepared. The percentage reloading of the β-blocker at a drug-to-polymer bead ratio of 1:1 for a NIP composite E-RS100 fiber (control) and a MIP composite Eudragit-RS100 fiber is indicated in Fig. 8. The results reveal that PPL can be bound at a higher extent and faster rate in the MIP composite fiber than in the NIP composite fiber. Moreover, PPL showed a higher affinity to the MIP than those of the other β-blockers. This result indicates that the MIP composite E-RS100 fiber had a higher selectivity for PPL than to the other β-blockers. The percentage drug reloading of NIP composite E-RS100 fiber was nearly 0%, which indicated that NIP is not selective for any of the tested drugs. Thus, the MIP composite E-RS100 fiber was selective for PPL.
Fig. 8.
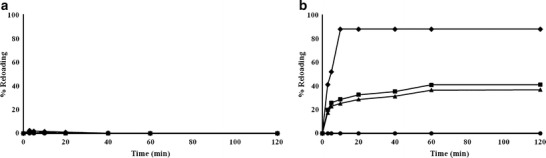
The percentage reloading of β-blocker drugs at a drug to polymer bead ratio of 1:1 for a 50% (w/w) NIP2 composite Eudragit-RS100 fiber mats and b 50% (w/w) MIP8 composite Eudragit-RS100 fiber mats; circle ATE, square MET, triangle TIM, diamond PPL
CONCLUSION
Molecularly imprinted microspheres selective for PPL were successfully synthesized by o/w emulsion polymerization and were characterized. The microspheres were incorporated at up to 50% (w/w) in Eudragit-RS100 electrospun nanofiber. The MIP composite Eudragit-RS100 fiber showed higher selectivity to PPL than to other β-blockers. Therefore, this membrane can be further developed for various applications in pharmaceutical and other affinity separation fields.
Acknowledgments
The authors would like to acknowledge Commission of Higher Education (Thailand) and the Thailand Research Funds through the Golden Jubilee Ph.D. Program (grant no. PHD/0092/2554) and the Silpakorn University Research and development institute for the financial support (grant no. SURID55/02/12).
References
- 1.Ma Z, Kotaki M, Ramakrishna S. Surface modified nonwoven polysulphone (PSU) fiber mesh by electrospinning: a novel affinity membrane. J Membr Sci. 2006;272(1–2):179–87. doi: 10.1016/j.memsci.2005.07.038. [DOI] [Google Scholar]
- 2.Ma Z, Kotaki M, Ramakrishna S. Electrospun cellulose nanofiber as affinity membrane. J Membr Sci. 2005;265(1–2):115–23. doi: 10.1016/j.memsci.2005.04.044. [DOI] [Google Scholar]
- 3.Borcherding H, Hicke H, Jorcke D, Ulbricht M. Surface functionalized microfiltration membranes for affinity separation. Desalination. 2002;149(1–3):297–302. doi: 10.1016/S0011-9164(02)00804-4. [DOI] [Google Scholar]
- 4.Mosbach K, Ramstrom O. The emerging technique of molecular imprinting and its future impact on biotechnology. Nat Biotechnol. 1996;14:163–70. doi: 10.1038/nbt0296-163. [DOI] [Google Scholar]
- 5.Ye L, Haupt K. Molecularly imprinted polymers as antibody and receptor mimics for assays, sensors and drug discovery. Anal Bioanal Chem. 2004;378:1887–97. doi: 10.1007/s00216-003-2450-8. [DOI] [PubMed] [Google Scholar]
- 6.Ye L, Weiss R, Mosbach K. Synthesis and characterization of molecularly imprinted microspheres. Macromolecules. 2000;33:8239–45. doi: 10.1021/ma000825t. [DOI] [Google Scholar]
- 7.Vlatakis G, Andersson LI, Miller R, Mosbach K. Drug assay using antibody mimics made by molecular imprinting. Nature. 1993;361:645–7. doi: 10.1038/361645a0. [DOI] [PubMed] [Google Scholar]
- 8.Yan H, Row KH. Characteristic and synthetic approach of molecularly imprinted polymer. Int J Mol Sci. 2006;7:155–78. doi: 10.3390/i7050155. [DOI] [Google Scholar]
- 9.Lubke C, Lubke M, Whitcombe MJ, Vulfson EN. Imprinted polymers prepared with stoichiometric template–monomer complexes: efficient binding of ampicillin from aqueous solutions. Macromolecules. 2000;33:5098–105. doi: 10.1021/ma000467u. [DOI] [Google Scholar]
- 10.Yao LD, Tang YW, Huang ZF. Nicotinic acid voltammetric sensor based on molecularly imprinted polymer membrane-modified electrode. Anal Lett. 2007;40:677–88. doi: 10.1080/00032710601017755. [DOI] [Google Scholar]
- 11.Haupt K, Mosbach K. Molecularly imprinted polymers and their use in biomimetic sensors. Chem Rev. 2000;100:2495–504. doi: 10.1021/cr990099w. [DOI] [PubMed] [Google Scholar]
- 12.Kriz O, Ramstrom O, Mosbach K. Molecular imprinting: new possibilities for sensor technology. Anal Chem. 1997;69:345A–9. doi: 10.1021/ac971657e. [DOI] [Google Scholar]
- 13.Ansell RJ, Kriz D, Mosbach K. Molecularly imprinted polymers for bioanalysis: chromatography, binding assays and biomimetic sensors. Curr Opin Biotechnol. 1996;7:89–94. doi: 10.1016/S0958-1669(96)80101-7. [DOI] [PubMed] [Google Scholar]
- 14.Liu HY, Row KH, Yan GL. Monolithic molecularly imprinted columns for chromatographic separation. Chromatographia. 2005;61:429–32. doi: 10.1365/s10337-005-0531-x. [DOI] [Google Scholar]
- 15.Hwang CC, Lee WC. Chromatographic resolution of the enantiomers of phenylpropanolamine by using molecularly imprinted polymer as the stationary phase. J Chromatogr B. 2001;765:45–53. doi: 10.1016/S0378-4347(01)00397-8. [DOI] [PubMed] [Google Scholar]
- 16.Martin PD, Jones GR, Stringer F, Wilson ID. Comparison of normal and reversed-phase solid phase extraction methods for extraction of β-blockers from plasma using molecularly imprinted polymers. Analyst. 2003;128:345–50. doi: 10.1039/b211787h. [DOI] [PubMed] [Google Scholar]
- 17.Lavignac N, Allender CJ, Brain KR. Current status of molecularly imprinted polymers as alternatives to antibodies in sorbent assays. Anal Chim Acta. 2004;510:139–45. doi: 10.1016/j.aca.2003.12.066. [DOI] [Google Scholar]
- 18.Ye L, Mosbach K. Molecularly imprinted microspheres as antibody binding mimics. React Funct Polym. 2001;48:149–57. doi: 10.1016/S1381-5148(01)00050-5. [DOI] [Google Scholar]
- 19.Chianella I, Lotierzo M, Piletsky SA, Tothill IE, Chen B, Karim K, et al. Rational design of a polymer specific for microcystin-LR using a computational approach. Anal Chem. 2002;74:1288–93. doi: 10.1021/ac010840b. [DOI] [PubMed] [Google Scholar]
- 20.Son SH, Jegal J. Chiral separation of D, L-serine racemate using a molecularly imprinted polymer composite membrane. J Appl Polym Sci. 2007;104:1866–72. doi: 10.1002/app.25845. [DOI] [Google Scholar]
- 21.Zaidi SA, Cheong WJ. Robust open tubular layer of S-ketoprofen imprinted polymer for chiral LC separation. J Sep Sci. 2008;31:2962–70. doi: 10.1002/jssc.200800160. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimatsu K, Ye L, Lindberg J, Chronakis IS. Selective molecular adsorption using electrospun nanofiber affinity membranes. Biosens Bioelectron. 2008;23:1208–15. doi: 10.1016/j.bios.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Bhardwaj N, Kundu SC. Electrospinning: a fascinating fiber fabrication technique. Biotechnol Adv. 2010;28:325–47. doi: 10.1016/j.biotechadv.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Bamford CH, Al-Lamee KG, Pm-brick MD, Wear TJ. Studies of a novel membrane for affinity separations. I. Functionalisation and protein coupling. J Chromatogr. 1992;606:19–31. doi: 10.1016/0021-9673(92)85253-P. [DOI] [Google Scholar]
- 25.Ongun MZ, Ertekin K, Gocmenturk M, Ergun Y, Suslu A. Copper ion sensing with fluorescent electrospun nanofibers. Spectrochim Acta A Mol Biomol Spectrosc. 2012;90:177–85. doi: 10.1016/j.saa.2012.01.042. [DOI] [PubMed] [Google Scholar]
- 26.Charernsriwilaiwat N, Opanasopit P, Rojanarata T, Ngawhirunpat T. Lysozyme-loaded, electrospun chitosan-based nanofiber mats for wound healing. Int J Pharm. 2012;427:379–84. doi: 10.1016/j.ijpharm.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimatsu K, Reimhult K, Krozer A, Mosbach K, Sode K, Ye L. Uniform molecularly imprinted microspheres and nanoparticles prepared by precipitation polymerization: the control of particle size suitable for different analytical applications. Anal Chim Acta. 2007;584:112–21. doi: 10.1016/j.aca.2006.11.004. [DOI] [PubMed] [Google Scholar]