

Abstract
Poly(ε-caprolactone) implants containing etoposide, an important chemotherapeutic agent and topoisomerase II inhibitor, were fabricated by a melt method and characterized in terms of content uniformity, morphology, drug physical state, and sterility. In vitro and in vivo drug release from the implants was also evaluated. The cytotoxic activity of implants against HeLa cells was studied. The short-term tolerance of the implants was investigated after subcutaneous implantation in mice. The original chemical structure of etoposide was preserved after incorporation into the polymeric matrix, in which the drug was dispersed uniformly. Etoposide was present in crystalline form in the polymeric implant. In vitro release study showed prolonged and controlled release of etoposide, which showed cytotoxicity activity against HeLa cells. After implantation, good correlation between in vitro and in vivo drug release was found. The implants demonstrated good short-term tolerance in mice. These results tend to show that etoposide-loaded implants could be potentially applied as a local etoposide delivery system.
KEY WORDS: etoposide, implant, poly(ε-caprolactone), prolonged release
INTRODUCTION
In recent decades, cancer has become an evident public health problem worldwide. The World Health Organization estimates that in the year 2030, 27 million incidents of cancer and 17 million cancer deaths can be expected (1). Several efforts have been made to obtain more effective treatments for malignant tumors, especially solid cancer, which accounts for about half of all cancer cases worldwide. In addition, these tumors are characterized by not responding well to conventional systemic chemotherapy or radiotherapy, more so when the cancers are large or poorly vascularized (2).
Treatment of solid tumors through locoregional therapy has been widely studied, and usage of polymeric implants containing the anticancer drug acts as an advantageous alternative. The implants are a sustained drug delivery system that can be inserted in the region where the tumor is located or within it. This increases the tumor’s exposure to the drug and also limits systemic toxicity. In addition, local maintenance of the therapeutic levels for a long period of time optimizes the chemotherapy regimen by reducing the number of doses to be administered (2).
Currently, some successful drug delivery systems are already commercially available, such as Gliadel® Wafer, a biodegradable polyanhydride wafer containing the chemotherapeutic carmustine, used clinically to treat and prevent glioblastoma, a brain cancer. Vantas®, another drug delivery system containing histrelin acetate, is used for the palliative treatment of advanced prostate cancer (3).
The implant can be a preformed device or an in situ forming polymeric system. The in situ forming polymeric systems are low viscous formulations that are injected and solidify in situ to form solid or semi-solid drug depots. These implants are formed from different mechanisms and are classified into: in situ cross-linked polymer systems, in situ solidifying organogels, and in situ phase separation systems (4,5). Kang et al. (6) verified that an injectable drug depot for doxorubicin prepared with poly(ethylene glycol)-b-polycaprolactone diblock copolymer gel showed a good in vivo efficacy after intratumoral injection. This system was more efficacious in inhibiting the growth of the B16F10 tumor implanted subcutaneously on mice than the single injection of the pure drug solution, and the biodistribution results implied fewer off-target side effects. New applications and strategies for the development of injectable biomaterials that form three-dimensional structures in situ have been studied. Wang et al. (7) employed oppositely charged poly(d,l-lactic-co-glycolic acid) nanoparticles to create a cohesive colloidal gel used as an injectable dexamethasone-loaded filler to promote healing in bone defects. A colloidal system also constituted by oppositely charged nanoparticles was developed for the controlled release of an immunomodulating peptide for the treatment of autoimmune encephalomyelitis (8).
Preformed implants are administered by a special application device or need a minor surgery to be inserted (4). These implants can be prepared from both biodegradable and non-biodegradable polymers. Biodegradable polymeric systems are advantageous over non-biodegradable systems since there is no need for surgical removal of the implant after the complete release of the drug. Polycaprolactone (PCL), a biodegradable and biocompatible polymer, is suitable for controlled drug delivery due to its high permeability to several drugs, its ability to be fully excreted from the body, and the possibility of a long, sustained drug release rate (9).
Etoposide is a semisynthetic derivative of podophyllotoxin, a compound extracted from the roots and rhizomes of the plants Podophyllum peltatum and Podophyllum emodi (10). It is a cytotoxic drug and its mechanism of action is believed to be the inhibition of the topoisomerase II enzyme. Etoposide is widely used in chemotherapy for various solid tumors, including small cell lung carcinoma, testicular tumor, stomach cancer, ovarian cancer, and retinoblastoma (11). Since the aqueous solubility of etoposide is very low, this drug is commercialized in the form of non-aqueous parenteral solutions for intravenous use and oral soft gelatin capsules. However, both of these formulations have disadvantages. Etoposide precipitates from the parenteral solution when diluted with infusion fluids. In addition, cases of hypotension resulting from the rapid infusion of the drug and hypersensitivity reactions related to excipients of the formulation (ethanol, benzyl alcohol, polysorbate 80, and polyethylene glycol) have also been reported (12,13). The oral administration of capsules containing a solution of etoposide in a solvent mixture exhibits low and variable bioavailability due, in part, to the inactivation of the drug in gastrointestinal fluids (13,14).
Attempts have been made to overcome the limitations of the formulations available in the market. Several reports have described the development of drug delivery systems containing etoposide, such as polymeric nanoparticles (13,15,16), microemulsion (17), solid lipid nanoparticles (18,19), and microspheres (20,21). However, polymeric implants consisting of etoposide and PCL have not been reported to date. For this study, an etoposide-loaded PCL implant was developed and characterized using analytical techniques, such as scanning electron microscopy (SEM), differential scanning calorimetry (DSC), Fourier transform infrared spectroscopy (FTIR), X-ray diffraction analysis (XRD), content uniformity, and sterility. The in vitro release of etoposide from the implant and preliminary in vitro bioactivity were also studied. Additionally, the in vivo release profile of the drug and the short-term tolerance of the implants were evaluated through their subcutaneous implantation on the back of mice.
MATERIALS AND METHODS
Materials
Poly-ε-caprolactone (PCL; molecular weight, 14,000) was purchased from Sigma-Aldrich Chemicals (USA). Etoposide was offered by Quiral Química (Brazil) and the etoposide chemical reference substance was purchased from the United States Pharmacopoeia (USA). Ultrapure water was provided by a Milli-Q® purification system (Millipore, USA). HPLC grade acetonitrile was purchased from Merck® (Brazil). The other solvents and reagents used were of analytical grade.
Preparation of the Implants Containing PCL and Etoposide
PCL was melted at 60°C over a water bath and etoposide was thoroughly dispersed in the polymer melt. The resulting mixture of PCL and etoposide (1:1) was allowed to cool at room temperature and molded in cylinders at 60°C.
Characterization
Fourier Transform Infrared Spectroscopy
Infrared spectra were generated with an FTIR spectrophotometer (model IR-Prestige 21, Shimadzu). Measurements were carried out using the attenuated total reflectance technique. Each spectrum was a result of 32 scans with a resolution of 4 cm−1.
Thermal Analysis
DSC analysis was carried out with TA Instruments (model 2910, modulated DSC) using aluminum pans closed with perforated lids. About 5 mg of the samples was used, under nitrogen atmosphere (50 mL min−1), at a heating rate of 10°C min−1 and a temperature range of 30 at 300°C. Thermogravimetry (TG) curves were obtained using similar conditions as those for the DSC, except at the temperature range of 30 at 500°C.
X-Ray Diffraction Analysis
XRD patterns were obtained using an X-ray diffractometer (model PW 3710, Philips) with a target Cu-Kα radiation (l = 1.54 Ǻ) and equipped with a nickel filter. Diffraction patterns were obtained over a 2θ range of 1 at 90° at the rate of 1°/min.
Scanning Electron Microscopy
SEM was performed using a JEOL microscope (model JSM-6360LV) operating at 15 kV. All micrographs were obtained from the fracture surfaces coated with gold. The surfaces of the implants were observed at ×100–5,000 magnification. The photomicrographs were adjusted using the software packages Adobe Photoshop 6.0 and Adobe Illustrator 9.01 (Adobe Systems Incorporated, 2000, USA).
Content Uniformity of the Etoposide-Loaded PCL Implants
Determination of the content uniformity of etoposide in the PCL implants was performed according to the method stated in the Brazilian Pharmacopeia (22). Ten implants were selected and weighed. Each implant was transferred to a 10-mL volumetric flask and dissolved in a mixture of acetic acid 4% (v/v) and acetonitrile (1:1). Subsequently, an aliquot of 3 mL of the obtained solution was transferred to a 10-mL volumetric flask and the volume was adjusted with phosphate-buffered saline (PBS; pH 7.4). The amount of etoposide was determined using high-performance liquid chromatography (HPLC).
HPLC Method for the Determination of Etoposide
The method described by Solano et al. (23) was used to determine the amount of etoposide in the implants and that released in the in vitro study. The samples were analyzed using a Thermo Surveyor System (USA) that included a quaternary pump, an autosampler, a diode array detector, and ChromQuest 4.2 software. The Ace C18 column (250 × 4.6-mm i.d., 5-μm particle size) from ACT was used and maintained at 25°C. The mobile phase comprised acetic acid 4% (v/v) and acetonitrile (70:30) at a flow rate of 2 mL/min. The injection volume was 25 μL and detection was performed at 285 nm.
Sterilization and Sterility Test for Implants
The implants were sterilized by exposing them to UV radiation at 254 nm for 30 min (24). The direct inoculation method, as described in the Brazilian Pharmacopeia (22), was used to test the sterility of the irradiated implants.
In Vitro Release of Etoposide from the PCL Implants
In vitro etoposide release was carried out in quintuplicate in the release medium (PBS, pH 7.4) under sink conditions. The sink conditions are “defined as the volume of medium at least three times that required in order to form a saturated solution of drug substance” (25), then each implant was immersed in 30 mL of PBS since the solubility of etoposide in PBS at 37°C is 125.93 μg/mL (26). The tubes containing the implant and PBS were kept in an incubator at 37°C and 30 rpm for 12 months. At predetermined time points, 15 mL of the release medium was taken out and replaced with 15 mL of fresh medium. Etoposide concentration in the release medium was determined by HPLC and expressed as the cumulative percentage of etoposide released in the medium. The average of the obtained measurements each time was calculated and used to plot the in vitro release profile curve.
Mechanism of Drug Release
Various mathematical models to describe the mechanisms of drug release from polymeric systems have been reported in the literature (27,28). The release data were evaluated by model-dependent (curve fitting) and two theoretical models were used: Higuchi and Korsmeyer–Peppas models.
Higuchi describes drug release as a diffusion process based on Fick’s law, according to the Eq. 1.
| 1 |
where Mt/M∞ is the fractional drug release at time t and KH is Higuchi’s dissolution constant. According to this model, a straight line is expected for the plot of Mt/M∞versus the square root of time if the drug release from the matrix is based on a diffusion mechanism.
The Korsmeyer–Peppas model considers that the drug release mechanism may deviate from Fick’s law and follow an anomalous behavior, described by Eq. 2.
| 2 |
where Mt/M∞ is the fractional drug release at time t, K is the kinetic constant, and n is the diffusional exponent characteristic of the release mechanism. The value of n is related to the geometrical shape of the polymeric system and determines the release mechanism conformation, shown in Table I for cylindrical devices.
Table I.
Release Exponent n and Drug Release Mechanism from Cylindrical Polymeric Delivery Systems
| Exponent (n) | Drug release mechanism |
|---|---|
| 0.45 | Fickian diffusion |
| 0.45 < n < 0.89 | Anomalous transport |
| 0.89 | Polymer swelling |
To calculate the kinetic parameters of both models, the first 60% drug release data were used since only this portion of the release curve should be used for the determination of parameter n of the Korsmeyer–Peppas model (27). The determination coefficient (r2) was used to define the best fit between the two models. Thus, the model that provided r2 closest to 1 was considered more adequate.
Preliminary Study of In Vitro Bioactivity
Evaluation of in vitro cytotoxicity activity was carried out with a cervical cancer line (HeLa). HeLa cells were maintained in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum and were cultured in a CO2 incubator containing 5% CO2 at 37°C. The cells were inoculated in 96-well microtiter plates and then pre-incubated for 24 h. Then, the old medium was removed and the testing solutions were added to each well. For the preparation of the testing solutions, etoposide-loaded PCL or PCL-only implant was placed in the culture medium and incubated at 37°C for 7 days. In addition, a solution of etoposide was prepared at the same etoposide concentration released from the PCL implant after incubation for 7 days. After adding the test solutions, the cells were incubated again for 48 h and cell viability was determined using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. MTT reagent (Sigma-Aldrich) was added to each well and the plates were incubated for 3 h at 37°C. Intracellular formazan crystals were solubilized in dimethyl sulfoxide and color intensity was measured at 550 nm. The antiproliferative effect of the different treatments was calculated as a percentage of cell growth with the control (wells containing the culture medium as a testing solution).
In Vivo Profile
Twenty-five female Swiss mice (6–8 weeks old) from the Centro de Bioterismo of the Federal University of Minas Gerais were maintained in individual cages, with food and water ad libitum, and controlled temperature and humidity in the animal house of the School of Pharmacy of the Federal University of São João Del Rei (UFSJ). The experiments were approved by the Ethics Committee in Animal Experimentation at UFSJ. The animals were randomly divided into five groups. Five implantation periods were used: 5, 10, 15, 20, and 25 days. Animals were anesthetized with an intraperitoneal injection of a mixture of xylazine (10 mg/kg) and ketamine hydrochloride (100 mg/kg). Their dorsal hair was shaved and the skin disinfected with 70% ethanol. A 1-cm incision was made in the dorsal lumbar region and sterile implant was aseptically inserted in the dorsal subcutaneous space. Subsequently, the incision was sutured. After the implantation procedure, the animals were housed individually, with water and food given ad libitum. The light/dark cycle was 12/12 h, with lights switched on at 7:00 am and switched off at 7:00 pm. Postoperatively, the animals were monitored regularly for complications related to surgery, as signs of infection at the operative site, or signs of discomfort, or distress. Any animals presenting such signs were immediately killed. At each time point, the mice (n = 5) were euthanized. Then, the implants, surrounding skin and subcutaneous tissue, heart, lung, spleen, liver, and the kidney were carefully retrieved. The implants were dissolved in a fixed volume of acetonitrile and the amount of etoposide remaining in the implants was measured using the HPLC method described earlier. The drug content obtained was used to calculate the amount of drug released within the subcutaneous tissue of the mice.
The in vitro data and in vivo results obtained were correlated by plotting the in vitro against the in vivo drug release. The regression coefficient was calculated using Minitab®.
Histological Evaluation
For histological evaluation, the tissue was separated from the implant and processed for histology. The tissue was fixed in neutral buffered formalin (10%) and then dehydrated in a graded ethanol series, cleared in xylene, and embedded in paraffin using standard techniques. Paraffin sections were stained with hematoxylin–eosin and evaluated under a light microscope.
RESULTS AND DISCUSSION
In cancer chemotherapy, polymeric implants have been investigated for drug delivery since these systems may improve the treatment regimen by increasing the local concentration and decreasing the incidence of side effects (2). In the present study, PCL implants containing etoposide were prepared using the melt method. This procedure is favored by a low melting temperature (60°C) and a low glass transition temperature (−60°C) of PCL (29). The cylindrical implants produced showed 2.26 ± 0.08 mg in average weight, 6.34 ± 0.18 mm length, and 0.62 ± 0.02 mm diameter (n = 10).
As previously reported, the rate of drug release from polymeric devices depends on the environmental conditions, physicochemical properties of the polymer, drug characteristics, the micro-organization of the components in the system, the shape and size of polymeric devices, and drug loadings on drug release (29,30). Thus, SEM, thermal analysis (DSC and TG), FTIR, and XRD were used to investigate the physicochemical properties of the implants and its components.
FTIR spectroscopy was performed to ensure that no chemical interaction between the drug and the polymer had occurred. The FTIR spectra of pure etoposide, etoposide-loaded PCL implant, physical mixture of etoposide and PCL (1:1), and PCL implant are shown in Fig. 1a–d, respectively. A characteristic band at 3,446 cm−1 related to the stretching of the phenolic –OH group; bands at 1,614, 1,504, and 1,459 cm−1 corresponding to C=C aromatic group stretching; and a band for the lactone group at 1,760 cm−1 can be seen in the infrared spectrum of etoposide. Similar types of bands were also observed earlier (13). The FTIR spectrum of PCL showed bands at 2,944 and 2,865 cm−1 which can be attributed to –CH2 group stretching and an intense band at 1,724 cm−1 that is due to the presence of an ester carbonyl group in the polymer. Typical absorption bands of the functional groups of etoposide and PCL were observed in the FTIR spectra of the physical mixture of etoposide and PCL (1:1) and etoposide-loaded PCL implant, indicating the absence of chemical interactions between the polymer and the drug during the manufacturing process of the implants and the presence of the drug as a molecular dispersion in the polymer matrix.
Fig. 1.
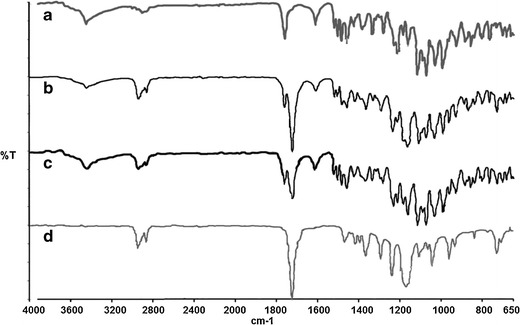
FTIR spectra of etoposide (a), etoposide-loaded PCL (b), physical mixture of etoposide and PCL (1:1) (c), and PCL (d) implants
Thermal analyses, as well as DSC and TG, have been used to determine the physical state of the polymer and the drug in the formulation and to evaluate the possibility of any interactions between the drug and the polymer within the polymeric matrix (30–32). Thus, the thermal behaviors of etoposide, PCL, and etoposide-loaded implants were analyzed using DSC and TG. The DSC curve of etoposide (Fig. 2b) shows a broad endothermic peak between 41.8°C and 121°C, attributed to a dehydration reaction, which was confirmed clearly in the TG thermogram (Fig. 3). This endothermic event also was observed by Jasti et al. (33), Mohanty et al. (34), and Wu et al. (35). After drug dehydration, there was no significant weight loss until the decomposition observed above 290°C (292–412°C), as shown in Fig. 3 (13,33). In Fig. 2a, it is possible to observe a second endothermic peak in the range of 173 at 187°C, centered at about 183°C, which appears to be related to a possible melting of the drug (34,36). The third event corresponds at an exothermic peak (213–230°C), which is probably related to crystallization to a different polymorphic form since the occurrence of an endothermic event, followed by an exothermic event, is usually associated with polymorphic transitions due to melting followed by recrystallization (30). Jasti et al. (33) studied the polymorphic forms of etoposide and reported that the dehydrated drug, after melting, recrystallizes to a different polymorphic form at 206°C. A third endothermic event in the range of 270 at 286°C may be attributed to the melting point of the newly formed etoposide, as reported previously by Shah et al. (13) and Jasti et al. (33). Drug decomposition occurs just after its melting, which can be confirmed by the TG thermogram (Fig. 3a) (13).
Fig. 2.
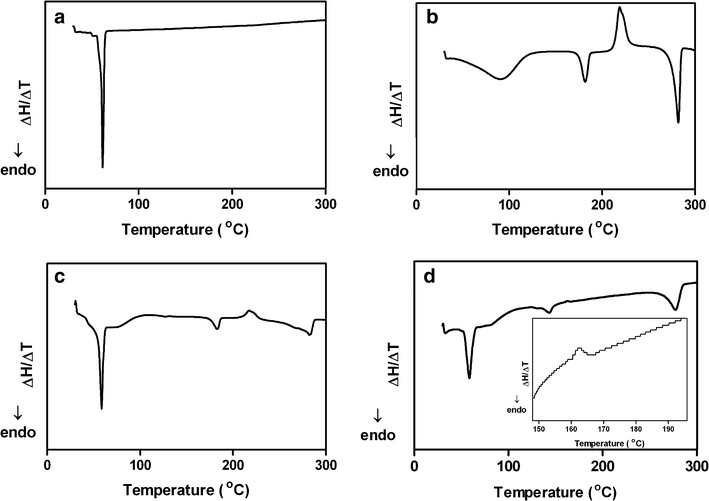
DSC curves of PCL (a), etoposide (b), physical mixture of etoposide and PCL (c), and etoposide-loaded PCL (d) implants
Fig. 3.
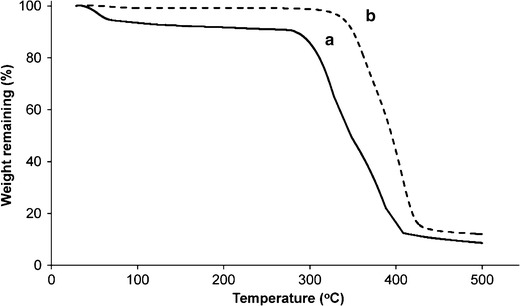
TG curves of etoposide (a) and etoposide-loaded PCL (b) implants
From the DSC curve of PCL (Fig. 2a), it is possible to observe a melting peak between 54.8°C and 63.5°C, centered at about 61°C (20,31). Shown in Fig. 2c is the DSC curve for the physical mixture of the drug and the polymer with an identical proportion to the drug-loaded implant. A similar thermal behavior to that identified for pure drug and polymer was observed, with the first event between 51.5°C and 63.3°C (approximately at 59°C) due to the melting of the PCL in the physical mixture. The second endothermic peak (174–188°C) is attributed to the melting of the drug, followed by the exothermic event due to polymorphic transition occurring in a temperature range of about 209–230°C. Finally, the event between 261°C and 288°C is assigned to the melting point of the polymorphic form of etoposide.
The DSC curve for the etoposide-loaded implant (Fig. 2d) showed the original peak of PCL (52.8–64.3°C, approximately at 58°C). A broad endothermic peak (135–150°C) and an exothermic event (160–165°C) of low intensity were attributed to the melting and crystallization of etoposide, respectively. The melting point and the phase transition of the drug occurred at lower temperatures than that identified for the neat etoposide. These results may be due to the possible dissolution of part of the drug into the melting PCL during the fabrication process. A similar thermal behavior has been reported previously in the literature by Cheng et al. (31). Marsac et al. (37) verified that a drug identified as miscible with a polymeric carrier polymer showed a melting point depression of the drug in the drug–polymer mixture. However, systems considered as immiscible showed little or no melting point depression of the drug since the drug remained unaltered once embedded in the carrier polymer. The endothermic event that occurred in the range of about 261–285°C may be attributed to the melting point of the newly formed etoposide.
The TG thermogram of the etoposide-loaded implant (Fig. 3b) showed a single well-defined event corresponding to the thermal degradation of the device that occurred after 342°C (342–430°C). These data confirm an increment in the etoposide thermal stability in the polymeric implant. Enhancement in the thermal stability of the drug by the presence of the polymer was reported in the literature by Oliveira et al. (30), which verified that the decomposition of methotrexate that encapsulated into PLGA nanoparticles occurred in a temperature above that observed for the pure drug.
The thermal analysis results are in agreement with the FTIR data, indicating that no significant interaction between the etoposide and the PCL were observed in the implants.
XRD analysis was performed with the aim of complementing the results obtained from the thermal analysis. Thus, XRD was used to characterize the crystalline state of the drug and the polymer in the implant. Figure 4a–c shows the XRD patterns for etoposide, PCL implant, and the etoposide-loaded PCL implant, respectively. PCL displays two characteristic peaks at 21.21° and 23.61°, confirming its semi-crystalline structure (38). The sharpness and the intensity of the peaks in the diffraction pattern of etoposide confirmed its crystalline nature. Etoposide showed six principal peaks at 10.53°, 16.53°, 19.65°, 21.09°, 23.55°, and 25.35°. The aforementioned peaks are seen in the XRD pattern of the etoposide-loaded PCL implant, but with lessened intensity than that of the pure drug and polymer. This suggested that a certain fraction of the etoposide crystals was dissolved into the melting PCL during the fabrication process. These data confirmed the results obtained with thermal analysis.
Fig. 4.
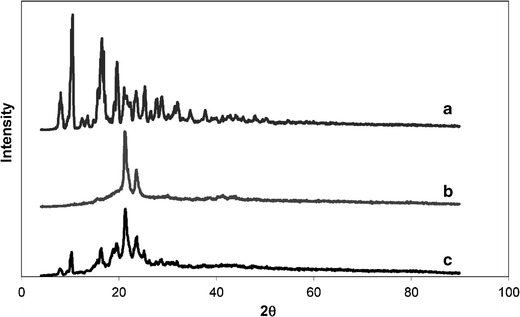
XRD patterns for etoposide (a), PCL (b), and etoposide-loaded PCL (c) implants
One possible disadvantage associated with the melting fabrication process of the implants is the non-uniform dispersion of the drug in the polymeric matrix, due principally to a short time of mixing of the components (39). Determination of the content uniformity of etoposide in the PCL implants was carried out to evaluate the mixing phase during the fabrication process. All tested units (n = 10) had etoposide content almost equal to the predefined value of etoposide in the formulation (50%, w/w). In addition, the variation limit of the etoposide content of the tested implants (7.86%) was lower than the pharmacopeic specification (15%) (22). These results confirm that etoposide presents a uniform distribution in the PCL implants.
UV radiation, used to sterilize the polymeric implants, caused no changes in the chemical structure of etoposide and PLC in the implant since no change was observed in the FTIR spectrum of sterile implants. In addition, there was no significant difference (p > 0.05) between the content of etoposide present in sterile (100.78 ± 0.95%) and non-sterile implants (101.07 ± 0.67%). Microbial growth was not observed in any tube containing the implant and the culture medium, confirming the sterility of the implants exposed to UV radiation.
The morphology of the implant is an important characteristic for the drug release rate that the system can supply. SEM was used to evaluate the microstructure of the polymeric drug delivery system. The surface of the implant was found to be smooth and homogeneous, with no evidence of pores or channels, as shown in Fig. 5a. However, the cross-section of the etoposide-loaded implant revealed a few pores (Fig. 5b).
Fig. 5.
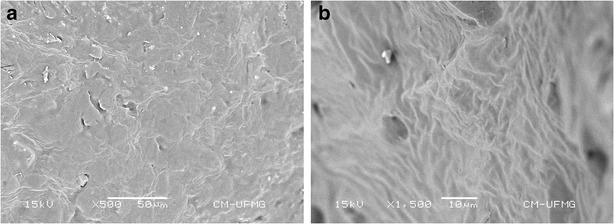
SEM images of the external surface (a) and cross-section (b) of the etoposide-loaded implant
The cumulative release of etoposide from the implant is shown in Fig. 6. The release profile demonstrated a small burst effect phase, followed by slow release over a prolonged period. During the first 15 days, approximately 12% of the etoposide was released from the implant. This initial fast release was considered to be a result of the fast dissolution and diffusion of the drug at the solid–liquid interface. In the second phase, the drug release rate gradually slowed down; approximately 66% of the etoposide was released from the implant. The slow and constant rate of drug release probably was dominated by the diffusion of etoposide from the polymer since PCL is characterized by a very low hydrolysis rate, which can extend over a period of more than 1 year (9). In addition, the low water solubility of etoposide may make its release to the medium difficult, making its diffusion very slow.
Fig. 6.
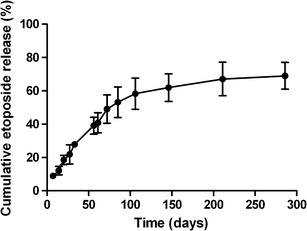
In vitro release profile of etoposide from the manufactured implants. Results represent the mean ± standard deviation (n = 5 for each time)
Several preclinical and clinical findings suggest that the duration of exposure of neoplastic cells to etoposide is important in producing maximal antitumor activity (40–42). This fact may be due to etoposide being a phase-specific cytotoxic drug. Etoposide inhibits topoisomerase II that is most active during the late S and G2 phases of the cell cycle. This drug induces the stabilization of the enzyme–DNA complex, causing double- and single-strand breaks in the DNA and, thus, leading to cell cycle arrest in G2 and subsequent triggering of apoptosis (43,44). Enzymatic inhibition is reversible, and the dissociation of the DNA–topoisomerase II–etoposide complex allows DNA repair and consequently reduces the cytotoxic activity of the drug. Thus, prolonged exposure to the drug could produce longer periods of enzyme inhibition, resulting in the increased cytotoxicity of etoposide (43,45). Then, the fact that the PCL implant is able to control the drug release for a prolonged period may be considered an important advantage of the evaluated system.
To investigate the kinetics of drug release from the implant, two theoretical models describing drug release from polymeric systems were considered: the Higuchi and Korsmeyer–Peppas models. To calculate the kinetic parameters of both models, the first 60% drug release data were used since only this portion of the release curve should be used for the determination of parameter n of the Korsmeyer–Peppas model (27). The determination coefficient (r2) and kinetic parameters of each model are listed in Table II. The best fit for the drug release profile of the etoposide-loaded implant was obtained using the Korsmeyer–Peppas model, which had a higher coefficient of determination compared to the other model. The magnitude of the release exponent n (0.691) in the Korsmeyer–Peppas model indicated that the mechanism that led to the release of etoposide was an anomalous transport since the value of n was between 0.45 and 0.81 (28). Thus, drug release probably occurs by an overlapping of different phenomena, including drug diffusion and polymer swelling.
Table II.
Data of Drug Release Profiles Fitted by Two Kinetic Models
| Kinetic model | r 2 | k | n |
|---|---|---|---|
| Higuchi | 0.9898 | 6.780 ± 0.243 | – |
| Korsmeyer–Peppas | 0.9922 | 0.876 ± 0.079 | 0.691 ± 0.022 |
Studies show that etoposide presents cytotoxic activity against HeLa cells (46,47). Figure 7 shows the in vitro cytotoxicity of the etoposide-loaded implants against this cell line. Samples containing PCL implant showed no significant cytotoxic effect on HeLa cells, as expected since PCL is a biocompatible polymer. Samples containing etoposide released from the polymeric implant after incubation in the culture medium for 7 days showed significant cytotoxic activity. In order to compare the activity observed with that expected for the polymeric system after 7 days of incubation, the concentration of etoposide present in the medium was calculated based on the in vitro release profile. Then, a solution of this concentration was prepared and evaluated for bioactivity. There was no significant difference (p > 0.05) between the activity of the freshly prepared etoposide solution (37 μg/mL) and the drug released from the implants. This result proves that etoposide remains active after the process of preparation of the implants and for a period of 7 days of incubation in the culture medium.
Fig. 7.
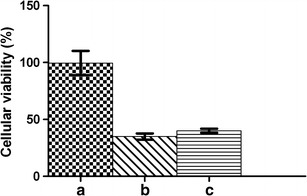
Viability of HeLa cells treated with different samples: culture medium in which the PCL implants (a) and etoposide-loaded implants (b) were incubated for 7 days and the etoposide solution of 37 μg/mL (c). Data are presented as the mean of the survival relative to the untreated control ± standard deviation of three independent experiments
The etoposide-loaded implants promoted the sustained release of drug within the subcutaneous tissue of female mice over a period of 25 days (Fig. 8). These implants showed a similar behavior under in vivo and in vitro conditions. The polymeric systems exhibited an initial burst release, where approximately 6% of the etoposide was released over the first 5 days. The burst effect in the initial stage could be due to a faster dissolution of the drug deposited on the surface of the implant. The rate of drug release markedly decreased from the fifth to the tenth day and remained almost constant until the 25th day, indicating that the implants promoted the sustained release of the antitumoral activity in the subcutaneous tissue of mice. During the period between the 6th and the 25th day, approximately 19% of the drug was released from the implant, which was found to be nearly 1.2-fold lower than the in vitro release.
Fig. 8.
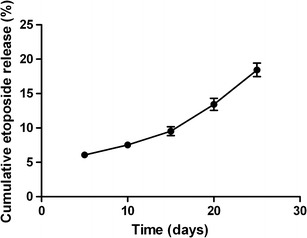
In vivo cumulative release profile for etoposide-loaded implants inserted in the subcutaneous space of mice. Data are shown as the mean ± standard deviation (n = 5 for each time)
In vitro studies constitute a useful approximation to the in vivo model, although it is difficult, in many cases, to predict the in vivo behavior based on them. However, in this present study, a good correlation between in vitro and in vivo drug release was found, as evidenced by the high correlation coefficient value (0.9732).
The Higuchi and Korsmeyer–Peppas models were adopted to predict the drug release kinetic. The in vivo release profile was best fitted with the Korsmeyer–Peppas model (r2 = 0.9908), and the n value (n = 0.6624 ± 0.1298) obtained from the equation of this model indicated that etoposide release occurs by an overlapping of different phenomena, including drug diffusion and polymer swelling. These data corroborate the results obtained by the in vitro study.
An important vantage of the developed system was the sustained release of etoposide from the implants in the in vivo experimental model. As previously reported, prolonged exposure of cancerous cells to anticancer drugs appears to be essential for the success of chemotherapy treatment. Thus, in many treatment regimens, etoposide is administered intravenously or orally, on five or more days per cycle (45). Therefore, polymeric implants may be considered an alternative to a multi-dose chemotherapy regimen. In addition, the release of low doses of etoposide from the implant may also be considered an advantageous condition of the polymer system since some studies suggest that the efficacy of etoposide in cancer patients was related to prolonged maintenance of extremely low plasma concentrations; otherwise, the plasmatic concentration of etoposide becomes toxic to the patients. In the case of small cell lung cancer, a low steady-state plasma etoposide concentration (0.5–1 μg/mL) was associated with tumor cytotoxicity and higher plasma concentrations (>3–10 μg/mL) produced greater toxicity, but not necessarily with more tumor cytotoxic activity (42,48,49). However, new investigations should be performed to evaluate the cytotoxic activity of the etoposide-loaded PCL implant in an in vivo experimental model.
During the in vivo study, no toxic clinical symptoms were observed. In addition, the animals presented constant body weight, responsiveness, and normal bowel habits. No macroscopic signs of infection were observed at the area of implantation.
Figure 9 shows the histopathology of the representative tissue sections taken from the vicinity of the implant inserted in the subcutaneous space of mice. Evaluation of the tissue alterations that occur at the implantation site is a variable to be studied since these changes may contribute to failure of the drug delivery system (50,51). After 5 days of implantation, the subcutaneous tissue exhibited a cellular immunogenic response characterized by a diffuse inflammatory process which is predominantly mononuclear, located in the deep dermis, which was established due to tissue injuries during the implantation and the presence of a foreign body—the implant (Fig. 9a, b) (50,52). A decrease in the number of inflammatory cells was observed after 15 days of implantation, as shown in Fig. 9c. Twenty-five days after surgery, the subcutaneous tissue near the implantation site showed aspects of a normal tissue, in which the presence of muscle segments, connective tissue, and blood vessels was observed (Fig. 9d). At 25 days, the implants were encapsulated within a thin fibrotic capsule, which typically has been considered a sign of the biocompatibility of the material (31,52). The fibrous encapsulation of the drug-loaded implant was previously studied and was considered as a characteristic event of the chronic phase of the inflammatory process (31,50).
Fig. 9.
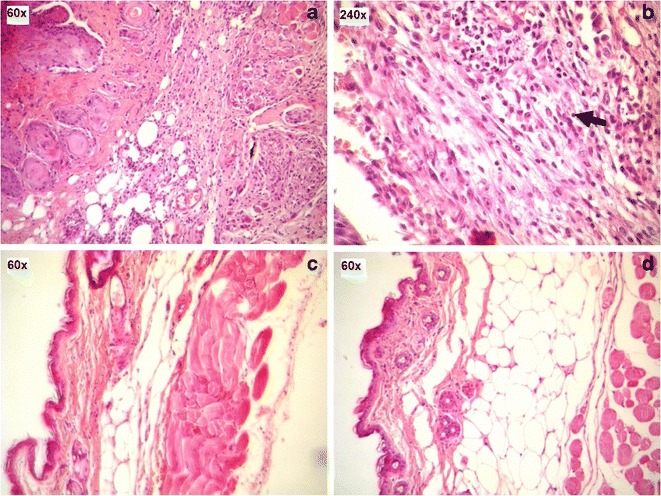
Light micrographs of H&E-stained sections of the subcutaneous tissue surrounding etoposide-loaded implants after 5, 15, and 25 days of implantation: a 5 days. b The infiltrate which is predominantly mononuclear after 5 days of implantation. c 15 days. d 25 days
In the study of tolerance of the implants, histological evaluation of different organs was carried out; no histological changes were observed in the heart, lung, spleen, liver, and the kidney of mice after 5, 15, and 25 days of implantation. These observations show that etoposide implants have good short-term tolerance.
Considering the favorable results obtained for the etoposide-loaded PCL implants, new studies may be performed to verify the possibility to employ the devices in the treatment of ocular tumors (such as retinoblastoma) and brain cancers (such as malignant glioma). The low bioavailability of the drug into the site of disease may be the major reason for the failure of systemic chemotherapy of retinoblastoma and brain tumors (53–55). Thus, in the case of treatment of retinoblastoma, etoposide-loaded PCL implants can be inserted into the vitreous cavity, promoting drug release directly at the target site. In addition, polymeric devices can be inserted into the episclera, avoiding the inner blood–retinal barrier restriction and maintaining the concentration of the drug in the segment posterior of the eye, within the therapeutic level for a longer period. Drug delivery systems, similar to PCL implants, employed in the treatment of diseases of the posterior segment of the eye have already been proven for their efficacy and safety. Among them, Ozurdex®, Retisert®, and Vitrasert® are already commercially available (56). In the treatment of brain cancers, polymeric devices containing etoposide can be implanted after surgical resection of a tumor into the remaining cavity, promoting the release of doses of the drug into the site of tumor resection over a sustained period. These strategies discussed previously can potentially reduce systemic toxicity and increase the exposure of tumor cells to the drug since etoposide is administered locally.
CONCLUSION
In the present work, etoposide-loaded PCL implants were successfully fabricated using the melt method. Etoposide was dispersed uniformly in the polymeric matrix and kept stable during the sterilization process by UV radiation. FTIR data showed that the incorporation of the drug into the PCL implant did not promote changes in the chemical structure of the components of formulation. The XRD results demonstrated that etoposide was present in crystalline form in the polymeric implant. These results were confirmed by thermal analysis. The in vitro release profile showed that the PCL implant allowed a prolonged and controlled release of the drug, which followed the Peppas model. The etoposide released from the implants had significant cytotoxic activity against HeLa cell. In the in vivo study, good short-term tolerance after the implantation was obtained. In addition, good correlation between the in vitro and in vivo drug release was found.
ACKNOWLEDGMENTS
The authors would like to thank Quiral Química do Brasil S.A. for etoposide donation and CNPq, FAPEMIG, and Brazilian Pharmacopoeia for the financial support.
REFERENCES
- 1.Instituto Nacional de Câncer José Alencar Gomes da Silva. Estimativa 2012: Incidência de Câncer no Brasil. http://www.inca.gov.br/estimativa/2012/. Accessed 12 December 2012.
- 2.Weinberg BD, Blanco E, Gao J. Polymer implants for intratumoral drug delivery and cancer therapy. J Pharm Sci. 2008;97:1681–1702. doi: 10.1002/jps.21038. [DOI] [PubMed] [Google Scholar]
- 3.Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers. J Control Release. 2012;159:14–26. doi: 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kempe S, Mäder K. In situ forming implants—an attractive formulation principle for parenteral depot formulations. J Control Release. 2012;161:668–679. doi: 10.1016/j.jconrel.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal P, Rupenthal ID. Injectable implants for the sustained release of protein and peptide drugs. Drug Discov Today. 2013;18:337–349. doi: 10.1016/j.drudis.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 6.Kang YM, Kim GH, Kim JI, Kim DY, Lee BN, Yoon SM, et al. In vivo efficacy of an intratumorally injected in situ-forming doxorubicin/poly-(ethylene glycol)-b-polycaprolactone diblock copolymer. Biomaterials. 2011;32:4556–4564. doi: 10.1016/j.biomaterials.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Wang Q, Wang J, Lu Q, Detamore MS, Berkland C. Injectable PLGA based colloidal gels for zero-order dexamethasone release in cranial defects. Biomaterials. 2010;31:4980–4986. doi: 10.1016/j.biomaterials.2010.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Büyüktimkin B, Wang Q, Kiptoo P, Stewart JM, Berkland C, Siahaan TJ. Vaccine-like controlled-release delivery of an immunomodulating peptide to treat experimental autoimmune encephalomyelitis. Mol Pharm. 2012;9:979–985. doi: 10.1021/mp200614q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodruff MA, Hutmacher DW. The return of a forgotten polymer—polycaprolactone in the 21st century. Prog Polym Sci. 2010;35:1217–1256. doi: 10.1016/j.progpolymsci.2010.04.002. [DOI] [Google Scholar]
- 10.Shirazi FH, Bahrami G, Stewart DJ, Tomiak E, Delorme F, Noel D, et al. A rapid reversed phase high performance liquid chromatographic method for determination of etoposide (VP-16) in human plasma. J Pharm Biomed Anal. 2001;25:353–356. doi: 10.1016/S0731-7085(00)00520-3. [DOI] [PubMed] [Google Scholar]
- 11.Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. EJC. 1998;34:1514–1521. doi: 10.1016/S0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 12.Reif S, Kingreen D, Kloft C, Grimm J, Siegert W, Schunack W, et al. Bioequivalence investigation of high dose etoposide and etoposide phosphate in lymphoma patients. Cancer Chemother Pharmacol. 2001;48:134–140. doi: 10.1007/s002800100280. [DOI] [PubMed] [Google Scholar]
- 13.Shah S, Pal S, Gude R, Devi S. A novel approach to prepare etoposide-loaded poly(n-vinyl caprolactam-co-methylmethacrylate) copolymeric nanoparticles and their controlled release studies. J Appl Polym Sci. 2012;127:4991–4999. doi: 10.1002/app.38107. [DOI] [Google Scholar]
- 14.Reif S, Nicolson MC, Bisset D, Reid M, Kloft C, Jaehde U, et al. Effect of grapefruit juice intake on etoposide bioavailability. Eur J Clin Pharmacol. 2002;58:491–494. doi: 10.1007/s00228-002-0495-9. [DOI] [PubMed] [Google Scholar]
- 15.Snehalatha M, Venugopal K, Saha RN, Babbar AK, Sharma RK. Etoposide loaded PLGA and PCL nanoparticles II: biodistribution and pharmacokinetics after radiolabeling with Tc-99m. Drug Deliv. 2008;15:277–287. doi: 10.1080/10717540802006500. [DOI] [PubMed] [Google Scholar]
- 16.Tang BC, Fu J, Watkins N, Hanes J. Enhanced efficacy of local etoposide delivery by poly(ether-anhydride) particles against small cell lung cancer in vivo. Biomaterials. 2010;31:339–344. doi: 10.1016/j.biomaterials.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain J, Fernandes C, Patravale V. Formulation development of parenteral phospholipid-based microemulsion of etoposide. AAPS PharmSciTech. 2010;11:826–831. doi: 10.1208/s12249-010-9440-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patlolla RR, Vobalaboina V. Folate-targeted etoposide-encapsulated lipid nanospheres. J Drug Target. 2008;16:269–275. doi: 10.1080/10611860801945400. [DOI] [PubMed] [Google Scholar]
- 19.Reddy LH, Adhikari JS, Dwarakanath BSR, Sharma RK, Murthy RR. Tumoricidal effects of etoposide incorporated into solid lipid nanoparticles after intraperitoneal administration in Dalton’s lymphoma bearing mice. AAPS J. 2003;8:E254–E262. doi: 10.1007/BF02854895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhanaraju MD, Sathyamoorthy N, Sundar VD, Suresh C. Preparation of poly(epsilon-caprolactone) microspheres containing etoposide by solvent evaporation method. Asian J Pharm Sci. 2010;5:114–122. [Google Scholar]
- 21.Schaefer JM, Singh J. Effect of isopropyl myristic acid ester on the physical characteristics and in vitro release of etoposide from PLGA microspheres. AAPS PharmSciTech. 2000;1:49–54. doi: 10.1208/pt010432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brazilian Pharmacopeia. 5th ed. Brasília: ANVISA; 2010.
- 23.Solano AGR, Silva GR, Fialho SL, Cunha-Junior AS, Pianetti GA. Development and validation of a high performance liquid chromatographic method for determination of etoposide in biodegradable polymeric implants. Qui Nova. 2012;35:1239–1243. doi: 10.1590/S0100-40422012000600032. [DOI] [Google Scholar]
- 24.Patel DH, Patel MP, Patel MM. Formulation and evaluation of drug free ophthalmic films prepared by using various synthetic polymers. J Young Pharm. 2009;1:116–120. doi: 10.4103/0975-1483.55742. [DOI] [Google Scholar]
- 25.United States Pharmacopeia. 32nd ed. Rockville: The United States Pharmacopeial Convention; 2009.
- 26.Shah JC, Chen JR, Chow D. Preformulation study of etoposide: identification of physicochemical characteristics responsible for the low and erratic oral bioavailability of etoposide. Pharm Res. 1898;6:408–412. doi: 10.1023/A:1015935532725. [DOI] [PubMed] [Google Scholar]
- 27.Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–133. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 28.Siepmann J, Siepmann F. Mathematical modeling of drug delivery. Int J Pharm. 2008;364:328–343. doi: 10.1016/j.ijpharm.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Li C, Cheng L, Zhang Y, Guo S, Wu W. Effects of implant diameter, drug loading and end-capping on praziquantel release from PCL implants. Int J Pharm. 2010;386:23–29. doi: 10.1016/j.ijpharm.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 30.Oliveira AR, Molina EF, Mesquisa PC, Fonseca JLC, Rossanezi G, Pedrosa MFF, et al. Structural and thermal properties of spray-dried methotrexate-loaded biodegradable microparticles. J Therm Anal Calorim. 2012;1:1–11. [Google Scholar]
- 31.Cheng L, Lei L, Guo S. In vitro and in vivo evaluation of praziquantel loaded implants based on PEG/PCL blends. Int J Pharm. 2010;387:129–138. doi: 10.1016/j.ijpharm.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Silva-Junior AA, Matos JR, Formariz TP, Rossanezi G, Scarpa MV, Egito EST, et al. Thermal behavior and stability of biodegradable spray-dried microparticles containing triamcinolone. Int J Pharm. 2009;368:45–55. doi: 10.1016/j.ijpharm.2008.09.054. [DOI] [PubMed] [Google Scholar]
- 33.Jasti BR, Du J, Vasavada RC. Characterization of thermal behavior of etoposide. Int J Pharm. 1995;118:161–167. doi: 10.1016/0378-5173(94)00325-Y. [DOI] [Google Scholar]
- 34.Mohanty AK, Dilnawaz F, Mohanty C, Sahoo SK. Etoposide-loaded biodegradable amphiphilic methoxy (poly ethylene glycol) and poly(epsilon caprolactone) copolymeric micelles as drug delivery vehicle for cancer therapy. Drug Deliv. 2011;17:330–342. doi: 10.3109/10717541003720688. [DOI] [PubMed] [Google Scholar]
- 35.Wu Z, Guo D, Deng L, Zhang Y, Yang Q, Chen J. Preparation and evaluation of a self-emulsifying drug delivery system of etoposide–phospholipid complex. Drug Dev Ind Pharm. 2011;37:103–112. doi: 10.3109/03639045.2010.495752. [DOI] [PubMed] [Google Scholar]
- 36.Yadav KS, Sawant KK. Formulation optimization of etoposide loaded PLGA nanoparticles by double factorial design and their evaluation. Curr Drug Deliv. 2010;7:51–64. doi: 10.2174/156720110790396517. [DOI] [PubMed] [Google Scholar]
- 37.Marsac PJ, Li T, Taylor LS. Estimation of drug–polymer miscibility and solubility in amorphous solid dispersions using experimentally determined interaction parameters. Pharm Res. 2009;26:136–151. doi: 10.1007/s11095-008-9721-1. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Guo S, Cheng L. Disodium norcantharidate loaded poly(ε-caprolactone) microspheres I. Preparation and evaluation. Int J Pharm. 2008;350:130–137. doi: 10.1016/j.ijpharm.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 39.Cheng L, Guo S, Wu W. Characterization and in vitro release of praziquantel from poly(ε-caprolactone) implants. Int J Pharm. 2009;377:112–119. doi: 10.1016/j.ijpharm.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Drewinko B, Barlogie B. Survival and cycle-progression delay of human lymphoma cells in vitro exposed to VP-16-213. Cancer Treat Rep. 1976;60:1295–1306. [PubMed] [Google Scholar]
- 41.Katoh O, Yamada H, Hiura K, Aoki Y, Kuroki S. Clinical pharmacology and toxicity of low daily administration of oral etoposide in advanced lung cancer patients. J Clin Pharmacol. 1991;31:1155–1160. doi: 10.1002/j.1552-4604.1991.tb03689.x. [DOI] [PubMed] [Google Scholar]
- 42.Clark PI, Slevin ML, Joel SP, Osborne RJ, Talbot DI, Johnson PWM, et al. A randomized trial of two etoposide schedules in small-cell lung cancer: the influence of pharmacokinetics on efficacy and toxicity. J Clin Oncol. 1994;12:1427–1435. doi: 10.1200/JCO.1994.12.7.1427. [DOI] [PubMed] [Google Scholar]
- 43.Hande KR. Topoisomerase II inhibitors. Updat Cancer Ther. 2008;3:13–26. doi: 10.1016/j.uct.2008.02.001. [DOI] [Google Scholar]
- 44.Montecucco A, Biamonti G. Cellular response to etoposide treatment. Cancer Lett. 2007;252:9–18. doi: 10.1016/j.canlet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 45.Toffoli G, Corona G, Basso B, Boiocchi M. Pharmacokinetic optimisation of treatment with oral etoposide. Clin Pharmacokinet. 2004;43:441–466. doi: 10.2165/00003088-200443070-00002. [DOI] [PubMed] [Google Scholar]
- 46.Kiliçay E, Demirbilek M, Türk M, Güven E, Hazer B, Denkbas EB. Preparation and characterization of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHBHHX) based nanoparticles for targeted cancer therapy. Eur J Pharm Sci. 2011;44:310–320. doi: 10.1016/j.ejps.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 47.Rello-Varona S, Gámez A, Moreno V, Stockert JC, Cristóbal J, Pacheco M, et al. Metaphase arrest and cell death induced by etoposide on HeLa cells. Int J Biochem Cell Biol. 2003;38:2183–2195. doi: 10.1016/j.biocel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 48.Clark PI, Cottier B. The activity of 10-, 14-, and 21-day schedules of single-agent etoposide in previously untreated patients with extensive small cell lung cancer. Semin Oncol. 1992;19(Suppl 14):36–39. [PubMed] [Google Scholar]
- 49.Carney DN, Grogan L, Smit EF, Harford P, Berendsen HH, Postmus PE. Single-agent oral etoposide for elderly small cell lung cancer patients. Semin Oncol. 1990;1(Suppl 2):49–53. [PubMed] [Google Scholar]
- 50.Bhardwaj U, Sura R, Papadimitrakopoulos F, Burgess DJ. PLGA/PVA hydrogel composites for long-term inflammation control following s.c. implantation. Int J Pharm. 2010;384:78–86. doi: 10.1016/j.ijpharm.2009.09.046. [DOI] [PubMed] [Google Scholar]
- 51.Patil SD, Papadmitrakopoulos F, Burgess DJ. Concurrent delivery of dexamethasone and VEGF for localized inflammation control and angiogenesis. J Control Release. 2007;117:68–79. doi: 10.1016/j.jconrel.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 52.Anderson JM. Biological responses to materials. Annu Rev Mater Res. 2001;31:81–110. doi: 10.1146/annurev.matsci.31.1.81. [DOI] [Google Scholar]
- 53.Angel M, Carcaboso AM, Bramuglia GF, Chantada GL, Fandiño AC, Chiappetta DA, et al. Topotecan vitreous levels after periocular or intravenous delivery in rabbits: an alternative for retinoblastoma chemotherapy. Invest Ophthalmol Vis Sci. 2007;48:3761–3767. doi: 10.1167/iovs.06-1152. [DOI] [PubMed] [Google Scholar]
- 54.Allhenna D, Boushehria MAS, Lamprechta A. Drug delivery strategies for the treatment of malignant gliomas. Int J Pharm. 2012;436:299–310. doi: 10.1016/j.ijpharm.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 55.Callewaert M, Dukic S, Gulick LV, Vittier M, Gafa V, Andry MC, et al. Etoposide encapsulation in surface-modified poly(lactide-co-glycolide) nanoparticles strongly enhances glioma antitumor efficiency. J Biomed Mater Res A. 2013;101:1319–1327. doi: 10.1002/jbm.a.34442. [DOI] [PubMed] [Google Scholar]
- 56.Kuno N, Fujii S. Biodegradable intraocular therapies for retinal disorders: progress to date. Drugs Aging. 2010;27:117–134. doi: 10.2165/11530970-000000000-00000. [DOI] [PubMed] [Google Scholar]