

Abstract
The objective of this study was to prepare celecoxib microcrystals using different stabilizers in order to evaluate the influence of microcrystal formulation on the in vitro dissolution rate and in vivo absorption after oral administration of celecoxib in rats. Three celecoxib microcrystals (MC1, MC2, and MC3) were prepared using solvent change method. Microcrystals were evaluated for morphology, particle size, crystallinity, solubility, in vitro dissolution, and in vivo absorption in rats. Scanning electron microscopy images showed distinct differences in the morphologies and dimensions of various celecoxib microcrystals. The particle size of all microcrystals was significantly (P < 0.05) reduced relative to plain celecoxib. The DSC and XRD results revealed that MC1 retain drug crystallinity relative to control crystals, MC2, and MC3. All microcrystals showed marked increase in the drug dissolution parameters particularly MC1 that exhibited a prompt drug release and significantly (P < 0.05) higher values of % dissolution efficiency as compared to control celecoxib and the other microcrystals. The influence of microcrystals on the in vivo absorption of celecoxib was studied in rats in comparison to plain drug. The results of in vivo absorption study in rats indicated that MC1 significantly improved the rate and extent of celecoxib absorption than plain celecoxib. The mean relative bioavailability of MC1 formulation to plain celecoxib was 157.55 ± 20.18%. In conclusion, microcrystal formulation of celecoxib results not only in an enhancement of dissolution parameters but also improves the bioavailability of celecoxib in rats.
KEY WORDS: celecoxib, dissolution, in vivo absorption, microcrystals, particle size
INTRODUCTION
Celecoxib as a selective cyclo-oxygenase 2 enzyme inhibitor is widely used for treatment of osteoarthritis, rheumatoid arthritis, and acute pain (1). It belongs to the BCS class II substance (2,3). Celecoxib is weakly acidic and has low aqueous solubility (3–7 μg/ml; 4). Low solubility of drugs in water results in poor bioavailability because the solubility of a drug is an important factor in determining its absorption rate (5). It has been reported that the bioavailability of the conventional celecoxib capsule dosage form ranges from 22% to 40% in dogs and the extent of drug absorption is limited by the dissolution rate (6,7). Increasing the solubility and dissolution rate of celecoxib has the potential to improve its overall oral bioavailability (6).
According to the Noyes–Whitney equation, the saturation solubility and dissolution rate of poorly water soluble drugs can be enhanced by reducing the particle size, which increases the total surface area (8–10). The common way for reducing the particle size is the milling of previously formed larger crystals. However, this technique is ineffective and shows disadvantages such as electrostatic effects, broad particle size distributions, and the tendency of the particles to grow (11). To overcome these problems, hydrophobic drugs can be microcrystallized.
In situ microcrystallization is a suitable method for the production of micron-sized drugs (12,13). During the in situ crystal formation, a new surface is formed that makes the system thermodynamically unstable. A stabilizer which has affinity to the crystal surface can cover the newly formed surfaces and in turn might reduce the surface energy, producing micron-sized particles stabilized against crystal growth by steric hindrance (11). Compared with milled products, drug properties are optimized, the particle size is more uniformly distributed, and the powder is less cohesive (14–16).
Several approaches have been attempted to produce micronized or nanosized celecoxib particles. Spherical agglomerates of celecoxib crystals prepared with polyvinylpyrrolidone exhibited improved micromeritic properties in addition to improving the solubility and dissolution rate (17). Nanosuspension of celecoxib had been prepared by the emulsion-diffusion method using three different stabilizers (Tween® 80, PVP K-30 and SLS) but with a broad particle size distribution (18). Margulis-Goshen et al. (19) have obtained celecoxib nanopowder by rapid evaporation of drug-loaded volatile microemulsions achieving high enhanced dissolution compared to the particulate drug in the presence of surfactants. However, relatively high amount of excipients in the microemulsions may increase the side effects and toxicity of the formulation.
The objective of this study was to prepare celecoxib microcrystals using different stabilizers and evaluate the influence of microcrystallization on the in vitro dissolution rate and in vivo absorption of celecoxib in rats.
MATERIALS AND METHODS
Materials
Celecoxib was a gift sample from Sedico pharmaceutical Co., Egypt. Methyl alcohol was obtained from Merck, Darmstadt, Germany. Water was used in double-distilled quality. Hydroxypropyl methylcellulose 4000 (HPMC) was obtained from Fluka Biochemika, Buchs, Switzerland. Cremophor EL was obtained from Sigma, Germany. Sodium lauryl sulphate (SLS) was purchased from El-Nasr pharmaceutical Chemical Co., Egypt. All other chemicals were of analytical grade used as received.
Methods
Preparation of Microcrystals
Celecoxib microcrystals were prepared using the solvent change method by instantaneously mixing two liquids in the presence of a stabilizing agent (20). Briefly, a weighed quantity of celecoxib (1.0 g) was dissolved in 30 ml of methanol. The resultant solution was rapidly injected by syringe into 100 ml of distilled water containing different stabilizers under constant stirring for about 10 min. A micron-sized dispersion was formed spontaneously. The obtained crystals were recovered by centrifugation and drying. Three celecoxib microcrystals (MC1, MC2, and MC3) were prepared using three different stabilizers SLS (0.2% w/v), HPMC (0.1% w/v) and Cremophor EL (1% w/v), respectively. In addition to microcrystallized celecoxib without stabilizer was prepared and used as a control. The obtained microcrystals were washed twice with 20 ml distilled water and dried at room temperature overnight.
Characterization of Celecoxib Microcrystals
Drug Content
An amount of 10 mg of the prepared microcrystals was weighed and dispersed in 10 ml methanol. The samples were centrifuged at 2,000 rpm for 5 min. Supernatant was diluted with suitable quantity of methanol and analyzed by UV spectrophotometer (Perkin Elmer, Type Lambda Ez 201, USA) at 254 nm. Each sample was tested in triplicate.
Saturation Solubility
Saturated aqueous solubility of pure celecoxib and various celecoxib microcrystals was carried out by adding an excess amount of samples to 10 ml distilled water in airtight screwcapped test tubes. The tubes were mounted on a water-bath shaker maintained at 25°C for 48 h to get equilibrium. The equilibrated samples were centrifuged at 3,000 rpm for 5 min. Aliquot portions of the supernatants were filtered through 0.2-μm-membrane filters and the filtrate was appropriately diluted with distilled water for quantification of celecoxib spectrophotometrically at 254 nm (17). Every sample was analyzed in triplicate and the mean values and standard deviations were reported.
Scanning Electron Microscopy
Electron micrographs of prepared microcrystals and the pure celecoxib were obtained using a scanning electron microscope (JEOL JSM-5500 LV-JEOL Ltd, Japan) by using high vacuum mode. The samples were coated by gold sputter coater (SPI–Module) prior to observation.
Particle Size Analysis
The mean particle size of celecoxib powder and various celecoxib microcrystals was determined, in triplicate, after dispersion in saturated aqueous solution of celecoxib at 25 ± 0.5°C using Zeta Sizer Nano-series (Nano ZS), Malvern, Worcestershire, UK.
Differential Scanning Calorimetry (DSC)
To detect any possible change in the physical state of celecoxib in microcrystalline form, DSC was performed on pure celecoxib powder and the prepared celecoxib microcrystals, using a thermal analysis system (DSC-60, Shimadzu, Japan). The samples (5 mg) were heated at a constant rate of 10°C/min, in an aluminum pan under a nitrogen atmosphere. A similar empty pan was used as the reference.
X-Ray Powder Diffraction
In order to further confirm the physical state of celecoxib, X-ray diffraction patterns of the prepared microcrystals as well as the pure drug powder samples were obtained using the X-ray diffractometer (X’Pert-PRO Diffractometer, PANalytical, the Netherlands) with Cu as tube anode. The diffractograms were recoded under the following conditions: voltage was 45 kV, the current was 30 mA, steps were 0.02º of (º2θ), and the counting rate was 0.5 s/step at room temperature. Data were collected from 4 to 40°2θ.
In Vitro Dissolution Test
The dissolution behaviors of celecoxib microcrystals were compared with pure celecoxib powder. The dissolution studies were performed according to the paddle method (USP) using Hanson dissolution apparatus (Hanson Research, California, USA). The paddles rotated at speed of 100 rpm, and the temperature was maintained at 37°C ± 0.5°C. The dissolution medium was 900 ml of distilled water containing 0.5% SLS to maintain sink conditions (3,21). Accurately weighed samples containing the equivalent of 50 mg celecoxib were placed in the dissolution medium. Samples of dissolution medium (5 ml) were withdrawn at different time intervals, filtered through a 0.2-μm-membrane filter, suitably diluted, and analyze spectrophotometrically for celecoxib content at 254 nm. Withdrawn samples were compensated by fresh medium. The dissolution experiments were conducted in triplicate.
In Vivo Absorption Study
Study Design
The in vivo absorption studies were carried out to evaluate the bioavailability of celecoxib MC1 in comparison to plain celecoxib powder. The selection of MC1 was based on its optimal in vitro dissolution results. Moreover, DSC and XRD results revealed that MC1 retain drug crystallinity relative to control crystals, MC2, and MC3.
The study was performed following administration of single oral dose in rats using two treatments, non-blind, randomized design. The protocol of the study was carried out according to a local ethics committee (Animal Ethics Committee of Faculty of Pharmacy, Helwan University). Guidelines of the ethics committee were followed for the study. Twelve adult male Wistar rats weighing 190–200 g were used in the study. All rats were housed and received similar diet. The rats were divided randomly into two groups; each was of 6 rats and all rats were fasted overnight for 12 h with free access to water. On the day of experiment, each rat in the first group received a single oral dose of 30 mg/kg (22) of plain celecoxib powder and rats in the second group received the same equivalent doses of celecoxib MC1. The doses were suspended in a solution of 1.0% carboxymethylcellulose in distilled water and given orally using an animal feeding needle. Blood samples (0.5 ml) were collected in heparinized tubes at 1 h pre-dose and at 1, 2, 3, 4, 5, 6, 8, 12, and 24 h post-dose. The blood samples were centrifuged at 4,000 rpm for 5 min, and the plasma was transferred to separate glass tubes to be kept frozen until analysis.
Analysis of Plasma Levels of Celecoxib
Celecoxib plasma concentration was quantified by a reported liquid chromatography-tandem mass spectrometry (LC-MS/MS) method (23) with slight modifications. Prior to analysis of plasma samples, aliquots of plasma (200 μl) spiked with 20 μg/ml hydrochlorothiazide (internal standard), were vortexed for 2 min with 400 μl of acetonitrile and centrifuged at 2,000 rpm for 10 min at room temperature. The supernatant was filtered and injected into the LC-MS/MS system.
LC-MS/MS System
An aliquot (5 μl) of the plasma extract was injected into an Inertsil ODS-3 C-18 (4.6 mm × 50 mm, 5 μm) column using an Agilent technologies 1200 LC system (Agilent, USA). Separation and elution were achieved using acetonitrile: 0.1% formic acid (75:25 v/v) as the mobile phase, at a flow-rate of 0.70 ml/min. The column temperature was kept at 40°C and the run time was 2.5 min. Mass spectrometric detection was performed using an Agilent technologies 6410 triple quadrupole mass spectrometer, equipped with an electrospray source. Nitrogen was used as desolvation gas (40 psi) with gas flow rate of 11 l/min and the temperature of the desolvation gas was 350°C. The electrospray source was operated in the negative ionization mode at 4,000 V, and multiple reaction monitoring mode (MRM), m/z 380.1 > 316 and m/z 296 > 205 were used for quantification of celecoxib and internal standard, respectively. The lower limit of quantification was 0.1 μg/ml. The standard calibration curve for celecoxib was linear (correlation coefficients were >0.9985) over the studied concentration range (0.1–10 μg/ml). Instrument control, data acquisition, and data evaluation were performed using Agilent MassHunter.
Assessment of Bioavailability
Assessment of bioavailability has relied on the comparison of rate and extent of drug absorption between products (24). The rate of absorption is commonly expressed by Cmax and Tmax, whereas the extent of absorption is expressed by the area-under-the-curve to the last quantifiable drug concentration (AUCt) and/or to time infinity (AUC∞).
Peak plasma concentration (Cmax) and the time to peak concentration (Tmax) were obtained directly from the individual plasma concentration versus time curve. The area under the plasma concentration–time curve from zero to the last measurable plasma concentration at time t (AUC0−t) was calculated using linear trapezoidal rule.
Statistical Analysis
In order to compare the results, Student’s t test (SPSS program; version 12.0) was used. Data were reported as means ± standard deviation (SD). A statistically significant difference was considered at P value < 0.05.
RESULTS AND DISCUSSION
Preparation of Microcrystals
In this study three different types of stabilizing agents (0.2% SLS, 0.1% HPMC, and 1% Cremophor EL) have been selected for preparation of celecoxib microcrystals using solvent change method. The selection of stabilizers was based on their higher affinity towards the newly formed surfaces of celecoxib crystals during precipitation to prevent the crystal growth and produce stable micron-sized crystals (25). The stabilizers (anionic surfactant, hydrophilic polymer, and nonionic surfactant) were investigated for their ability to produce micronized celecoxib and their influence of on the physical properties and in vivo absorption of celecoxib.
Drug Content
The drug content for the prepared celecoxib microcrystals MC1, MC2, and MC3 was found to be 99.32 ± 0.23%, 98.54 ± 0.34%, and 99.21 ± 0.12%, respectively. The higher drug content of the prepared microcrystals indicates that the used stabilizers have been removed to great extent after washing with distilled water.
Morphology and Particle Size
SEM images shown in Fig. 1 reveal distinct differences in the morphologies and dimensions of various celecoxib microcrystals. Plain celecoxib appears to be predominantly plate-like crystals with lengths ranged between 10 and 50 μm. The control crystals exhibited the same shape of plain celecoxib crystals but with 10–20 μm lengths. The MC1 and MC3 prepared using surfactants (SLS and Cremophor EL) as stabilizers appear uniform and plate-like with lengths ranged between 1 to 8 and 1 to 5 μm, respectively, while MC2 prepared by using HPMC as stabilizer showed irregular aggregated particles which is consisted of small microcrystal of length less than 1 μm.
Fig. 1.

SEM photographs of plain celecoxib, control crystals, MC1, MC2, and MC3 microcrystals
The aggregation of MC2 microcrystals may be due to HPMC may have a little affinity to the newly form surfaces or the concentration of HPMC (0.1% w/v) was not enough to form a protective layer on the crystal surface and reduces stickiness of the crystals formed. The polymer must have an affinity to the hydrophobic crystal surface (12).
The mean particle sizes of celecoxib powder and various celecoxib microcrystals were determined and presented in Table I. The mean crystal sizes of plain and control celecoxib were 5.68 ± 0.994 and 3.10 ± 0.13 μm, respectively. The particle size of all microcrystals was significantly (P < 0.05, using Student’s t test) reduced relative to the particle size of either plain or control celecoxib crystals. The obtained low values of polydispersity index (PDI) of MC1 and MC3 (Table I) indicating particle size distribution homogeneity of microcrystals prepared with SLS and Cremophor EL. While MC2 showed PDI value of 0.429 which indicate higher variation of the particle sizes that might be attributed most probably to the aggregation of microcrystals instead of crystal growth. This result indicates that the use of hydrophilic polymers as stabilizer may increase the chance of crystal aggregation in comparison to anionic or ionic surfactants.
Table I.
Stabilizers, Particle Size (μm), Polydispersity Index (PDI), and Saturated Aqueous Solubility (μg/ml) of Celecoxib from Different Microcrystal Formulations and Control
| Stabilizer | Particle size (μm ± SD, n = 3) | Mean PDI (±SD, n = 3) | Saturated aqueous solubility (μg/ml ± SD, n = 3) | |
|---|---|---|---|---|
| Plain celecoxib | – | 5.68 ± 0.99 | 0.175 ± 0.011 | 5.01 ± 0.92 |
| Control | – | 3.10 ± 0.50 | 0.161 ± 0.008 | 6.30 ± 1.21 |
| MC1 | SLS | 0.821 ± 0.13 | 0.158 ± 0.007 | 16.34 ± 1.34 |
| MC2 | HPMC | 0.721 ± 0.31 | 0.429 ± 0.036 | 18.67 ± 1.20 |
| MC3 | Cremophor | 1.10 ± 0.12 | 0.109 ± 0.011 | 9.45 ± 0.82 |
DSC Studies
DSC thermograms and data of plain celecoxib, control crystals, and the prepared microcrystals are shown in Fig. 2 and Table II. DSC thermogram of plain celecoxib exhibits a sharp endothermic peak at 161.58°C, Tm, corresponding to its melting point (17,26), while that of control crystals, MC1, MC2 and MC3 show endothermic peaks at 160.30°C, 160.93°C, 160.07°C, and 159.77°C, respectively. It is clear that the endothermic peaks of the prepared microcrystals of the drug in all the cases are almost the same as the peak of plain celecoxib but with a slight shift to lower Tm that does not seem to be significant. Moreover, a slight reduction in enthalpy changes of MC1 and MC3 microcrystals was noticed compared to the control crystals and the untreated drug. The slightly lower Tm and reduction of enthalpy indicate the absence of any important incompatibility between the drug and stabilizers, implying that the size reduction in microcrystals is responsible for these changes. Similar results have been reported by Varshosaz et al. (27). While, MC2 microcrystals showed a significant reduction in the enthalpy which could be attributed to a reduction in the particle size and or crystallinity of MC2 that can be confirmed by XRD.
Fig. 2.
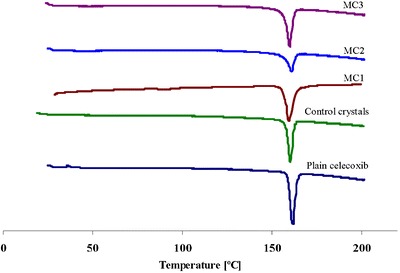
DSC thermograms of plain celecoxib, control celecoxib crystals, MC1, MC2, and MC3 microcrystals
Table II.
DSC Data of Plain Celecoxib, Control Crystals, and Prepared Microcrystals
| T m (melting point; °C) | ΔH (enthalpy of melting; J/g) | |
|---|---|---|
| Plain celecoxib | 161.58 | −86.21 |
| Control crystals | 160.30 | −75.64 |
| MC1 | 159.77 | −75.45 |
| MC2 | 160.93 | −49.99 |
| MC3 | 160.07 | −69.47 |
X-Ray Diffraction Studies
In order to investigate any polymorphic transformation, which occurs during the preparation of microcrystals, X-ray diffraction (Fig. 3) was performed for plain celecoxib, control crystals and the prepared microcrystals. It is clear that the diffractogram of the pure celecoxib showed two characteristic intensity reflections counts of 1617 and 1680 at diffraction angles of 2θ 16.09° and 21.49°, respectively. These peaks are present in the diffractograms of control microcrystals, MC1 and MC3 at the same diffraction angles but with sharp decrease in the reflections counts revealing a reduction in their crystallinity. The lower peak intensity was probably caused by particle size reduction. Usually, drugs with lower crystallinity and smaller size have higher dissolution rate and bioavailability (9,28). However, the characteristic crystalline peaks of MC2 appeared as less intense and broadened peaks as compared to the pure drug indicating possible partial transformation into an amorphous form.
Fig. 3.
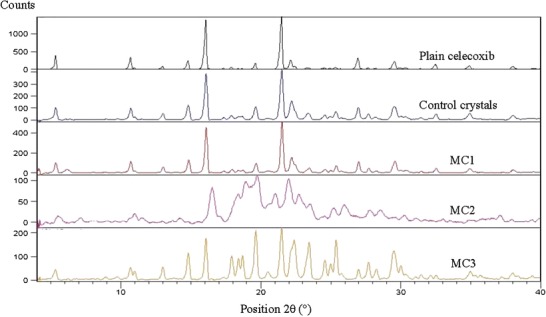
X-ray diffractograms of plain celecoxib, control celecoxib crystals, MC1, MC2, and MC3 microcrystals
Solubility and Dissolution Studies
Results of the saturated aqueous solubility studies are given in Table I. All microcrystals significantly (P < 0.05) increased celecoxib aqueous solubility compared to plain drug. This result seemed to be due the significant particle size reduction and decreased drug crystallinity. MC2 showed highest solubility (18.67 ± 1.20 μg/ml) as compared to the plain celecoxib (5.01 ± 0.92 μg/ml) and the other microcrystals. This result may due to the particle size reduction and the partial transformation of MC2 into an amorphous form.
As illustrated in Fig. 4, it is clear that all microcrystals exhibited higher dissolution characteristics than celecoxib control crystals in distilled water containing 0.5% SLS. The dissolution parameters including the percentage drug dissolved after 5 min (Q5%), 90 min (Q90%), % dissolution efficiency (%DE) at 5 min (%DE5 min) and 60 min (%DE60 min) were determined and presented in Table III. The %DE was calculated as the percent ratio of area under the dissolution curve up to time t to that of the area of the rectangle described by 100% dissolution at the same time (29).
Fig. 4.
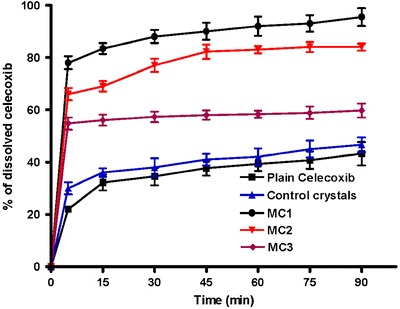
Dissolution profiles of plain celecoxib, control celecoxib crystals, MC1, MC2, and MC3 microcrystals in distilled water containing 0.5% SLS (n = 3, mean ± SD)
Table III.
The Percentage Drug Dissolved After 5 min (Q5%), 90 min (Q90%), and % Dissolution Efficiency at 5 min (%DE5 min) and 60 min (%DE60 min) of Celecoxib From Plain Powder, Control Crystals, and Different Celecoxib Microcrystals in Distilled Water Containing 0.5% SLS at 37°C ± 0.5°C
| Dissolution parameters | ||||
|---|---|---|---|---|
| Q5% | Q90% | %DE5 min | %DE60 min | |
| (mean ± SD, n = 3) | (mean ± SD, n = 3) | |||
| Plain celecoxib | 22.02 ± 1.01 | 43.23 ± 4.40 | 11.01 | 32.40 |
| Control crystals | 29.87 ± 2.30 | 46.65 ± 2.80 | 14.93 | 36.23 |
| MC1 | 78.12 ± 2.50 | 95.50 ± 3.38 | 39.06 | 83.15 |
| MC2 | 66.04 ± 3.40 | 84.00 ± 1.64 | 33.02 | 72.80 |
| MC3 | 54.75 ± 2.30 | 59.73 ± 2.65 | 27.37 | 54.61 |
All celecoxib microcrystals significantly (P < 0.05) increased the Q5%, Q90%, %DE5 min, and %DE60 min compared to the control drug crystals. In particular, MC1 prepared with 0.2% SLS, showed a prompt drug release and significantly (P < 0.05) higher values of %DE after 5 and 60 min (Table III) as compared to control celecoxib and the other microcrystals.
MC2, prepared with 0.1% w/v HPMC as stabilizer, and has obviously smaller particle size than the other microcrystals (Table I), failed to achieve comparable dissolution parameters relative to MC1. This could be attributed to possible aggregation of MC2 microcrystals during preparation as evidenced by the higher values of polydispersity index and SEM photographs. This result is dissimilar to that reported by (20,30). The dissolution results may be attributed to both improvement of drug wettability and reduction of crystal size of celecoxib to the nano-scale which increase drug surface area available for dissolution medium which consequently increased the dissolution velocity as described by Noyes–Whitney equation (31).
In Vivo Absorption Study
The mean plasma celecoxib concentration-time profiles following administration of single oral doses (30 mg/kg) of plain celecoxib and MC1 to fasted rats are shown in Fig. 5. Pharmacokinetic parameters (Cmax, Tmax, and AUC0–24) are calculated individually on the basis of celecoxib plasma concentration–time data. From individual pharmacokinetic parameters, their mean values ± SD were determined and tabulated in Table IV.
Fig. 5.
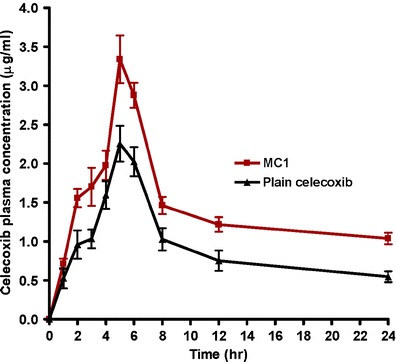
Mean (±SD) plasma concentration–time curves of celecoxib in rats (n = 6) after administration of a single oral dose (30 mg/kg) of plain celecoxib and MC1
Table IV.
Pharmacokinetic Parameters of Celecoxib in Rats (n = 6) After Administration of a Single Oral Dose (30 mg/kg) of Plain Celecoxib and MC1
| Parameter | Plain celecoxib | MC1 |
|---|---|---|
| C max (μg/ml) | 2.26 ± 0.23 | 3.34 ± 0.30* |
| T max (h) | 5.00 ± 0.00 | 5.00 ± 0.00 |
| AUC0–24 (μg h/ml) | 21.77 ± 2.24 | 33.95 ± 1.20* |
Each value represents the mean ± standard division of six rats
*P < 0.05, statistically significant difference based on student t test
Statistical analysis of the bioavailability parameters AUC0–24 and Cmax data obtained for MC1 showed significantly (P < 0.05, student t test) higher values compared to plain celecoxib powder. However, there was no significant difference between the Tmax of both celecoxib formulations. This indicates that microcrystal formulation of celecoxib significantly improved the rate and extent of drug absorption from the gastrointestinal tract of rats in comparison to plain celecoxib.
The individual AUC0–24 values for MC1 were compared to those for plain celecoxib to determine the relative bioavailability. The mean relative bioavailability of MC1 to the plain celecoxib was 157.55 ± 20.18%. This result indicated that 57.55% increase in the oral bioavailability of celecoxib was achieved by microcrystal formulation. These results were attributed to the pronounced effect of microcrystal formulation on the solubility and dissolution of celecoxib.
CONCLUSION
In conclusion, the results reported here suggest that microcrystal formulation of celecoxib prepared with SLS as stabilizer results not only in an enhancement of the celecoxib dissolution rate but also improves the bioavailability of celecoxib in terms of rate and extent of absorption in rats.
Acknowledgments
The author thanks Sedico pharmaceutical Co. for kindly supplying celecoxib powder.
References
- 1.Davies NM, McLachlan AJ, Day RO, Williams KM. Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclo-oxygenase-2 inhibitor. Clin Pharmacokinet. 2000;38:225–242. doi: 10.2165/00003088-200038030-00003. [DOI] [PubMed] [Google Scholar]
- 2.Löbenberg R, Amidon GL. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur J Pharm Biopharm. 2000;50:3–12. doi: 10.1016/S0939-6411(00)00091-6. [DOI] [PubMed] [Google Scholar]
- 3.Tan A, Simovic S, Davey AK, Rades T, Prestidge CA. Silica-lipid hybrid (SLH) microcapsules: a novel oral delivery system for poorly soluble drugs. J Control Release. 2009;134:62–70. doi: 10.1016/j.jconrel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Reddy MN, Rehana T, Ramakrishna S, Chowdary KPR, Diwan PV. Β cyclodextrin complexes of celecoxib: molecular-modeling characterization, and dissolution studies. AAPS PharmSci. 2004;6(1):1–9. doi: 10.1208/ps060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orienti I, Bigucci F, Luppi B, Cerchiara T, Zuccari G, Giunchedi P, et al. Polyvinylalcohol substituted with triethyleneglycolmonoethyl ether as a new material for preparation of solid dispersion of hydrophobic drugs. Eur J Pharm Biopharm. 2002;54:229–233. doi: 10.1016/S0939-6411(02)00055-3. [DOI] [PubMed] [Google Scholar]
- 6.Paulson SK, Vaughn MB, Jessen SM, Lawal Y, Gresk CJ, Yan B, et al. Pharmacokinetics of celecoxib after oral administration in dogs and humans: effect of food and site of absorption. J Pharmacol Exp Ther. 2001;297:638–645. [PubMed] [Google Scholar]
- 7.Shono Y, Jantratid E, Janssen N, Kesisoglou F, Mao Y, Vertzoni M, et al. Prediction of food effects on the absorption of celecoxib based on biorelevant dissolution testing coupled with physiologically based pharmacokinetic modeling. Eur J Pharm Biopharm. 2009;73:107–114. doi: 10.1016/j.ejpb.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Dressmann JB, Reppas C. In vitro–in vivo correlations for lipophilic, poorly water soluble drugs. Eur J Pharm Sci. 2000;11:73–80. doi: 10.1016/S0928-0987(00)00181-0. [DOI] [PubMed] [Google Scholar]
- 9.Sarkari M, Brown J, Chen X, Swinnea S, William RO, Johnston KP. Enhanced drug dissolution using evaporative precipitation into aqueous solution. Int J Pharm. 2002;243:17–31. doi: 10.1016/S0378-5173(02)00072-8. [DOI] [PubMed] [Google Scholar]
- 10.Kesisoglou F, Panmai S, Wu Y. Nanosizing-oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;5916:31–44. doi: 10.1016/j.addr.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Rasenack N, Steckel H, Müller BW. Preparation of microcrystals by in situ micronization. Powder Technol. 2004;143–144:291–296. doi: 10.1016/j.powtec.2004.04.021. [DOI] [Google Scholar]
- 12.Rasenack N, Müller BW. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm Res. 2002;19:1894–1900. doi: 10.1023/A:1021410028371. [DOI] [PubMed] [Google Scholar]
- 13.Kim ST, Kwon JH, Lee JJ, Kim CW. Microcrystallization of indomethacin using a pH-shift method. Int J Pharm. 2003;263:141–150. doi: 10.1016/S0378-5173(03)00358-2. [DOI] [PubMed] [Google Scholar]
- 14.Williams RO, 3rd, Brown J, Liu J. Influence of micronization method on the performance of a suspension triamcinolone acetonide pressurized metered-dose inhaler formulation. Pharm Dev Technol. 1999;4(2):167–179. doi: 10.1081/PDT-100101351. [DOI] [PubMed] [Google Scholar]
- 15.Zhang HX, Wang JX, Zhang ZB, Le Y, Shen ZG, Chen JF. Micronization of atorvastatin calcium by antisolvent precipitation process. Int J Pharm. 2009;374(1–2):106–113. doi: 10.1016/j.ijpharm.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Huang QP, Wang JX, Chen GZ, Shen ZG, Chen JF, Yun J. Micronization of gemfibrozil by reactive precipitation process. Int J Pharm. 2008;360:58–64. doi: 10.1016/j.ijpharm.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 17.Gupta VR, Mutalik S, Patel MM, Jani GK. Spherical crystals of celecoxib to improve solubility, dissolution rate and micromeritic properties. Acta Pharma. 2007;57:173–184. doi: 10.2478/v10007-007-0014-8. [DOI] [PubMed] [Google Scholar]
- 18.Dolenc A, Kristl J, Baumgartner S, Planinsek O. Advantages of celecoxib nanosuspension formulation and transformation into tablets. Int J Pharm. 2009;376:204–212. doi: 10.1016/j.ijpharm.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 19.Margulis-Goshen K, Kesselman E, Danino D, Magdassi S. Formation of celecoxib nanoparticles from volatile microemulsions. Int J Pharm. 2010;393:230–237. doi: 10.1016/j.ijpharm.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Rasenack N, Hartenhauer H, Müller BW. Microcrystals for dissolution rate enhancement of poorly water-soluble drugs. Int J Pharm. 2003;254:137–145. doi: 10.1016/S0378-5173(03)00005-X. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Sun C, Haob Y, Jiang T, Zheng L, Wang S. Mechanism of dissolution enhancement and bioavailability of poorly water soluble celecoxib by preparing stable amorphous nanoparticles. J Pharm Pharm Sci. 2010;13(4):589–606. doi: 10.18433/j3530j. [DOI] [PubMed] [Google Scholar]
- 22.Niederberger E, Tegeder I, Vetter G, Schmidtko A, Schmidt H, Euchenhofer C, et al. Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF kappa B. FASEB J. 2001;15(9):1622–1624. doi: 10.1096/fj.00-0716fje. [DOI] [PubMed] [Google Scholar]
- 23.Park MS, Shim WS, Yim SV, Lee KT. Development of simple and rapid LC-MS/MS method for determination of celecoxib in human plasma and its application to bioequivalence study. J Chromatogr B Anal Technol Biomed Life Sci. 2012;902:137–141. doi: 10.1016/j.jchromb.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 24.U.S. Food and Drug Administration . Title 21 Code of Federal Regulations (CFR), Part 320.1. Washington, DC: Office of Federal Register, National Archives and Records Administration, U.S. Government Printing Office; 2010. [Google Scholar]
- 25.Zhang JY, Shen ZG, Zhong J, Hu TT, Chen JF, Ma ZQ, et al. Preparation of amorphous cefuroxime axetil nanoparticles by controlled nano-precipitation method without surfactants. Int J Pharm. 2006;323:153–160. doi: 10.1016/j.ijpharm.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 26.Chawla G, Gupta P, Thilagavathi R. Characterization of solid-state forms of celecoxib. Eur J Pharm Sci. 2003;20:305–317. doi: 10.1016/S0928-0987(03)00201-X. [DOI] [PubMed] [Google Scholar]
- 27.Varshosaz J, Talari R, Mostafavi SA, Nokhodchi A. Dissolution enhancement of gliclazide using in situ micronization by solvent change method. Powder Technol. 2008;187:222–230. doi: 10.1016/j.powtec.2008.02.018. [DOI] [Google Scholar]
- 28.Zhong J, Shen Z, Yang Y, Chen J. Preparation and characterization of uniform nanosized cephradine by combination of reactive precipitation and liquid anti-solvent precipitation under high gravity environment. Int J Pharm. 2005;301:286–293. doi: 10.1016/j.ijpharm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;28:48–49. doi: 10.1111/j.2042-7158.1975.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 30.Li XS, Wang JX, Shen ZG, Zhang PY, Chen JF, Yun J. Preparation of uniform prednisolone microcrystals by a controlled microprecipitation method. Int J Pharm. 2007;342:26–32. doi: 10.1016/j.ijpharm.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 31.Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19:930–934. doi: 10.1021/ja02086a003. [DOI] [Google Scholar]