

Abstract
Colloidal solid dispersion is an innovative breakthrough in the pharmaceutical industry that overcomes the solubility-related issue of poorly soluble drugs by using an amorphous approach and also the stability-related issue by means of a complex formation phenomenon using different carrier materials. In the present study, a newly developed adsorption method is introduced to incorporate a high-energy sulfathiazole–polyvinylpyrrolidone (Plasdone® K-29/32) solid dispersion on porous silicon dioxide (Syloid® 244FP). Different ternary systems of sulfathiazole–Plasdone® K-29/32–Syloid® 244FP were prepared (1:1:2, 1:1:3, and 1:2:2) and categorized depending on the mechanism by which Syloid® 244FP was incorporated. Modulated differential scanning calorimetry (MDSC), X-ray diffraction, Fourier transform infrared spectroscopy, and in vitro dissolution studies were conducted to characterize the ternary systems. The X-ray diffraction and MDSC data showed a lack of crystallinity in all internal and external ternary systems, suggesting a loss of the crystallinity of sulfathiazole compared to the physical mixtures. USP apparatus II was used to measure the in vitro dissolution rate of the prepared systems at 75 rpm in different media. The dissolution rate of the optimum ratio (1:2:2) containing an internal ternary solid dispersion system was found to be three times higher than that of the external and physical systems. Thus, the porous silicon dioxide incorporated into the conventional binary solid dispersion acted as a carrier to disperse the complex and increase the dissolution rate.
Electronic supplementary material
The online version of this article (doi:10.1208/s12249-013-9947-z) contains supplementary material, which is available to authorized users.
KEY WORDS: amorphous, colloidal solid dispersion, FTIR, porous silicon dioxide, ternary solid dispersion
INTRODUCTION
Recent data suggest that approximately 40% of the marketed active pharmaceutical ingredients (API’s) and 90% of the new chemical entities (NCE’s) under development are classified as Biopharmaceutics Classification System (BCS) class II (high permeability and low solubility) and class IV (low permeability and low solubility) drugs, suggesting issues associated with solubility and ultimately bioavailability in situ (1). Since the concept of bioavailability came into existence, it became clear that, when a drug is administered orally, it has to first dissolve in the alimentary tract fluid, and the rate of this dissolution becomes a fundamental factor for the rate of absorption (2). The rate of solution is the rate-determining step for the bioavailability of orally administered poorly water-soluble drugs (3). Various methods are available for improving the rate of dissolution, including salt formation, the use of cosolvents, complexation, the use of surfactants, particle size reduction, and the prodrug approach (4).
Early researchers proved that solid dispersion is one of the most successful techniques for improving the effective surface area of poorly water-soluble drugs such as sulfathiazole, indomethacin, and griseofulvin (5). According to the modified Noyes and Whitney equation for slightly water-soluble drugs (0.1 mg/ml), surface area is one of the most promising parameters for improving the diffusion rate and ultimately the absorption of poorly water-soluble drugs (6). Some of the working principles of solid dispersion that make it one of the most efficient techniques for improving solubility are as follows: (a) When a solid dispersion of a drug and an inert carrier (highly water-soluble) comes into contact with water, the water-soluble carrier dissolves, leaving the drug suspension in a microcrystalline state that is rapidly absorbed (7). (b) The surface area can be increased and the crystalline form of a drug can be transformed into the amorphous form (8).
Despite all the advantages associated with solid dispersion formulations, researchers have revealed various associated caveats including stickiness or tackiness due to the water-soluble polymeric carrier (9), the need for improving the flow properties as well as the bulk properties of the solid dispersion granules (10), the lack of stability of a prepared solid dispersion during storage (11,12), and the difficulty associated with high drug loading (13). These problems might be the reason for the small number of commercially available products using solid dispersion.
Each formulation requires a different individualized approach for successful development. After the evolution of surface-modified solid dispersions, both water-soluble carriers (14) and water-insoluble carriers (15) have been used to improve the dissolution rate profiles and stabilities. Water-soluble carriers are moisture-sensitive and might create more handling problems compared to water-insoluble carriers. Water-insoluble carriers, such as microcrystalline cellulose and maize starch, are required in higher ratios, making them unsuitable for use as a high-dose drug (16).
Surface-modifying carriers like fine silica particles are gaining attention in the pharmaceutical world for improving solid dispersion technology. A great deal of research has been conducted using porous and nonporous, hydrophilic and hydrophobic fine silica particles as carriers for solid dispersions (17,18). The porous silica materials showed higher dissolution rates because of their ability to decrease the crystallinity of the drugs compared to nonporous silica materials (19). In addition, the porous silica materials formed more stable and well-distributed solid dispersion systems (20), although it has been found that incorporating fine silica materials alone without any high-molecular-weight polymer resulted in rapid crystallization from the supersaturated drug solution.
The objective of the present work is to apply the concept of a triple “C” solid dispersion system to the binary solid dispersion system of sulfathiazole and polyvinylpyrrolidone (PVP), which had been studied previously for its stability and dissolution characteristics (21,22). The term triple “C” solid dispersion system, called an internal ternary solid dispersion (ITSD) system herein, represents an innovative way to combine the advantages of both conventional solid dispersion and surface solid dispersion by utilizing the adsorptive nature of porous fine silica materials to improve the particle size and by modifying the model drug into an optimal molecular form. The authors are eager to describe this newly coined term triple “C” solid dispersion system as “coherent colloidal carrier”-mediated solid dispersion technology. Figure 1 is a schematic representation of the basic concept involved in making the triple “C” solid dispersion system. Specifically, a synthetic amorphous silicon dioxide (Syloid® 244FP) has been selected with a smaller particle size (2.5–3.7 μm) that provides a larger internal surface area (300 m2/g), enabling it to improve the dissolution rate profile in conjunction with its highly adsorptive nature and high pore volume (1.6 ml/g). The adsorptive carrier was incorporated in multiple ways to evaluate its effect on the dissolution rate profile.
Fig. 1.

Basic concept of the triple “C” solid dispersion system
MATERIALS AND METHODS
Materials
Sulfathiazole (molecular weight, 255.32; lot no. TF1502) was obtained from Spectrum Chemical Mfg. Corp. (New Brunswick, New Jersey, USA). It is slightly soluble in water (<0.6 mg/L at 37°C) and has a melting point of 202°C. Chemically, sulfathiazole is 4-amino-N-2-thiazolylbenzenesulfonamide. PVP (Plasdone® K-29/32; molecular weight, 58,000; viscosity of 2.5 cP at 37°C) was supplied by ISP Technologies Inc. (Wayne, New Jersey, USA). A synthetic amorphous silicon dioxide (Syloid® 244FP; average particle size of ∼2.5–3.7 μm) was obtained from W.R. Grace & Co.-Conn (Baltimore, Maryland, USA). Ethyl alcohol (ethanol) at 190 proof was supplied by Pharmco Products Inc. (Brookfield, Connecticut, USA).
Methods
Preparation of the Binary Solid Dispersion Systems
Conventional binary solid dispersion systems were prepared using two different ratios (1:1 and 1:2) of drug and polymer by the solvent evaporation method. A homogeneous clear solution of the drug and polymer in ethanol was prepared and poured into a Petri dish. Excessive solvent was removed by vacuum drying at 70–80°C. Each pulverized binary solid dispersion system was sieved through a #30 mesh stainless steel (SS) screen and collected into an airtight glass vial. Each system was stored in a dehumidifying chamber at room temperature.
Preparation of the Internal Ternary Solid Dispersion Systems
A homogeneous clear solution of drug and polymer in ethanol were prepared using two different ratios (1:1 and 1:2). This clear solution was heated gently in a water bath until half of the solvent evaporated. At that point, the carrier was suspended on top of the clear concentrated solution containing the drug and polymer to make the final ITSD systems in three different drug/polymer/carrier ratios (1:1:2, 1:1:3, and 1:2:2). After suspending the carrier, the systems were mixed thoroughly and the excess solvent was removed by gently heating the systems in a water bath. Once dried, the fine particles were collected and sieved through a #30 mesh SS screen. Each ITSD system was collected and placed into an airtight glass vial and stored in a dehumidifying chamber at room temperature.
Preparation of the External Ternary Solid Dispersion Systems
Binary solid dispersion systems of drug and polymer in two different ratios (1:1 and 1:2) were prepared as described earlier. The carrier was added externally into the respective binary solid dispersion system and mixed uniformly using a mortar and pestle to obtain the final external ternary solid dispersion (ETSD) systems in three different ratios (1:1:2, 1:1:3, and 1:2:2) of drug/polymer/carrier. Each ETSD system was passed through a #30 mesh SS screen and collected into an airtight glass vial. The glass vials were stored in a dehumidifying chamber at room temperature.
Preparation of Physical Mixtures
Physical mixtures (PM) with three different drug/polymer/carrier ratios (1:1:2, 1:1:3, and 1:2:2) were prepared by uniformly mixing proportional amounts of drug, polymer, and carrier with a mortar and pestle. Each PM system was passed through a #30 sieve and collected into an airtight glass vial. The glass vials were stored in a dehumidifying chamber at room temperature.
Modulated Differential Scanning Calorimetry
A thermal analysis of all samples was conducted using modulated differential scanning calorimetry (MDSC) (DSC Q 100, TA Instruments, New Castle, Delaware, USA). Accurately weighed test samples (approximately 5–10 mg) and reference samples (empty) were hermetically sealed in aluminum pans and analyzed using a heat–cool–heat cycle at a rate of 5°C/min from 10°C to 230°C using a nitrogen gas flow of 50 ml/min.
X-Ray Diffraction
Samples were analyzed at room temperature (25°C) to determine the crystalline characteristics of all the systems. A scanning X-ray diffractometer (Advanced Diffraction System, Scintag Inc. Model X1, Cupertino, California, USA), controlled by a computer with Diffraction Management System software for Windows NT, was used for this study. The radiation used was generated by a CuKα filter with a wavelength of 1.54 Å at 45 kV and 40 mA. Powder samples were scanned over a range from 10° to 35° 2θ degrees, using a scan rate of 2°/min and a step scan of 0.02°.
Fourier Transform Infrared Spectroscopy
Solid-state interactions were characterized by Fourier transform infrared spectroscopy (FTIR). The IR absorbance spectra were obtained with a Mattson Galaxy 5020 FTIR spectrometer equipped with a DTGS detector. The sample pellets were prepared by making an approximately 10–15% PM with dry KBr (IR grade). Powders were triturated in a small-sized mortar and pestle until the powder mixture was fine and uniform. Sixty-four scans were collected for each sample at a resolution of 4 cm−1 over the frequency region of 4,000–400 cm−1.
In Vitro Release Profile
A USP apparatus II (paddle apparatus) was used to obtain the in vitro release profile of the drug from all the systems under different dissolution conditions. Dissolution experiments were conducted using three different media (900 ml of water, 900 ml of phosphate buffer, pH 7.4, and 250 ml hydrochloric acid buffer, pH 1.2) at 75 rpm and 37 ± 0.5°C. Samples corresponding to 150 mg of sulfathiazole were placed in each vessel. A 5-ml aliquot was withdrawn at regular time intervals and replaced with an equal volume (5 ml) of dissolution media. Samples were filtered through Acrodisc® syringe filters (0.45 μm) and assayed using a Schimadzu UV spectrophotometer at 283 nm.
Physical Stability of the Prepared Binary Solid Dispersions and Internal Ternary Solid Dispersions
The physical stability studies of the prepared binary solid dispersions and ITSDs were conducted under accelerated stability conditions (60°C/75% relative humidity [RH]) for 5 days. A thermal analysis of the stability samples was conducted using MDSC (DSC Q 100, TA Instruments, New Castle, Delaware, USA) to evaluate physical stability.
RESULTS AND DISCUSSION
Dissolution Rate Studies
The effectiveness of the prepared systems was evaluated using in vitro dissolution in three different media. In purified water, as shown in Fig. 2, the dissolution rate varied depending on the incorporation of the carrier in the final ternary system. The internal system showed the highest dissolution rate profile followed by the ETSD and the physical ternary mixture. As shown in Fig. 3, 1:1:3 ITSD and 1:1:2 ITSD showed 9 to 18 times faster dissolution rate profiles in 5 min compared to the respective conventional binary solid dispersion (1:1 SD). The 1:2:2 ITSD showed an improved dissolution rate profile with 100% released in the first 5 min, which was 12 times faster than the conventional binary solid dispersion (1:2 SD). Increasing the polymer fraction in the 1:1:2 ITSD improved the dissolution rate profile to almost double the profile of 1:2:2 ITSD. This can be explained by the phenomenon of the increasing solubility of a drug at the molecular level with an increasing amount of polymer (23). All internal systems dispersed uniformly in the media due to Syloid® 244FP, which may be the reason for the improved dissolution rate without precipitation of the drug. In the ITSDs, Syloid® 244FP was dispersed at a molecular level, whereas in the PMs and ETSDs, it was dispersed physically only. The physical dispersion alone does not help the system to disperse uniformly in the dissolution media. This observation indicates the importance of incorporating the carrier into the final ternary system.
Fig. 2.
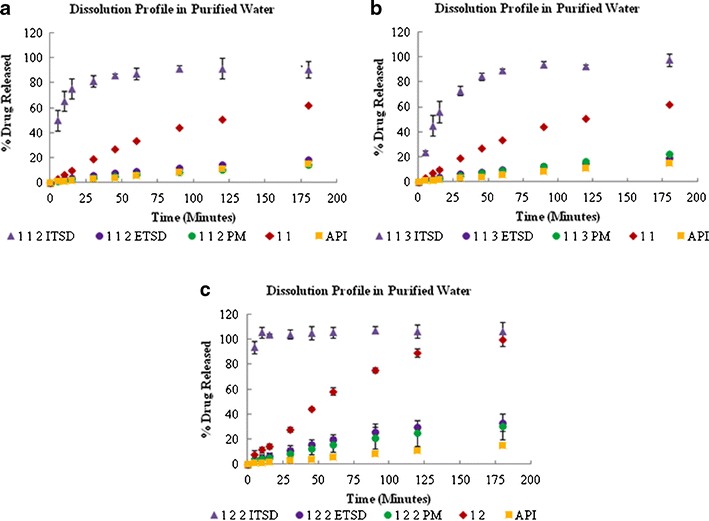
Dissolution profiles of the 1:1:2 (a), 1:1:3 (b), and 1:2:2 (c) ternary solid dispersion systems in purified water
Fig. 3.
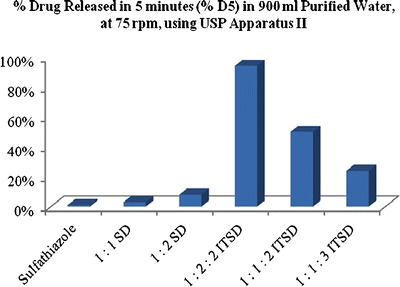
Comparative chart representing the percent drug released in 5 min for the prepared ITSDs in purified water
The dissolution rate was hindered by the amount of carrier used for the final ternary system, and it was discriminated using phosphate buffer (pH 7.4) media, as shown in Fig. 4 where the dissolution rate followed the trend of 1:2:2 > 1:1:2 > 1:1:3. As shown in Fig. 4b, increasing the carrier fraction in the 1:1:2 ITSD resulted in a slower dissolution rate profile in the 1:1:3 ITSD compared to the conventional binary solid dispersion (1:1 SD). Thus, the optimum amount of carrier is needed to form an effective ITSD system. As shown in Fig. 4c, the sudden increase in the dissolution rate is followed by a decrease. This change might be explained by the fact that, as the complex of the drug and polymer is released from the carrier, some of the complex remains in the solid phase in an attempt to maintain equilibrium with the quantity of drug being released from and reabsorbed into the carrier. After reaching equilibrium, this phenomenon is no longer observed.
Fig. 4.
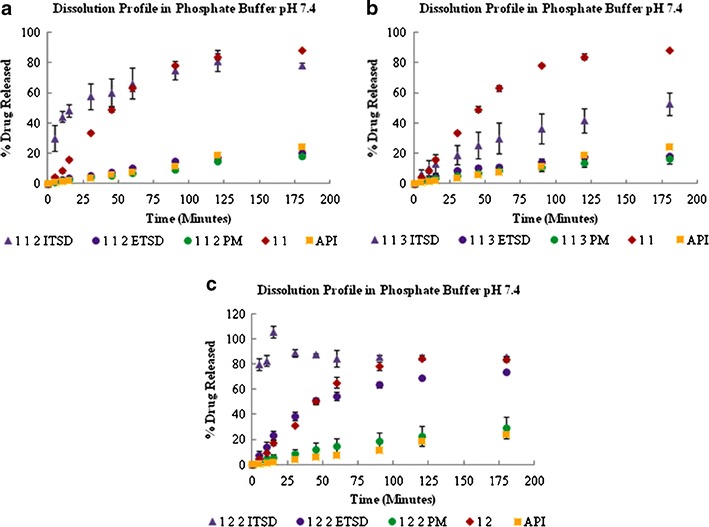
Dissolution profiles of the 1:1:2 (a), 1:1:3 (b), and 1:2:2 (c) ternary solid dispersion systems in phosphate buffer, pH 7.4
To see the effect of acidic pH on the release profile of all prepared systems, hydrochloric acid buffer (pH 1.2) was used. As shown in Fig. 5, the dissolution rate of all ITSD systems was higher than that of their respective conventional binary solid dispersions. The plain sulfathiazole showed a higher dissolution rate compared to the physical and external systems because it has higher solubility in an acidic medium. Sulfathiazole, both in the conventional binary solid dispersions and the ITSD systems, showed a similar extent of drug release at recovery (Fig. 5).
Fig. 5.
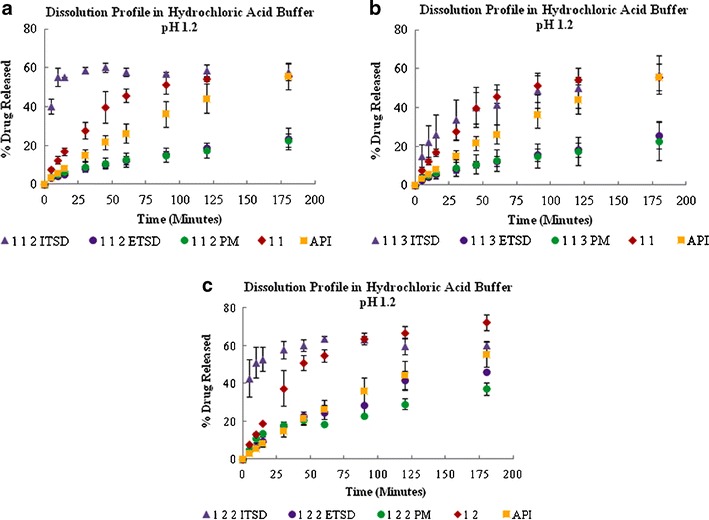
Dissolution profiles of the 1:1:2 (a), 1:1:3 (b), and 1:2:2 (c) ternary solid dispersion systems in hydrochloric acid buffer, pH 1.2
Modulated Differential Scanning Calorimetry
The MDSC thermograms of the pure drug, polymer, porous carrier, and all experimental binary and ternary systems are shown in Figs. 6 and 7. In the past, different polymorphic forms of sulfathiazole have been prepared and molecularly characterized (24). The MDSC thermogram of sulfathiazole showed two different characteristic endothermic peaks at approximately 175°C and 203°C representing polymorphic form III and form I, respectively. As shown in Fig. 7, all binary solid dispersions, ITSDs, and ETSDs showed an absence of the characteristic melting endotherm of sulfathiazole, confirming a solid solution of an amorphous form of sulfathiazole dispersed in the polymeric porous carrier matrix at the molecular level (25–27). While preparing the PM, a partial disorientation of the organized crystalline structure of sulfathiazole occurs due to the size reduction, which partially disrupts the crystalline structure (28,29). This phenomenon is confirmed by the low intensity endothermic peak at 175°C in the DSC thermograph of the PM and the absence of a second endothermic peak, as shown in Fig. 6. Decreasing the drug-to-Syloid® 244FP ratio in the ternary PM system with a 1:1:3 ratio resulted in the lowest intensity endothermic peak in the DSC thermograph at 175°C, whereas the ternary PM systems with the 1:1:2 and 1:2:2 ratios showed equal intensity endothermic peaks at 175°C.
Fig. 6.
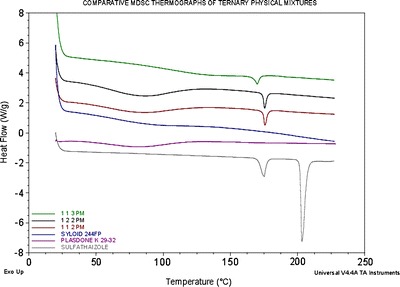
Comparative MDSC thermogram of sulfathiazole, Plasdone® K-29/32, Syloid® 244FP, and ternary PM of sulfathiazole–Plasdone® K-29/32–Syloid® 244FP (1:1:3, 1:2:2, and 1:1:2)
Fig. 7.
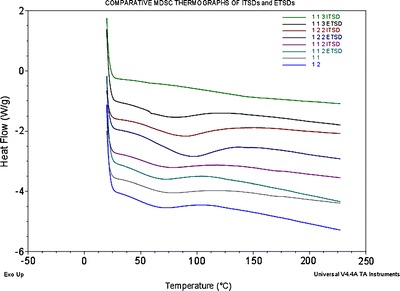
Comparative MDSC thermogram of the binary solid dispersion systems of sulfathiazole–Plasdone® K-29/32 (1:1 and 1:2) and the ITSDs and ETSDs of sulfathiazole–Plasdone® K-29/32–Syloid® 244FP (1:1:3, 1:2:2, and 1:1:3)
X-Ray Powder Diffraction Study
The X-ray powder diffraction (XRPD) patterns of the individual components of the ternary systems with all experimental systems showed similar trends for the respective ratios, as shown in Fig. 8. Sulfathiazole showed remarkably sharp and intense diffraction peaks at 20.5°, 22.5°, and 25.9°, indicating a highly organized crystalline structure. The XRPD patterns of the binary solid dispersions, ITSDs, and ETSDs support the DSC results because the characteristic X-ray diffraction peaks of sulfathiazole were absent in all systems, which indicates a loss of crystallinity. The PM showed distinct diffraction peaks with a marked decrease in the intensity at 22.5°, indicating the presence of partially crystalline structure of sulfathiazole.
Fig. 8.

X-ray diffractogram of sulfathiazole, Plasdone® K-29/32, Syloid® 244FP, binary solid dispersion system of sulfathiazole–Plasdone® K-29/32 (1:2), and ternary PM, ITSD, and ETSD of sulfathiazole–Plasdone® K-29/32–Syloid® 244FP (1:2:2)
Fourier Transform Infrared Spectroscopy
An FTIR study was conducted to further investigate the intermolecular interactions between the drug, polymer, and porous carrier. For comparison purposes, the IR spectra of the individual components of the ternary system were also collected. The acquired spectra were overlapped for each ratio of the binary solid dispersions, ITSDs, and PMs for easier comparison, as shown in Figs. 9 and 10. The IR spectra of pure sulfathiazole showed two medium intensity bands in the frequency range of 3,300–3,500 cm−1 for the stretching vibration of the –NH2 and N–H groups (30). The medium intensity band at 1,600–1,700 cm−1 indicates the presence of –N–H bending. A band characteristic of an asymmetric –SO2 group was observed at a frequency of 1,300 cm−1, and a symmetric –SO2 band was observed at approximately 1,140 cm−1. For the aromatic benzene ring, C–H stretching and C=C stretching were observed in the form of a medium intensity band in the frequency ranges of 3,000–3,100 and 1,400–1,600 cm−1, respectively. Plasdone® K-29/32 showed a characteristic strong intensity band of –C=O stretching in the frequency range of 1,600–1,800 cm−1. A strong intensity band of C–H stretching was observed at 2,850–3,000 cm−1, and a medium intensity band of –C–H bending was observed at 1,350–1,500 cm−1. Due to the hygroscopic nature of Plasdone® K-29/32, a characteristic broad band was observed at 3,500 cm−1, indicating the presence of water (31). Syloid® 244FP showed an intense Si–O linkage band at 1,106.5 cm−1, representing a silanol group. All binary solid dispersions, ITSDs, and ETSDs showed the absence of N–H stretching vibration bands between 3,300 and 3,500 cm−1 and the presence of a new band near 3,200 cm−1, confirming the intermolecular hydrogen bond formation between the amine (N–H) group of sulfathiazole and the carbonyl (–C=O) group of Plasdone® K-29/32 (32,33). In addition, the characteristic symmetric –SO2 peak at approximately 1,140 cm−1 disappeared in all ITSDs and ETSDs, where the porous carrier (Syloid® 244FP) was incorporated in the final ternary system. Rather, the ITSDs and ETSDs showed a relatively intense band at 1,106.5 cm−1 that confirmed the presence of a hydrogen bond between the silanol (Si–OH) groups of Syloid® 244FP and a symmetric –SO2 group of sulfathiazole (34). The IR spectra of the physical ternary mixture should represent a summation of all the individual spectra of sulfathiazole, Plasdone® K-29/32, and Syloid® 244FP. However, this was not the case, as the combined spectra showed the disappearance of a symmetric –SO2 at approximately 1,140 cm−1, as was seen in the ITSDs and ETSDs, and the presence of a distinct band at 1,106.5 cm−1 that confirmed hydrogen bond formation. All PM showed the presence of two medium intensity bands between 3,300 and 3,500 cm−1, confirming the presence of nonreacted –NH2 and N–H groups. This observation supports the DSC and X-ray patterns for the physical ternary mixtures with respect to the partially crystalline sulfathiazole. Despite the hydrogen bond formation, the physical ternary mixtures did not improve the solubility of sulfathiazole. When the weight ratio of the porous carrier (Syloid® 244FP) was increased in the physical ternary mixtures, the FTIR spectra of the respective ratios (1:1:2 and 1:1:3) showed an absence of the –C=O stretching of the Plasdone® K-29/32 between 1,600 and 1,800 cm−1, representing intermolecular interaction between the extra silanol groups of Syloid® 244FP and the nonreacted carbonyl group of Plasdone® K-29/32. While increasing the polymer (Plasdone® K-29/32) weight ratio for the 1:2:2 physical ternary mixture, the –C=O stretching was relatively prominent. This can be explained by the fact that the optimum amount of polymer and porous carrier is required to obtain the desired PM with a favorable intermolecular interaction between the drug and porous carrier or polymer and the porous carrier or drug, polymer, and porous carrier.
Fig. 9.
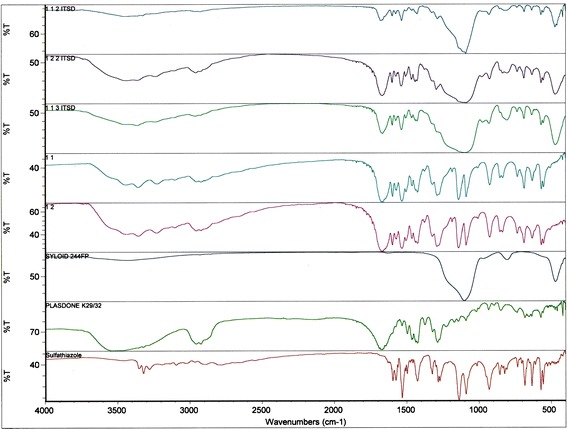
Comparative FTIR spectra of sulfathiazole, Plasdone® K-29/32, Syloid® 244FP, binary solid dispersion systems of sulfathiazole–Plasdone® K-29/32 (1:1 and 1:2), and ITSDs of sulfathiazole–Plasdone® K-29/32–Syloid® 244FP (1:1:2, 1:2:2, and 1:1:3)
Fig. 10.
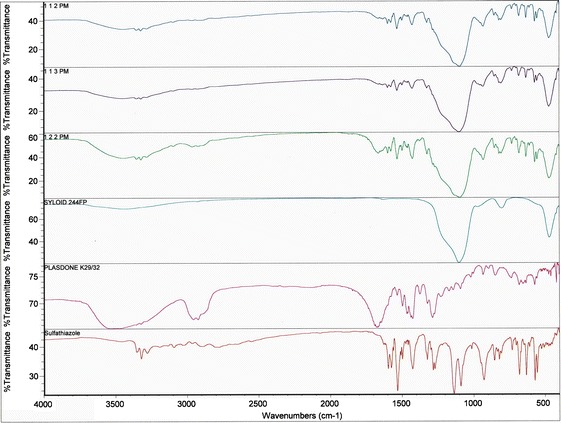
Comparative FTIR spectra of all ternary PMs of sulfathiazole–Plasdone® K-29/32–Syloid® 244FP (1:1:2, 1:2:2, and 1:1:3)
Thermal Analysis of the Stability Samples
As shown in Fig. 11, the MDSC data confirmed an amorphous form of sulfathiazole in the ITSD systems stored under accelerated stability conditions (60°C/75% RH) for 5 days. Thus, there was no significant change in the molecular level of sulfathiazole in the ITSDs, which confirmed the stable formulation of the ITSDs.
Fig. 11.
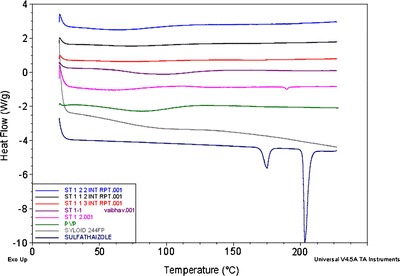
Comparative MDSC thermogram of the stability samples of the binary solid dispersion systems of sulfathiazole–Plasdone® K-29/32 (1:1 and 1:2) and all ternary ITSDs of sulfathiazole–Plasdone® K-29/32–Syloid® 244FP (1:1:2, 1:2:2, and 1:1:3) stored under accelerated stability conditions (60°C/75% RH) for 5 days
CONCLUSION
The method of incorporating Syloid® 244FP into the binary dispersion of sulfathiazole and Plasdone® K-29/32 is a key factor in preparing an effective triple “C” solid dispersion system. One of the common drawbacks of high-energy solids is recrystallization upon storage (35). This technology helps prevent this phenomenon by forming strong hydrogen bonds between the drug and polymer complex, which is adsorbed on the porous carrier with further stabilization effects of the silanol groups present on the pore surfaces. The ratio of all three components (sulfathiazole, Plasdone® K-29/32, and Syloid® 244FP) significantly affects the solubility and dispersibility of the final triple “C” solid dispersion system, thus requiring the optimum ratio of all three components. The incorporation of the porous carrier (Syloid® 244FP) is attributed to the sound art and design of the triple “C” solid dispersion system and brings formulation science technology to a new level for improving the solubility of poorly water-soluble drugs by increasing the effective surface area, improving the wettability, and transforming a crystalline structure into an amorphous form. During the dissolution process, due to the large specific surface area, as soon as the system touches water, the particles dispersed immediately. This phenomenon had prevented the crystal growth during dissolution, and a higher dissolution rate was rapidly achieved.
ELECTRONIC SUPPLEMENTARY MATERIAL
Below is the link to the electronic supplementary material.
(PDF 9113 kb)
REFERENCES
- 1.Arnum P. Solubilizing the insoluble. Pharm Technol. 2010;34(11):50–6. [Google Scholar]
- 2.Dare J. Particle size in relation to formulation. Australas J Pharm. 1964;45:S58–65. [Google Scholar]
- 3.Kai T, Akiyama Y, Nomura S, Sato M. Oral absorption improvement of poorly soluble drug using solid dispersion technique. Chem Pharm Bull. 1996;44:568–71. doi: 10.1248/cpb.44.568. [DOI] [PubMed] [Google Scholar]
- 4.Serajuddin A. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–66. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 5.Chiou W, Riegelman S. Pharmaceutical application of solid dispersion systems. J Pharm Sci. 1971;60:1281–302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 6.Noyes A, Whitney W. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19:930–4. doi: 10.1021/ja02086a003. [DOI] [Google Scholar]
- 7.Sekiguchi K, Obi N. Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem Pharm Bull. 1961;9:866–72. doi: 10.1248/cpb.9.866. [DOI] [Google Scholar]
- 8.Vasconcelos T, Sarmento B, Costa P. Solid dispersion as the strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2010;12:1068–75. doi: 10.1016/j.drudis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Leuner C, Dressman J. Improving drug solubility using solid dispersion. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 10.Gupta M, Goldman D, Bogner R, Tseng Y. Enhanced drug dissolution and bulk properties of solid dispersions granulated with a surface adsorbent. Pharm Dev Technol. 2001;6:563–72. doi: 10.1081/PDT-120000294. [DOI] [PubMed] [Google Scholar]
- 11.Prabhu S, Brocks D, Betageri G. Enhancement of dissolution of ethopropazine using solid dispersions prepared with phospholipid and/or polyethylene glycol. Drug Dev Ind Pharm. 2001;27:413–8. doi: 10.1081/DDC-100104316. [DOI] [PubMed] [Google Scholar]
- 12.Hirasawa N, Ishise S, Miyata H, Danjo K. An attempt to stabilize nilvadipine solid dispersion by the use of ternary systems. Drug Dev Ind Pharm. 2003;29:997–1004. doi: 10.1081/DDC-120025456. [DOI] [PubMed] [Google Scholar]
- 13.Okonogi S, Puttipipatkhachorn S. Dissolution improvement of high drug-loaded solid dispersion. AAPS PharmSciTech. 2006;7:E1–6. doi: 10.1208/pt070252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chutimaworapan S, Ritthidej G, Yonemochi E, Oguchi T, Yamamoto K. Effect of water-soluble carriers on dissolution characteristics of nifedipine solid dispersions. Drug Dev Ind Pharm. 2000;26(11):1141–50. doi: 10.1081/DDC-100100985. [DOI] [PubMed] [Google Scholar]
- 15.Lalitha Y, Lakshmi P. Enhancement of dissolution of nifedipine by surface solid dispersion technique. Int J Pharm Pharm Sci. 2011;3(Suppl 3):41–6. [Google Scholar]
- 16.Elbary A, Salem H, Maher M. In vitro and in vivo evaluation of glibenclamide using surface solid dispersion (SSD) approach. Br J Pharmacol Toxicol. 2011;2(1):51–62. [Google Scholar]
- 17.Wang L, Cui F, Sunada H. Preparation and evaluation of solid dispersions of nitrendipine prepared with fine silica particles using the melt-mixing method. Chem Pharm Bull. 2006;54:37–43. doi: 10.1248/cpb.54.37. [DOI] [PubMed] [Google Scholar]
- 18.Kim K, Frank M, Henderson N. Application of differential scanning calorimetry to the study of solid drug dispersions. J Pharm Sci. 1985;74(3):283–9. doi: 10.1002/jps.2600740312. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi H, Nagira S, Yamamoto H, Kawashima Y. Solid dispersion particles of amorphous indomethacin with fine porous silica particles prepared by using spray-drying method. Int J Pharm. 2005;293:155–64. doi: 10.1016/j.ijpharm.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi H, Nagira S, Yamamoto H, Kawashima Y. Solid dispersion particles of tolbutamide prepared with fine silica particles by the spray-drying method. Powder Technol. 2004;141:187–95. doi: 10.1016/j.powtec.2004.03.007. [DOI] [Google Scholar]
- 21.Simonell A, Mehta S, Higuchi W. Inhibition of sulfathiazole crystal growth by polyvinylpyrrolidone. J Pharm Sci. 1970;59:633–44. doi: 10.1002/jps.2600590512. [DOI] [PubMed] [Google Scholar]
- 22.Simonelli A, Mehta S, Higuchi W. Dissolution rate of high energy polyvinylpyrrolidone (PVP)–sulfathiazole coprecipitates. J Pharm Sci. 1969;58:538–49. doi: 10.1002/jps.2600580503. [DOI] [PubMed] [Google Scholar]
- 23.Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm. 2004;272:1–10. doi: 10.1016/j.ijpharm.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 24.Bakara M, Nagya Z, Rielly C, Dann S. Investigation of the riddle of sulfathiazole polymorphism. Int J Pharm. 2011;414:86–103. doi: 10.1016/j.ijpharm.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Tantishaiyakul V, Kaewnopparat N, Ingkatawornwong S. Properties of solid dispersions of piroxicam in polyvinylpyrrolidone K-30. Int J Pharm. 1996;143:59–66. doi: 10.1016/S0378-5173(96)04687-X. [DOI] [PubMed] [Google Scholar]
- 26.Karavas E, Ktistis G, Xenakis A, Georgarakis E. Miscibility behavior and formation mechanism of stabilized felodipine–polyvinylpyrrolidone amorphous solid dispersions. Drug Dev Ind Pharm. 2005;31(6):473–89. doi: 10.1080/03639040500215958. [DOI] [PubMed] [Google Scholar]
- 27.Drooge D, Hinrichs W, Visser M, Frijlink H. Characterization of the molecular distribution of drugs in glassy solid dispersions at the nano-meter scale, using differential scanning calorimetry and gravimetric water vapour sorption techniques. Int J Pharm. 2006;310:220–9. doi: 10.1016/j.ijpharm.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Feng T, Pinal R, Carvjal MT. Process induced disorder in crystalline materials: differentiating defective crystals from the amorphous form of griseofulvin. J Pharm Sci. 2008;88:1058–66. doi: 10.1002/jps.21219. [DOI] [PubMed] [Google Scholar]
- 29.Bahl D, Bogner RH. Amorphization alone does not account for the enhancement of solubility of drug co-ground with silicate: the case of indomethacin. AAPS PharmSciTech. 2008;9(1):146–53. doi: 10.1208/s12249-007-9013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benchaabane H, Ozer Y, Ozalp M, Kilic E, Polat M, Korkmaz M. Gamma radiation studies on sulfathiazole (powder and model-ophthalmic solution) FABAD J Pharm Sci. 2003;28(2):93–106. [Google Scholar]
- 31.Mooter G, Augustijns P, Blaton N, Kinget R. Physico-chemical characterization of solid dispersions of temazepam with polyethylene glycol 6000 and PVP K30. Int J Pharm. 1998;164:67–80. doi: 10.1016/S0378-5173(97)00401-8. [DOI] [Google Scholar]
- 32.Boldyrev V, Shakhtshneider T, Burleva L, Severtsev V. Preparation of the disperse system of sulfathiazole–polyvinylpyrrolidone by mechanical activation. Drug Dev Ind Pharm. 1994;20(6):1103–14. doi: 10.3109/03639049409038355. [DOI] [Google Scholar]
- 33.Hirasawa N, Ishise S, Miyata H, Danjo K. Physicochemical characterization and drug release studies of nilvadipine solid dispersions using water-insoluble polymer as a carrier. Drug Dev Ind Pharm. 2003;29(3):339–44. doi: 10.1081/DDC-120018207. [DOI] [PubMed] [Google Scholar]
- 34.Chauhan B, Shimpi S, Paradkar A. Preparation and evaluation of glibenclamide-polyglycolized glycerides solid dispersions with silicon dioxide by spray drying technique. Eur J Pharm Sci. 2005;26:219–30. doi: 10.1016/j.ejps.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Zhu L, Wong L, Yu L. Surface-enhanced crystallization of amorphous nifedipine. Mol Pharm. 2008;5(6):921–6. doi: 10.1021/mp8000638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 9113 kb)