

Abstract
The nucleotide-binding domain, leucine-rich repeat (NLR) proteins are a recently discovered family of intracellular pathogen and danger signal sensors. NLRs have emerged as important contributors to innate immunity in animals. The physiological impact of these genes is increasingly evident, underscored by the genetic association of variant family members with an array of inflammatory diseases. The association of mutations in NLR genes with autoinflammatory diseases indicates an important function of these genes in inflammation in vivo. This review summarizes the role of the inflammasome NLR proteins in innate immunity and inflammatory diseases and explores the possible utility of some of these NLRs as pharmacological targets.
The nucleotide-binding domain (NBD), leucine-rich repeat (LRR) (NLR) gene family is an evolutionarily conserved family of genes, important for immune function in animals (1–3). There are >20 NLR genes in humans. The NLR gene family members were discovered by their structural similarity to the MHC class II gene master regulator CIITA and other NBD-LRR-containing proteins (4). NLR genes encode cytoplasmic proteins with a tripartite domain structure that is conserved with a subclass of plant disease resistance genes (3). The tripartite structure of NLRs consists of a variable N-terminal effector domain, a central NBD, and a variable number of C-terminal LRRs. Fig. 1 provides schematics of the domain structures of the NLR proteins described in this review. The NLRs are responsible for rapid sensing of pathogen-associated molecular patterns (PAMPs) such as the bacterial cell wall components LPS, lipoproteins, and flagellin (5–11), bacterial and viral nucleic acids (12–15), and the fungal cell wall components zymosan and mannan (16). In addition, NLRs also sense damage-associated molecular patterns (DAMPs) such as ATP (17), uric acid (18, 19), amyloid-β (20), asbestos (21, 22), silica (21), hyaluronan, and heparan sulfate (23). However a major unresolved issue in the field is how an NLR acts as a sensor, because direct evidence of NLR proteins binding to a specific pathogen- or non-pathogen derived ligand is lacking. Regardless of the mechanism, the sensing of PAMPs and DAMPs by NLR proteins can result in the assembly of a caspase-1 activating multiprotein complex referred to as the “inflammasome” (2). This is similar to the cytoplasmic multiprotein complexes assembled for the activation of caspase-9 and caspase-8 referred to as the apoptosome (containing Apaf-1) (24) and the death-inducing signaling complex (Fas/CD95-DISC) (25), respectively. The protein components of the caspase-activating platforms are present as inactive monomers that oligomerize on exposure to the activating PAMP or DAMP signal. Inflammasome formation results in the cleavage of caspase-1 from its inactive proprotein form to its active mature form. This active caspase-1 then processes the cleavage of pro-IL-1 β and pro-IL-18 into mature IL-1 β and IL-18, respectively. Although IL-1 β and IL-18 are the most widely studied targets of caspase-1, two recent studies have identified >70 new targets of caspase-1 ranging from chaperones, cytoskeletal and translation machinery, and glycolysis and immune proteins (26, 27). There are several studies and related reviews analyzing the role of the NLR gene family in infectious diseases, but this review focuses on the role of the inflammasome NLRs in inflammatory diseases.
FIGURE 1.
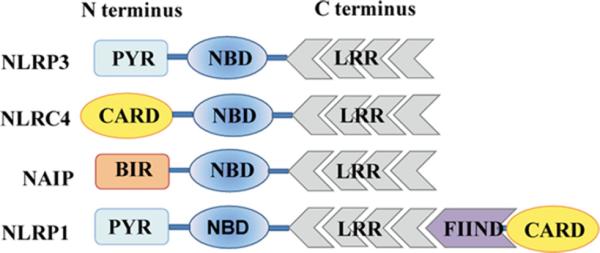
Domain organization of inflammasome NLRs. NLR proteins have a conserved tripartite structure consisting of an N-terminal effector domain, a central NBD, and a variable number of C-terminal LRRs. Abbreviations not defined elsewhere: BIR, baculovirus inhibitor of apoptosis repeat; FIIND, function to find domain; PYR, pyrin domain.
NLR gene family and NLR inflammasomes
The well known inflammasomes, the NLRP1, NLRP3, NLRC4, and NAIP5 inflammasome complexes, and their key component proteins will be discussed in brief in this section. Fig. 2 depicts the triggering PAMPs and DAMPs and the key component proteins of the four inflammasomes discussed below.
FIGURE 2.

NLR Inflammasomes. In response to PAMPs or DAMPs, the NLRs are activated to form multiprotein caspase-activating platforms referred to as inflammasomes. The NLRP1 inflammasome when activated by MDP or anthrax lethal toxin can recruit procaspase-1 via direct CARD-CARD interactions and cause its autocatalytic cleavage to mature caspase-1. The activated caspase-1 can then process IL-1 β and IL-18 from their inactive proproteins to mature active forms. The NLRP3 inflammasome is activated in response to several PAMPs and DAMPs, including but not restricted to nucleic acids (12–15), LPS (13), lipooligosaccharide (29),MDP(30), ATP(17), uric acid crystals (18), hyaluronan and heparan sulfate (31), amyloid- β (20), and asbestos and silica (21, 32). NLRP3forms a multiprotein inflammasome complex with the adaptor protein ASC and procaspase-1. Association of NLRP3 with ASC is required for recruitment of procaspase-1. The CARD domain of ASC is used to recruit procaspase-1 by CARD-CARD interactions, thus leading to the processing of procaspase-1 into active caspase-1. Caspase-1 is in turn critical for the processing and release of IL-1 β and IL-18. The NLRC4 inflammasome is a cytosolic sensor of flagellin and pathogens such as S. typhimurium, S. flexneri, and L. pneumophila (5–9, 11, 34). NLRC4 forms a homo-oligomeric inflammasome with caspase-1. The C-terminal, 35-aa fragment of flagellin is sensed by NAIP5 leading to a NAIP5-dependent cell death whereas full-length flagellin induces NAIP5-independent but NLRC4-dependent cell death and IL-1 β release (10). BIR, baculovirus inhibitor of apoptosis repeat; FIIND, function to find domain; PYR, pyrin domain.
The NLRP3 inflammasome
The NLR family, pyrin domain-containing (NLRP) 3 (NLRP3; also called Cryopyrin, NALP3, PYPAF1, CIAS1, and CLR1.1) inflammasome is activated by the presence of pathogen products such as nucleic acids (12–15), LPS (12, 13, 18, 28), lipooligosaccharide (29), and muramyldipeptide (MDP) (30); certain toxins such as nigericin (Streptomyces hygroscopicus) and maitotoxin (marine dinoflagellates) (17); cellular danger signals such as ATP (17), uric acid crystals (18), hyaluronan and heparan sulfate (31), and amyloid-β (20), environmental danger signals such as asbestos and silica (21, 32); and alum and other particulate adjuvants (32, 33). NLRP3 forms a multiprotein complex, referred to as the NLRP3 inflammasome, with the adaptor protein apoptosis-associated speck-like protein containing a caspase activating and recruitment domain (ASC) (28) and procaspase-1. Association of NLRP3 with ASC is required for recruitment of procaspase-1 (34). The caspase activating and recruitment domain (CARD) of ASC is used to recruit procaspase-1 by CARD-CARD interactions, thus leading to the processing of procaspase-1 into active caspase-1 (35).
The NLRP1 inflammasome
The human NLRP1 (also called CARD7, DEFCAP, and CLR17.1) inflammasome was the first caspase-1-activating inflammasome to be identified (36). There is only one Nlrp1 gene in humans in contrast to three paralogues in mice; Nlrp1a, Nlrp1b, and Nlrp1c (37). The NLRP1 protein in humans consists of an N-terminal pyrin domain central, an NBD, an NBD-associated domain (NAD), LRR and function to find domains (FIIND), and a C-terminal CARD domain. The mouse counterparts vary in structure from the human protein; Nlrp1a lacks the N-terminal pyrin domain, Nlrp1b lacks both the pyrin and NAD domains, and Nlrp1c lacks all but the NBD and LRR domains. Initial studies on NLRP1 using cell extracts suggested that the NLRP1 inflammasome in humans consisted of NLRP1, caspase-1, caspase-5 (not present in mice), and ASC (38, 39). Even though the presence of ASC is not required for processing of caspase-1 by the NLRP1 inflammasome, it does augment this function (40). The mouse Nlrp1b inflammasome is activated in response to Bacillus anthracis (41) and specifically to the lethal toxin. Faustin et al. used a cell-free system with recombinant NLRP1 inflammasome components to show inflammasome assembly and caspase-1 activation in response to the peptidoglycan component MDP (40). Hsu et al. showed that MDP stimulation of macrophages leads to association of NLRP1 with nucleotide oligomerization domain (NOD) 2 (41). Gel filtration experiments revealed a complex consisting of NLRP1, NOD2, and caspase-1. Moreover, Bacillus anthracis infection also induces NOD2- and caspase-1-dependent IL-1 β secretion. These results suggest the existence of a NLRP1- and NOD2-containing inflammasome and the potential for MDP to activate both NLRP1 and NOD2. However there is no data to show that MDP binds to either NLRP1 or NOD2; thus, how MDP activates this pathway is unclear.
The NLRC4 inflammasome
NLR family, CARD-containing (NLRC) 4 (NLRC4; also called IPAF, NOD27, and CLR16.1) is a cytosolic sensor of flagellin, flagellated pathogens such as Salmonella typhimurium (6, 7, 34) and Legionella pneumophila (5), and nonflagellated pathogens such as Shigella flexneri (9), and Pseudomonas aeruginosa (11). NLRC4 forms a homo-oligomeric inflammasome with caspase-1 (34). Initial characterization of NLRC4 in human tissues and cell lines demonstrated its direct association with the CARD domain of procaspase-1 through CARD-CARD interactions (42, 43). This interaction can cause autocatalytic processing of procaspase-1 into caspase-1 (43). A constitutively active NLRC4 could cause autocatalytic processing of procaspase-1 leading to caspase-1-dependent apoptosis in transfected cells (43). In macrophages, caspase-1 activation and IL-1 β release by cytoplasmic flagellin requires NLRC4 (6, 7, 34). NLRC4 can interact directly with procaspase-1 through CARD-CARD interaction; however, direct interaction of ASC with NLRC4 has not been demonstrated. Nonetheless, ASC-deficient macrophages show defective caspase-1 activation and IL-1 β release in response to Salmonella, Shigella, and Pseudomonas infection, indicating that the function of NLRC4 is ASC dependent (9, 11, 34).
The NAIP5 inflammasome
The NLR apoptosis-inhibitory protein (NAIP) 5 (also called BIRC1 and NLRB1) is also a cytosolic sensor of flagellin. Although the human genome has one Naip5 gene, there are seven paralogues of NAIP, Naip1-7, in mice (44). Based on coimmunoprecipitation studies using overexpressed Myc-tagged NAIP and hemagglutinin-tagged NLRC4 in HEK293 cells, these two proteins can coassociate, suggesting that they can be part of the same caspase-1-activating inflammasome (45). Recently, Lightfield et al. reported a novel role for NAIP5 in inflammasome activation in response to the C terminus of flagellin and L. pneumophila infection (10). Interestingly, whereas transduction of macrophages with a C-terminal 35-aa fragment of flagellin led to NAIP5-dependent cell death, full-length flagellin induced NAIP5-independent but NLRC4-dependent cell death and IL-1β release, suggesting a separation of duty for NAIP5 and NLRC4. Moreover, because NLRC4 can sense some nonflagellated bacteria (9, 11), this might point to a mechanism for differential sensing of bacteria via the regulation of inflammasome components. However, NAIP5 has no caspase domain and needs NLRC4 to activate procaspase 1. Thus, NAIP5 appears to possess NLRC4-dependent and -independent functions.
Inflammasome NLRs and inflammatory disease
NLR-related inflammatory diseases can be classified into three categories based on disease resulting from the following: 1) mutation of core components of the inflammasome complexes (intrinsic inflammasomopathies); 2) aberrant activation of the inflammasome complex (acquired or complex inflammasomopathies); and 3) mutation of accessory or regulatory proteins upstream or downstream of the inflammasome complex (extrinsic inflammasomopathies) such as pyrin or the proline serine threonine phosphatase interaction protein PSTPIP1 (46). The first two will be discussed in this review, but readers can refer to excellent reviews on the last group of proteins because they do not directly involve NLR proteins (46, 47). Table I provides a list of the disease-associated mutations discussed in this section.
Table I.
Disease-associated mutations
| NLR | Mutation(s) (Amino acid change) | Disease Association | Ref. |
|---|---|---|---|
| NLRP3 | A439V, V198M, E627G, A352V | FCAS and MWS | 57 |
| R260W, D303N, T348M, A439T, and G569R | FCAS and MWS | 54 | |
| F575S, Q306L, T436N, H358R, M662T, D303N, F309S | CINCA | 58 | |
| L264H, D303N, A374N, Y570C, F523L | CINCA | 55 | |
| L353P | FCAS | 56 | |
| T348M, E354D, L632N, R260L, R260P, D303N, D303G, F309S, T405P, T436I, Y570C | CINCA | 51 | |
| NLRP1 | L155H | Vitiligo | 63 |
| NLRP12 | R284X, V635T | Guadeloupe variant periodic fever syndrome | 66 |
Intrinsic inflammasomopathies
Cryopyrin-associated periodic syndromes
Autosomal dominant mutations in NLRP3 in humans leads to three autoinflammatory syndromes collectively referred to as cryopyrin-associated periodic syndromes (CAPS; also called cryopyrinopathies) (48–51). Gain-of-function mutations of NLRP3 cause a lowered activation threshold that leads to IL-1 β secretion even in the absence of a stimulus in vitro (36, 52, 53). All CAPS are characterized by increased levels of IL-1 β in the absence of infection. CAPS consist of a spectrum of diseases ranging from the mild, such as familial cold autoinflammatory syndrome (FCAS), to the intermediate, such as Muckle-Wells syndrome (MWS), to the severe, such as chronic infantile neurological, cutaneous and articular (CINCA) syndrome, also known as neonatal-onset multisystem inflammatory disease (NOMID). All three syndromes present with fever, urticaria-like rash, and varying degrees of arthropathy and neurological manifestations (4, 54–56). FCAS consists of the mildest symptoms, including cold-induced urticaria and mild arthralgia. MWS is characterized by spontaneous urticaria (not cold-induced), sensorineural hearing loss, arthralgia, and in some cases renal amyloidosis. CINCA is the most severe, with spontaneous urticaria, deforming arthropathy, sensorineural hearing loss, and chronic aseptic meningitis.
Hoffman et al. sequenced a region within chromosome 1q44 that was previously known to contain mutations that lead to FCAS and MWS (57). This screening approach led to the discovery of four distinct mutations in exon 3 of a nine-exon gene that segregated with the disorder in three families with FCAS and one family with MWS. This gene is now referred to as NLRP3. NLRP3 is a cytoplasmic protein that is expressed in monocytes, macrophages, granulocytes, dendritic cells, nonkeratinized epithelial cells, osteoblasts, and uroepithelial cells (58, 59). NLRP3 is composed of three distinct domains: an N-terminal pyrin domain, a central NBD, and C-terminal LRRs. All 84 of the disease-associated mutations lie within exon 3, which encodes the central NBD of NLRP3 (60). The pyrin domain of NLRP3 is essential for homotypic interactions with the pyrin domain of other proteins. The NBD is thought to be involved in the oligomerization of NLRP3 to form the inflammasome complex. The LRR domain is suggested to mediate interaction with intracellular or extracellular PAMPs or DAMPs, albeit no evidence has been reported. Additional studies reported the role of NLRP3 in response to bacterial RNA, dsRNA, viral RNA, uric acid crystals, TLR ligands, bleomycin, and ATP using Nlrp3-deficient mice (12, 13, 18, 28). Recently, the Strober and Hoffman laboratories separately generated mice expressing mutations corresponding to human FCAS or MWS mutations (61, 62). Meng et al. generated a mouse expressing the R258W mutation corresponding to the human R260W substitution (62). Brydges et al. generated two lines of mice carrying the A350V and L351P mutations that correspond to the human A352V and L353P mutations downstream of a LoxP – flanked neomycin resistance cassette in reverse orientation (61). When these mice were crossed with Cre recombinase-expressing mice they would express the mutated NLRP3 protein in all tissues (CreZ), in myeloid cells only (CreL), or after exposure to tamoxifen (CreT). The mice generated by both studies developed severe cutaneous lesions associated with inflammatory cell infiltrates, recapitulating some of the urticaria-like skin lesions in MWS patients. Interestingly, both studies could recapitulate human disease by either expressing mutant NLRP3 only in myeloid cells (61) or by the generation of bone marrow chimeras with the mutant R258W protein in bone marrow cells (62).
Vitiligo
This is an autoimmune disease resulting from destruction of melanocytes causing patches of depigmented skin in patients. Vitiligo patients are at a higher risk for the development of other autoimmune diseases such as rheumatoid arthritis, diabetes, lupus, and thyroid disease. Fine scale association analyses of patients with vitiligo identified Nlrp1 variants that are associated with the development of vitiligo alone (63). The mechanism by which NLRP1 leads to skin hypopigmentation in vitiligo remains unknown.
Complex or acquired inflammasomopathies
Gout/pseudogout
Gout and pseudogout are rheumatic diseases caused by deposition of monosodium urate (MSU) and calcium pyrophosphate dihydrate crystals respectively, in joints and periarticular tissues. This deposition can lead to acute or chronic inflammation of the joints. MSU and calcium pyrophosphate dihydrate crystals increase caspase-1 activation and IL-1 β release from murine macrophages in an NLRP3- and ASC-dependent manner (18). The importance of IL-1 β in gout studied in mice was further supported by the resistance of mice deficient in the IL-1 and TLR signaling adaptor protein MyD88 to MSU-induced inflammation (64). Although TLR deficient mice still showed inflammation, the IL-1 β receptor deficient mice did not, thus indicating a specific role for IL-1 signaling in the pathology. Bone marrow reconstitution experiments established that IL-1R expression in nonhematopoietic and hematopoietic cells is required for the initiation of inflammation upon MSU stimulation, indicating IL-1 β engagement to its receptor in this model.
Asbestosis and silicosis
Prolonged inhalation of asbestos and silica leads to two environmentally induced forms of pulmonary fibrosis referred to as asbestosis and silicosis, respectively. Alveolar macrophages from individuals with prolonged exposure to asbestos exhibit enhanced IL-1 β release (22). Moreover, Nlrp3- deficient mice show decreased IL-1 β release in response to asbestos and silica (21, 32), indicating a role for NLRP3 in the immune response to asbestos and silica. Silica crystals, once phagocytosed, can cause lysosomal damage leading to release of the lysosomal protease, cathepsin B, which can activate the NLRP3 inflammasome. Inhibition of phagosomal acidification or cathepsin B impairs NLRP3 inflammasome activation (32). In the bleomycin-induced lung injury model of fibrosis, the NLRP3 inflammasome is triggered by local uric acid release in response to DNA damage and degradation after bleomycin injury, suggesting that uric acid may be one of the triggering DAMPs in lung fibrosis and disease (65).
Guadeloupe variant periodic fever syndrome (FCAS2)
This syndrome was first reported in two families in Guadeloupe and thus named the Guadeloupe variant periodic fever syndrome (66). Based on the similarities in symptoms to FCAS, this syndrome is also referred to as FCAS2. Individuals with this syndrome present with cold-induced heterogeneous symptoms including fever, arthralgia, myalgia, sensorineural hearing loss, aphthous ulcers, and lymphadenopathy.
Genetic studies in patients with the Guadeloupe variant periodic fever syndrome revealed two missense mutations, one nonsense mutation, and one deletion mutation in the Nlrp12 gene. The nonsense mutation caused a truncation within the NBD of the protein whereas the splice mutation caused a deletion of the C-terminal LRRs. NLRP12 was recognized as one of the few NLR proteins that can suppress NF-κB signaling (67, 68). Both of the missense mutations in Nlrp12 caused a reduction in the suppression of NF-κB signaling by NLRP12, whereas the NBD mutation caused a more significant impact on normal NLRP12-induced NF-κB signaling as compared with the LRR mutation.
NLRs as potential pharmacological targets
Activation of the various inflammasome complexes discussed in this review leads to activation of caspase-1 and production of the proinflammatory cytokines IL-1 β and IL-18. Although specific drugs that interfere with inflammasome components are under development, there have been several clinical studies exploring the modification of the IL-1 β pathway owing to its central role in several diseases (69). Modulation of IL-1 β function has been approached at three levels: firstly, the release of IL-1 β can be blocked by the inhibition of upstream pathways (70, 71); secondly, the released cytokine can be neutralized or its receptor blocked to prevent downstream signaling (70, 72); and finally, the signaling mechanisms in the target cells can be blocked by disrupting further downstream signaling pathways (73–78). A detailed list of the available drugs targeting the above mentioned steps of regulation for the IL-1 β pathway along with their mechanisms of action is provided in Table II. There are some caveats in the use of some of the inhibitors because they can inhibit not only the IL-1 β but also the IL-18 pathways. A better understanding of the underlying mechanism for each disease would provide more accurate targets. Target specificity would enable a more accurate control of pathology. CAPS symptoms remain the gold standard, as they can be reversed by treatments with the IL-1R antagonist Kineret. Although some of these drugs are efficacious in relieving symptoms (72, 74–78), several others are in clinical trials or remain to be tested in humans, awaiting further studies of their mode of action (70, 71).
Table II.
Pharmacological inhibitors
| Action | Target | Drug (Company) | Description | Ref. |
|---|---|---|---|---|
| Suppression of IL-1β production | ||||
| Caspase-1 inhibition | Caspase-1 | Pranalcasan (Aventis/Vertex) | VX-740; VX-765 | 70 |
| IL-1 β posttranslational processing | Unknown | CP424174, CP412245 (Pfizer) | Diarylsulphonyl urea | 70 |
| IL-1 β production inhibitor | Unknown | CJ14877, CJ14897 (Pfizer) | Pyridine-2-carboxylates | 70 |
| Unknown | LL-Z1217a (Pfizer) | Terpenoid lactone | 70 | |
| Suppression of IL-1 β release | ||||
| IL-1 β release inhibitors | Unknown | CP424174 (Pfizer) | Diarylsulphonyl urea | 70 |
| Neutralization of secreted IL-1 β | IL-1 | Anakinra (Kineret, Amgen) | rhuIL-1Raa | 70 |
| IL-1 | IL-1trap (Regeneron/Novartis) | Human IL-1R1:IgG1 protein | 70 | |
| IL-1 | CDP-484 (Celltech) | PEGylated Ab | 70 | |
| Inhibition of IL-1R signal transduction | ||||
| MyD88 inhibitors | MyD88 | Hydrocinnamoyl-L-valyl pyrrolidine15 | MyD88 mimic | 70, 73 |
| ST2825 (Sigma-Tau)b | Peptidomimetic | 74 | ||
| IRAK-4 inhibitors | IRAK-4 | Names unavailableb | Amides, imidazo [1,2-a] pyridine compounds | 76-78 |
Recombinant human IL-1Ra.
May also inhibit IL-18R and TLR signal transduction.
Conclusions
The association of the NLRs with several immunological diseases suggests a role for these proteins in both innate and adaptive immunity. Recent studies are beginning to unfold the role of this family in immune regulation and dysregulation; however, a plethora of questions remain unanswered. Firstly, how is the diversity of PAMPs and DAMPs sensed and differentiated from self-molecules? Secondly, how does such a wide range of symptoms in CAPS arise from mutations that are relatively clustered in the NBD of NLRP3? Thirdly, is there a cross-talk between the different inflammasome pathways and do they compensate for each other? Finally, what are the DAMPs and PAMPs that might activate the inflammasome pathways in complex immune diseases such as type II diabetes, multiple sclerosis, and atherosclerosis? Considering the vibrant research in this field, significant progress is likely to resolve several of these issues.
Abbreviations used in this paper
- NBD
nucleotide-binding domain
- ASC
apoptosis associated speck-like protein containing a caspase recruitment domain
- CAPS
cryopyrin associated periodic syndrome
- CARD
caspase activating and recruitment domain
- CINCA
chronic infantile neurological, cutaneous and articular syndrome
- DAMP
damage-associated molecular pattern
- FCAS
familial code autoinflammatory syndrome
- LRR
leucine-rich repeat
- NAD
NBD-associated domain
- MDP
muramyl dipeptide
- MWS
Muckle-Well syndrome
- MSU
monosodium urate
- NAIP
NLR apoptosis inhibitory protein
- NLR
nucleotide-binding domain, leucine-rich repeat
- NLRC
NLR family, CARD containing (inflammasome)
- NLRP
NLR family, pyrin domain-containing (inflammasome)
- NOD
nucleotide oligomerization domain
- NOMID
neonatal onset multisystem inflammatory disease
- PAMP
pathogen-associate molecular pattern
Footnotes
This work was supported by National Institutes of Health Grant AI-067798 and a grant from the Radiation Countermeasures Center of Research Excellence (to J.P.-Y.T.).
Disclosures
The authors have no financial conflict of interest.
References
- 1.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 2.Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659–662. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu. Rev. Immunol. 2005;23:387–414. doi: 10.1146/annurev.immunol.23.021704.115616. [DOI] [PubMed] [Google Scholar]
- 4.Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, pyrin and hereditary immunological disorders. Nat. Rev. Immunol. 2006;6:183–195. doi: 10.1038/nri1788. [DOI] [PubMed] [Google Scholar]
- 5.Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, Nunez G. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 2006;281:35217–35223. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- 6.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 7.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1 β via Ipaf. Nat. Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 8.Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Nat. Acad. Sci. USA. 2008;105:2562–2567. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 2008;9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 13.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 14.Di Paolo NC, Miao EA, Iwakura Y, Murali-Krishna K, Aderem A, Flavell RA, Papayannopoulou T, Shayakhmetov DM. Virus binding to a plasma membrane receptor triggers interleukin-1 β-mediated proinflammatory macrophage response in vivo. Immunity. 2009;31:110–121. doi: 10.1016/j.immuni.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–107. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 16.Lamkanfi M, Malireddi RK, Kanneganti TD. Fungal zymosan and mannan activate the cryopyrin inflammasome. J. Biol. Chem. 2009;284:20574–20581. doi: 10.1074/jbc.M109.023689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 18.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 20.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid- β. Nat. Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perkins RC, Scheule RK, Hamilton R, Gomes G, Freidman G, Holian A. Human alveolar macrophage cytokine release in response to in vitro and in vivo asbestos exposure. Exp. Lung Res. 1993;19:55–65. doi: 10.3109/01902149309071080. [DOI] [PubMed] [Google Scholar]
- 23.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 24.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 25.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 26.Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J. Biol. Chem. 2007;282:36321–36329. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- 27.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 28.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Duncan JA, Gao X, Huang MT, O'Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF, Ting JP. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 2009;182:6460–6469. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marina-Garcia N, Franchi L, Kim YG, Miller D, McDonald C, Boons GJ, Nunez G. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J. Immunol. 2008;180:4050–4057. doi: 10.4049/jimmunol.180.6.4050. [DOI] [PubMed] [Google Scholar]
- 31.Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, Grant EP, Coyle AJ, Misaghi A, Hoffman HM, Gallo RL. NLRP3/Cryopyrin is necessary for interleukin-1 β (IL-1 β) release in response to Hyaluronan, an endogenous trigger of inflammation in response to injury. J. Biol. Chem. 2009;284:12762–12771. doi: 10.1074/jbc.M806084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum's adjuvant effect are mediated by NLRP3. J. Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 35.Li P, Allen H, Banerjee S, Seshadri T. Characterization of mice deficient in interleukin-1β converting enzyme. J. Cell. Biochem. 1997;64:27–32. doi: 10.1002/(sici)1097-4644(199701)64:1<27::aid-jcb5>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 36.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 37.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu ZL, Pio F, Xie Z, Welsh K, Krajewska M, Krajewski S, Godzik A, Reed JC. A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis. J. Biol. Chem. 2001;276:9239–9245. doi: 10.1074/jbc.M006309200. [DOI] [PubMed] [Google Scholar]
- 39.Hlaing T, Guo RF, Dilley KA, Loussia JM, Morrish TA, Shi MM, Vincenz C, Ward PA. Molecular cloning and characterization of DEFCAP- L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J. Biol. Chem. 2001;276:9230–9238. doi: 10.1074/jbc.M009853200. [DOI] [PubMed] [Google Scholar]
- 40.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 41.Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, Eckmann L, Powell JJ, Nizet V, Dixit VM, Karin M. A NOD2- NALP1 complex mediates caspase-1-dependent IL-1β secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geddes BJ, Wang L, Huang WJ, Lavellee M, Manji GA, Brown M, Jurman M, Cao J, Morgenstern J, Merriam S, et al. Human CARD12 is a novel CED4/Apaf-1 family member that induces apoptosis. Biochem. Biophys. Res. Commun. 2001;284:77–82. doi: 10.1006/bbrc.2001.4928. [DOI] [PubMed] [Google Scholar]
- 43.Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 2001;276:28309–28313. doi: 10.1074/jbc.C100250200. [DOI] [PubMed] [Google Scholar]
- 44.Endrizzi MG, Hadinoto V, Growney JD, Miller W, Dietrich WF. Genomic sequence analysis of the mouse Naip gene array. Genome Res. 2000;10:1095–1102. doi: 10.1101/gr.10.8.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, Kuida K, Mariathasan S, Dixit VM, Flavell RA, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 2006;7:318–325. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 46.Masters SL, Lobito AA, Chae J, Kastner DL. Recent advances in the molecular pathogenesis of hereditary recurrent fevers. Curr. Opin. Allergy Clin. Immunol. 2006;6:428–433. doi: 10.1097/ACI.0b013e3280109b57. [DOI] [PubMed] [Google Scholar]
- 47.Ryan JG, Kastner DL. Fevers, genes, and innate immunity. Curr. Top. Microbiol. Immunol. 2008;321:169–184. doi: 10.1007/978-3-540-75203-5_8. [DOI] [PubMed] [Google Scholar]
- 48.Boschan C, Witt O, Lohse P, Foeldvari I, Zappel H, Schweigerer L. Neonatal-onset multisystem inflammatory disease (NOMID) due to a novel S331R mutation of the CIAS1 gene and response to interleukin-1 receptor antagonist treatment. Am J. Med. Genet. A. 2006;140:883–886. doi: 10.1002/ajmg.a.31148. [DOI] [PubMed] [Google Scholar]
- 49.Lequerre T, Vittecoq O, Saugier-Veber P, Goldenberg A, Patoz P, Frebourg T, Loet XL. A cryopyrin-associated periodic syndrome with joint destruction. Rheumatology (Oxford) 2006;46:709–714. doi: 10.1093/rheumatology/kel399. [DOI] [PubMed] [Google Scholar]
- 50.Kone-Paut I, Sanchez E, Le Quellec A, Manna R, Touitou I. Autoinflammatory gene mutations in Behcet's disease. Ann. Rheum. Dis. 2007;66:832–824. doi: 10.1136/ard.2006.068841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neven B, Callebaut I, Prieur AM, Feldmann J, Bodemer C, Lepore L, Derfalvi B, Benjaponpitak S, Vesely R, Sauvain MJ, et al. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood. 2004;103:2809–2815. doi: 10.1182/blood-2003-07-2531. [DOI] [PubMed] [Google Scholar]
- 52.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 53.Gattorno M, Tassi S, Carta S, Delfino L, Ferlito F, Pelagatti MA, D'Osualdo A, Buoncompagni A, Alpigiani MG, Alessio M, et al. Pattern of interleukin-1 β secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum. 2007;56:3138–3148. doi: 10.1002/art.22842. [DOI] [PubMed] [Google Scholar]
- 54.Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, Meyrier A, Watts RA, Scott DG, Nicholls A, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am. J. Hum. Genet. 2002;70:1498–1506. doi: 10.1086/340786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, Stein L, Russo R, Goldsmith D, Dent P, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–3348. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffman HM, Gregory SG, Mueller JL, Tresierras M, Broide DH, Wanderer AA, Kolodner RD. Fine structure mapping of CIAS1: identification of an ancestral haplotype and a common FCAS mutation, L353P. Human Genet. 2003;112:209–216. doi: 10.1007/s00439-002-0860-x. [DOI] [PubMed] [Google Scholar]
- 57.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, de Saint Basile G. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manji GA, Wang L, Geddes BJ, Brown M, Merriam S, Al-Garawi A, Mak S, Lora JM, Briskin M, Jurman M, et al. PYPAF1, a PYRIN-containing Apaf1- like protein that assembles with ASC and regulates activation of NF-B. J. Biol. Chem. 2002;277:11570–11575. doi: 10.1074/jbc.M112208200. [DOI] [PubMed] [Google Scholar]
- 60.Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, Lesage S, Waterham H, Wise C, Sarrauste de Menthiere C, Touitou I. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum. Mut. 2008;29:803–808. doi: 10.1002/humu.20720. [DOI] [PubMed] [Google Scholar]
- 61.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, et al. Inflammasome mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–887. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–874. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR, Spritz RA. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 2007;356:1216–1225. doi: 10.1056/NEJMoa061592. [DOI] [PubMed] [Google Scholar]
- 64.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J. Clin. Invest. 2006;116:2262–2271. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, Couillin I. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2009;179:903–913. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- 66.Jeru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, Grimprel E, Landman-Parker J, Hentgen V, Marlin S, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA. 2008;105:1614–1619. doi: 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, Ting JP. Monarch-1 suppresses non-canonical NF-κB activation and p52- dependent chemokine expression in monocytes. J. Immunol. 2007;178:1256–1260. doi: 10.4049/jimmunol.178.3.1256. [DOI] [PubMed] [Google Scholar]
- 68.Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, Kurtz S, Coffield VM, Accavitti-Loper MA, Su L, et al. The CATERPILLER protein Monarch-1 is an antagonist of Toll-like receptor-, tumor necrosis factor α-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J. Biol. Chem. 2005;280:39914–39924. doi: 10.1074/jbc.M502820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 70.Braddock M, Quinn A. Targeting IL-1 in inflammatory disease: new opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 2004;3:330–339. doi: 10.1038/nrd1342. [DOI] [PubMed] [Google Scholar]
- 71.Ichikawa K, Hirai H, Ishiguro M, Kambara T, Kato Y, Kim YJ, Kojima Y, Matsunaga Y, Nishida H, Shiomi Y, et al. Cytokine production inhibitors produced by a fungus, Oidiodendron griseum. J. Antibiot. 2001;54:697–702. doi: 10.7164/antibiotics.54.697. [DOI] [PubMed] [Google Scholar]
- 72.Hoffman HM. Rilonacept for the treatment of cryopyrin-associated periodic syndromes (CAPS). Expert Opin. Biol. Ther. 2009;9:519–531. doi: 10.1517/14712590902875518. [DOI] [PubMed] [Google Scholar]
- 73.Bartfai T, Behrens MM, Gaidarova S, Pemberton J, Shivanyuk A, Rebek J., Jr. A low molecular weight mimic of the Toll/IL-1 receptor/resistance domain inhibits IL-1 receptor-mediated responses. Proc. Natl. Acad. Sci. USA. 2003;100:7971–7976. doi: 10.1073/pnas.0932746100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loiarro M, Capolunghi F, Fanto N, Gallo G, Campo S, Arseni B, Carsetti R, Carminati P, De Santis R, Ruggiero V, Sette C. Pivotal Advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leukocyte Biol. 2007;82:801–810. doi: 10.1189/jlb.1206746. [DOI] [PubMed] [Google Scholar]
- 75.Loiarro M, Sette C, Gallo G, Ciacci A, Fanto N, Mastroianni D, Carminati P, Ruggiero V. Peptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of NF-κB. J. Biol. Chem. 2005;280:15809–15814. doi: 10.1074/jbc.C400613200. [DOI] [PubMed] [Google Scholar]
- 76.Buckley GM, Gowers L, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, et al. IRAK-4 inhibitors. Part 1: a series of amides. Bioorg. Med. Chem. Lett. 2008;18:3211–3214. doi: 10.1016/j.bmcl.2008.04.058. [DOI] [PubMed] [Google Scholar]
- 77.Buckley GM, Fosbeary R, Fraser JL, Gowers L, Higueruelo AP, James LA, Jenkins K, Mack SR, Morgan T, Parry DM, et al. IRAK-4 inhibitors. Part III: a series of imidazo[1,2-a]pyridines. Bioorg. Med. Chem. Lett. 2008;18:3656–3660. doi: 10.1016/j.bmcl.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 78.Buckley GM, Ceska TA, Fraser JL, Gowers L, Groom CR, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, et al. IRAK-4 inhibitors. Part II: a structure-based assessment of imidazo[1,2-a]pyridine binding. Bioorg. Med. Chem. Lett. 2008;18:3291–3295. doi: 10.1016/j.bmcl.2008.04.039. [DOI] [PubMed] [Google Scholar]