

Abstract
The presence of 2 distinct populations of somatic or germline cells within a single individual harboring different genotypes is termed mosaicism. Recent reports suggest that parental mosaicism is involved in the heritability of type 1 Timothy syndrome (TS1), an extremely rare and life-threatening multisystem disorder characterized by severe QT interval prolongation, syndactyly, and several other complications. Although full TS1 is caused by a single missense mutation in the CACNA1C-encoded cardiac calcium channel, mosaic TS1 parents can display isolated syndactyly without additional phenotypic manifestations. A newborn boy presented with syndactyly at birth. The presence of syndactyly in his mother led to a diagnosis of benign familial syndactyly. However, at 9 months of age, during his first syndactyly-corrective surgery, intraoperative electrocardiograms revealed extreme QT prolongation and 2:1 atrioventricular block. A comprehensive cardiac evaluation was performed, and both mother and child were tested genetically, confirming a clinical suspicion of TS1. Only the patient tested positive for the TS1 mutation; however, more extensive molecular testing revealed a limited presence of the mutation in maternally-derived DNA. This case illustrates the potential of parental mosaicism to confound the diagnosis of potentially life-threatening genetic diseases, such as TS1. Here, a mother with a partial TS1 phenotype and genetically confirmed mosaicism transmitted the TS1-causative mutation to her son, resulting in fully expressive TS1. Thus, a shared partial phenotype should not be dismissed as a benign or insignificant finding, but should be evaluated further to rule out the possibility of parental mosaicism concealing a potentially fatal heritable disease.
Keywords: long QT syndrome, mosaicism, syndactyly, Timothy syndrome
Timothy syndrome is an extremely rare multisystem disorder characterized by a constellation of phenotypic abnormalities including syndactyly, cognitive defects, and a high risk of sudden death secondary to severe cardiac arrhythmias.1 Type 1 Timothy syndrome (TS1), the most common form of the disease, stems from gain-of-function mutations in the CACNA1C-encoded Cav1.2 calcium channel, canonically a single missense mutation (G406R) in CACNA1C’s exon 8a-containing splice variant.
Typically, individuals with TS present at birth with severe perturbations of the cardiac electrical system, including marked heart rate–corrected QT interval prolongation (QTc > 500 ms).2 The webbing of fingers (unilateral or bilateral variably affecting digits 2 through 5) and toes (bilateral between toes 2 and 3), known as syndactyly, is an additional clinical hallmark.1,3 Syndactyly is one of the most commonly observed congenital malformations, affecting an estimated 1 in 2000 to 2500 live births (0.04%–0.05%), with regional occurrence rates ranging between 0.06% to 0.12%.3–6
The genetic phenomenon of mosaicism, whereby 2 genetically distinct populations of cells coexist within an individual’s somatic cells or gametes, has been observed in TS1 individuals/families on at least 3 separate occasions.1,2,7 These individuals are classified as having somatic and/or germline mosaicism according to the source of the affected cells. Interestingly, mosaicism also contributes to the inheritance of other multisystem disorders, such as Klinefelter’s syndrome.8 With such a wide variety of phenotypic manifestations in TS1, somatic and germline mosaicism provides limitless possibilities for presenting and concealing TS1-causative mutations.
Case Report
Here, we present the case of a 17-month-old boy with TS1 who was born with bilateral syndactyly on both hands and feet (Fig 1A). Because of maternal preeclampsia, the patient was prematurely delivered at 33 weeks. As a result, the patient spent the first 3.5 weeks of life in the NICU.
FIGURE 1.
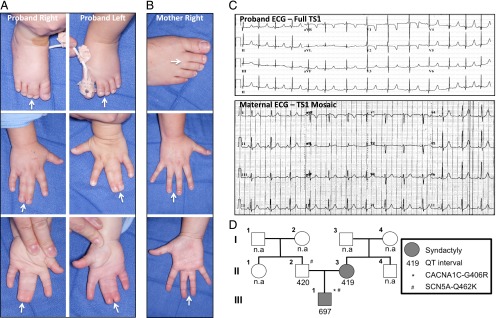
Clinical and electrocardiographic phenotypes of the proband and his mother. A, Proband with complete bilateral syndactyly of the fingers and toes. B, Mother with incomplete right-sided syndactyly of the fingers and toes. C, Proband’s 12-lead ECG demonstrating an extremely prolonged interval (QT/QTc of 430/697 ms with a heart rate of 158 beats per minute). The maternal ECG was completely normal (QT/QTc of 386/419 ms with a heart rate of 79 beats per minute). D, Pedigree structure indicating commercial genetic testing results and QT intervals for the proband and his parents.
While in the NICU, a genetics consultation was obtained, and the index case’s bilateral syndactyly was attributed to familial syndactyly secondary to the presence of maternal right-sided syndactyly (Fig 1B). At this time, rare, life-threatening, multisystem causes of syndactyly, such as TS1, were not evaluated further, and the index case was scheduled to follow-up with medical genetics in 6 months. However, during the infant’s first syndactyly-corrective surgery, the presence of 2:1 atrioventricular block and a prolonged QT interval were noted on an intraoperative electrocardiogram (ECG) and remained prolonged on subsequent follow-up 12-lead ECGs (Fig 1C).
Because of the presence of a prolonged QT interval in conjunction with bilateral syndactyly, TS1 was suspected and commercial genetic testing was initiated on all targeted exons and intron boundaries of the 13 established long QT syndrome susceptibility genes. While awaiting genetic testing results, the patient was referred to the Mayo Clinic for further evaluation. Genetic test results were positive for the canonical TS1-causative mutation (c.1216G>A in the alternatively spliced exon 8a of CACNA1C), resulting in the missense mutation annotated as p.G406R (Figs 1D and 2A), as well as a variant of unknown significance (p.G462K) in the SCN5A-encoded Nav1.5 sodium channel that was also identified in the patient’s father (Fig 1D). Although a contributory role of SCN5A-G462K cannot be ruled out, the normal QT interval of the patient’s father and presence of a clinical picture consistent with TS1 suggest that CACNA1C-G406R is responsible for the patient’s disease. Given the high risk of fatal arrhythmia associated with TS1, the patient underwent left cervical sympathetic denervation surgery and received an implantable cardiac defibrillator for primary prevention. Of note, the patient has had no documented episodes of torsades de pointes or torsades de pointes–triggered cardiac events, which is surprising given the symptomatic nature of the disease and average age of death of 2.5 years for TS1 individuals.1
FIGURE 2.

Proof of maternal somatic mosaicism for the canonical TS1-causative CACNA1C c.1216 G>A mutation. A, The G406R mutation is shown in the context of the alternatively spliced exon 8a of CACN1AC. B, Amplification of DNA using the same primers used in the discovery of TS1.1 A minor A peak can be recognized in the 1216 position in the mother’s tissue (outlined in red). After an Acil restriction enzyme digest and gel extraction, the A peak is exaggerated, indicating a successful, although incomplete, digestion. C, TS1 mutation (p.G406R, c.1216 G>A) causes the loss of a splice site on exon 8a for the restriction enzyme AciI.7 The mother’s digested DNA shows a faint mutant band (outlined in yellow).
Given the mother’s right-sided syndactyly, it was postulated that a genetic link might exist between the 2 cases, but without evidence of QT prolongation or a positive genetic test (Fig 1 C and D), a diagnosis of TS1 in the mother could not be rendered. At this point, the non-Mendelian genetic phenomenon of mosaicism was proposed to explain both the existence of syndactyly in the absence of a discernible cardiac phenotype and the suspected transmission of CACNA1C-G406R to her son. As such, we hypothesized that the presence of CACNA1C-G406R in a portion of the mother’s somatic cells (somatic mosaicism) accounted for the phenotypic overlap of syndactyly, whereas the presence of CACNA1C-G406R in some of her gametes (germline mosiacism) was responsible for passing the TS1 mutation to her son.
Because of this suspicion, buccal, lymphocyte, and left/right arm skin biopsy samples were acquired and genomic DNA (gDNA) extracted. Both left and right arm biopsies were taken to determine if a difference in CACNA1C-G406R copy number exists, providing a potential explanation for the mother’s unilateral syndactyly. Each sample was submitted for direct DNA sequencing and underwent an AciI restriction enzyme assay that selectively cuts CACNA1C-wild type (WT). The lymphocyte-derived gDNA was assessed for mutant and WT copy numbers, using a method previously described by Etheridge et al,8 to quantify the degree of mosaicism present in the mother.
Direct DNA sequencing of the mother’s DNA samples revealed traces of a G>A substitution at position 1216 (Fig 2B). To enhance the detection of the mutant CACNA1C-G406R allele in these samples, the aforementioned AciI restriction enzyme assay was used to cut CACNA1C-WT strands, leaving the 1216G>A mutant sequence intact that confirmed the presence of mosaicism (Fig 2C). The uncut band underwent a QIAGEN (Valencia, CA) gel extraction protocol and was sequenced, revealing a stronger “A” peak in all cases, indicating a successful, although not complete, digest and extraction of the uncut mutant allele (Fig 2B). To estimate the gene copy number and transcript levels in the mother’s blood, copies of the gene from lymphocyte-derived gDNA were TA cloned into Escherichia coli cells. Collectively, 32 colonies were sequenced, and of these, 2 colonies contained the mutation, 29 were WT, and 1 was inconclusive, probably owing to 2 separate colonies being picked up and sequenced together.
From these data, we estimated the percent mosaicism to be ∼6.5% (2/31), which corresponds to the estimated allelic frequency of the mutation in the mother’s lymphocytes. Assuming this allelic frequency is consistent throughout the body’s cells, we would expect 13% of diploid somatic cells and just 6.5% of haploid gametes to harbor the mutation.
Discussion
Non-Mendelian genetic interactions, such as mosiacism, can present a challenge for physicians and clinical geneticists. In this case, the neonatal diagnosis of potentially fatal TS1 was confounded by what was presumed to be benign familial syndactyly passed from mother to son. Luckily, in this case, the intraoperative detection of a prolonged QT interval triggered a subsequent, comprehensive cardiovascular and genetic investigation that led to the diagnosis of TS1 before the proband suffered a fatal cardiac event. After commercial genetic testing, the proband’s mother was classified as negative. However, on closer examination, our analyses revealed a low allelic frequency of CACNA1C-G406R (∼6.5%) throughout her body, suggesting that her syndactyly may be linked to TS1 somatic mosaicism. Given the absence of QT prolongation and history of cardiac events suggestive of a concealed long QT syndrome phenotype, we postulate that the low level of CACNA1C-G406R alleles in the mother’s heart fail to manifest as an overt or concealed cardiac phenotype. As such, the patient’s mother is believed to be at low risk for ventricular arrhythmias, and additional cardiac surveillance is deemed unnecessary.
From a genetic standpoint, the low allelic frequency of CACNA1C-G406R observed in the patient’s mother indicates a low chance of transmitting fully expressive TS1 to her progeny, assuming the degree of somatic and germline mosaicism are equivalent. However, recent studies of mosaicism in Klinefelter’s syndrome have shown that mosaicism may affect the germline preferentially, with a mutation frequency of <10% in blood lymphocytes and >36% in the gametes.7 This observation, coupled with the fact that commercial genetic testing was unable to detect low-level CACNA1C-G406R mosiacism, suggests that a negative test result in the parent(s) of children with rare genetic disorders associated with germline mosiacism should be viewed with healthy skepticism, as these tests are optimized to detect heterozygous mutations (ie, 50% allelic frequency), rather than the low frequency (ie, <10%) of the mutant allele present in the lymphocyte-derived gDNA of mosaic individuals. As such, clinicians may consider additional testing, particularly in cases in which an overlapping phenotype is observed, when using genetic test results to help parents make informed family planning decisions.
Despite the extreme rarity of TS1, mosaicism has now been reported in at least 4 separate cases of TS1.1,7 Given these findings, it is clear that mosaicism contributes to the inheritance of TS1. Although we acknowledge this is not the first case in which a parent harboring limited presence of the canonical TS1 mutation has been affected by syndactyly alone, without any cardiac phenotype, this is the first case in which the diagnosis of this potentially lethal disorder was confounded by mosaicism. Thus, isolated parental syndactyly unaccompanied by additional syndromic manifestations should not simply be assumed to be benign from a genetic or cardiologic standpoint. Although the vast majority of familial syndactyly cannot be attributed to multisyndromic disorders such as TS1, this case highlights the potential danger of neglecting further evaluation of newborns for potentially fatal disorders simply because a partial phenotypic overlap is observed between parent and child. In this case, a simple and inexpensive neonatal 12-lead ECG would have detected the index case’s extreme QT prolongation and prevented the year delay before a TS1 diagnosis was rendered. Luckily, the index case’s TS1 was discovered before he suffered a life-threatening cardiac event. Nevertheless, this case illustrates the need for physicians to be vigilant when dealing with an overlapping partial phenotype, which can confound a diagnosis, so that other infants are not less fortunate.
Glossary
- ECG
electrocardiogram
- gDNA
genomic DNA
- TS1
type 1 Timothy syndrome
- WT
wild type
Footnotes
Mr Dufendach attended patient consultations, carried out experimental analysis, and drafted the initial manuscript; Mr Giudicessi analyzed clinical data, aided in experimental design, and reviewed and revised the manuscript; Ms Boczek aided in experimental design and critically reviewed the manuscript; and Dr Ackerman cared for the patient, aided in experimental design, and reviewed and revised the manuscript. All authors approved the final manuscript as submitted.
FINANCIAL DISCLOSURE: Dr Ackerman is a consultant for and collects royalties from Transgenomic. Intellectual property derived from Dr Ackerman’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals and now Transgenomic). Mr Dufendach, Mr Giudicessi, and Ms Boczek have indicated they have no financial relationships relevant to this article to disclose.
FUNDING: Supported by the Windland Smith Rice Sudden Comprehensive Sudden Cardiac Death Program (to Dr Ackerman). Mr Giudicessi is supported by a National Institutes of Health/National Heart, Lung, and Blood Institute National Research Service Award Ruth L. Kirschstein individual predoctoral MD/PhD fellowship (F30-HL106993). Funded by the National Institutes of Health (NIH).
References
- 1.Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31 [DOI] [PubMed] [Google Scholar]
- 2.Splawski I, Timothy KW, Priori SG, Napolitano C, Bloise R. Timothy syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. Gene Reviews [Internet]. Seattle, WA: University of Washington; 2011
- 3.Dao KD, Shin AY, Billings A, Oberg KC, Wood VE. Surgical treatment of congenital syndactyly of the hand. J Am Acad Orthop Surg. 2004;12(1):39–48 [DOI] [PubMed] [Google Scholar]
- 4.Sun G, Xu ZM, Liang JF, Li L, Tang DX. Twelve-year prevalence of common neonatal congenital malformations in Zhejiang Province, China. World J Pediatr. 2011;7(4):331–336 [DOI] [PubMed] [Google Scholar]
- 5.Merz RD, Forrester MB. Hawaii Birth Defects Program 1986–2003 Statewide Data (internet). http://hawaii.gov/health/family-child-health/genetics/newpdf/databook1986to2003.pdf (2004).
- 6.Forrester MB, Merz RD. Rates for specific birth defects among offspring of Japanese mothers, Hawaii, 1986-2002. Congenit Anom (Kyoto). 2006;46(2):76–80 [DOI] [PubMed] [Google Scholar]
- 7.Etheridge SP, Bowles NE, Arrington CB, et al. Somatic mosaicism contributes to phenotypic variation in Timothy syndrome. Am J Med Genet A. 2011;155A(10):2578–2583 [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Quevedo L, Blanco J, Sarrate Z, Català V, Bassas L, Vidal F. Hidden mosaicism in patients with Klinefelter’s syndrome: implications for genetic reproductive counselling. Hum Reprod. 2011;26(12):3486–3493 [DOI] [PubMed] [Google Scholar]