

Abstract
Background. Infection with hepatitis C virus (HCV) is a burgeoning worldwide public health problem, with 170 million infected individuals and an estimated 20 million deaths in the coming decades. While 6 main genotypes generally distinguish the global geographic diversity of HCV, a multitude of closely related subtypes within these genotypes are poorly defined and may influence clinical outcome and treatment options. Unfortunately, the paucity of genetic data from many of these subtypes makes time-consuming primer walking the limiting step for sequencing understudied subtypes.
Methods. Here we combined long-range polymerase chain reaction amplification with pyrosequencing for a rapid approach to generate the complete viral coding region of 31 samples representing poorly defined HCV subtypes.
Results. Phylogenetic classification based on full genome sequences validated previously identified HCV subtypes, identified a recombinant sequence, and identified a new distinct subtype of genotype 4. Unlike conventional sequencing methods, use of deep sequencing also facilitated characterization of minor drug resistance variants within these uncommon or, in some cases, previously uncharacterized HCV subtypes.
Conclusions. These data aid in the classification of uncommon HCV subtypes while also providing a high-resolution view of viral diversity within infected patients, which may be relevant to the development of therapeutic regimens to minimize drug resistance.
Keywords: Hepatitis C virus, pyrosequencing, subtype classification, drug resistance mutations, viral diversity
Hepatitis C virus (HCV) is an enveloped, positive-strand RNA virus belonging to the Hepacivirus genus in the Flaviviridae family. Seven confirmed genotypes (1–7) are generally distinguished by phylogenetic methods and pair-wise distance calculations [1, 2]. On the basis of full genome nucleotide sequences, HCV genotypes diverged from each other by a pair-wise distance of >30%. Individual genotypes can be further divided into more closely related subtypes that diverged by a pair-wise distance of 15%–30%. All viral genotypes retain their repertoire of colinear structural and nonstructural genes, thereby facilitating preliminary genotype classification on the basis of partial genome sequences of short fragments (approximately 300–400 nucleotides) in the structural core/E1 region and the nonstructural NS5B region [3]. However, full genome sequences remain indispensible for the detection of genome recombination events.
While only recombinant HCV genotype 2 k/1b viruses have been found to actively circulate in the population so far [4–7], recombinants of genotypes 2/5, 2b/1b, 2b/1a, and 2i/6p have been detected in single isolates from humans [8–11]. Furthermore, the frequency of HCV intergenotype and intragenotype recombination may be underestimated because of the lack of robust detection methods [12, 13]. Although genotyping based on core/E1 or NS5B sequences has resulted in the provisional classification of a large number of subtype variants within each genotype, at least 1 but preferably ≥2 full genome sequences are required to confirm subtype designation [2, 12].
Accurate genotype and subtype classification is clinically important because major genotypes differ considerably in their response rates to treatment with pegylated interferon and ribavirin and with directly acting antiviral drugs (DAAs) that are designed mainly against genotype 1 isolates. The limited efficacy of DAAs against other genotypes has been shown for genotype 3 [14], and treatment response rates for drug regimens containing boceprevir (an NS3 protease inhibitor) or BMS-790052 (an NS5A inhibitor) were found to vary even between subtypes 1a and 1b [15, 16]. Different baseline frequencies of drug resistance mutations may account for these differences [17], and such variation might become more significant if interferon-free regimens containing 1 or more DAAs could indeed displace the current standard of care [18]. Until clinically tolerable DAAs with high barriers to resistance against the complete spectrum of HCV genotypes become available [18, 19], accurate subtype determination and, possibly, drug resistance profiling of the individual's viral population may guide the optimal choice of drugs. Full genome deep sequencing, as performed in this study, allows the identification of drug resistance mutations across the genome that may exist as dominant or minor variants in treatment-naive patients, thereby informing the design of therapeutic regimens.
METHODS
Sample Collection
Plasma samples obtained from HCV-infected patients in Canada and the United Kingdom were received with no accompanying patient-associated demographic or clinical information. This study was approved by the Massachusetts General Hospital Review Board and was granted exemption by the Massachusetts Institute of Technology and Massachusetts General Hospital Review Boards as discarded diagnostic samples. Preliminary HCV genotypes were assigned by the clinical centers, and all samples were thought to represent poorly characterized variants of genotypes 2–4.
Polymerase Chain Reaction (PCR) Amplification, Viral Sample Preparation, and Genome Sequencing
RNA isolation, complementary DNA generation, and PCR amplification within the 5′ untranslated region (UTR) and 3 small sequence islands, followed by amplification of 2 hemi-genome fragments that covered the entire protein coding region, were performed as previously described [20], using primers described in Supplementary Table 1. The 5′ UTR and small sequence islands underwent conventional Sanger sequencing. Pooled hemi-genome PCR products were sequenced by Roche/454, and a de novo assembly was generated as previously described [21]. Any ambiguities in 454 assemblies were resolved using Sanger sequencing. All full genome sequences used in this study have been submitted to Genbank (accession numbers JX227949–JX227979).
Alignments
All unique, nonrecombinant, full-length HCV genomes found in the Los Alamos Hepatitis C Sequence Database (http://hcv.lanl.gov), along with sequences generated in this study, were used for analyses. For core/E1-NS5B analyses, the Los Alamos National Laboratory (LANL) HCV database was searched for all isolates with available sequences for both core/E1 and NS5B (nucleotide positions 869–1292 and 8276–8615, according to genotype 1a reference strain H77). The intersection of isolates containing both of these sequences was examined on a genotype and subtype level.
Multiple alignments were performed in MUSCLE v3.8.31 [22], using default parameters with 5 iterative refinements. A total of 193 full-length genomes (excluding recombinants) were trimmed to the coding sequence producing a master alignment of 9323 bases (including gaps). Any gaps in the triplet of nucleotides underlying codons were removed from the entire alignment in a greedy fashion, thereby conservatively eliminating any alignment artifacts. For the genotyping fragment trees that included 300 viral strains for which both regions of core/E1 (869–1292) and NS5B (8276–8615) were available, these regions were concatenated into an alignment of 779 nucleotides (including gaps). No gap stripping was performed on the genotyping fragment sequences.
Identification of Recombinants
The recombination detection program (RDP) [23] was run on all full-length genome sequences representing all available genotypes and subtypes. Any sequences that produced consistently low P values among the RDP's multiple tests for recombination were subject to further analysis. These potential recombinant sequences were further validated with SimPlot v3.5.1 [24], in which all possible combinations of major and minor parent strains were run against the recombinant strain.
Construction of Phylogenetic Trees
The randomized axelerated maximum likelihood program [25] was run with 1000 bootstraps to construct a single tree for the 193 genomes mentioned above, using the cleaned alignment containing 3753 bases. The smaller genotyping trees of 300 concatenated fragments spanning 779 nucleotides were used in the same manner. The GTRGAMMA model was chosen as the model of nucleotide substitution. The single best tree from the 1000 bootstraps is shown, along with the total percentage (0%–100%) of bootstrap support for each branch.
Computation of Pair-Wise Intersubtype and Intrasubtype Divergence
Pair-wise distances for all 193 full-length nonrecombinant genomes were calculated in MEGA v.5.05 [26].
The PhyloPlace tool was used to compute the branching index over the length of our 2 putative subtype 4v genomes (http://hcv.lanl.gov/content/sequence/phyloplace/PhyloPlace.html).
Variant Calling in Mixed Populations
Prior to variant calling, correction of known error modes in Roche/454 read data was performed with the RC454 program [21]. Reliable and sensitive detection of variants in a mixed population and analysis of intrahost diversity was then accomplished using the VPhaser tool [27].
RESULTS
Phylogenetic Classification of HCV Genotypes Based on Full-Genome Sequences
To better characterize global HCV genotype diversity, sequences representing the entire HCV protein-coding region and portions of the 5′ and 3′ UTR were generated from plasma samples acquired from 54 patients in Canada or the United Kingdom. Preliminary HCV genotyping based on core/E1 or NS5B sequences indicated that these individuals were infected with poorly characterized subtypes of genotypes 2, 3, or 4. Use of a hemi-genome amplification approach [20], coupled with high-throughput pyrosequencing technology, enabled generation of sequence reads covering the entire coding region without the need for traditional primer-walking methods. The success rate for amplification of both hemi-genomes from the 54 samples attempted was 83%, with 31 samples being used for sequencing. A recently developed algorithm allowed de novo assembly of all 31 genomes, despite the lack of full-length reference sequences for these poorly characterized genotypes [21]. Genome sequences were aligned to all available full-length sequences from the National Center for Biotechnology Information and LANL public databases, including select representatives of the commonly found 1a or 1b subtypes.
Phylogenetic analysis revealed that several of the new sequences clustered with subtypes for which only 1 full-length genome sequence had been reported (2c, 2k, 3i, 4g, 4l, 4m, 4n, 4o, and 4r), thereby strengthening these phylogenetic groupings (Figure 1), with pair-wise distance measurements confirming their distinction from other subtypes (Table 1). Sequences provisionally assigned to the 2 m and 3 g subtypes on the basis of partial sequence grouped accordingly, formally establishing these as distinct HCV subtypes (Figures 1 and 2). Likewise, quantification of relatedness to all known subtypes by branching index analysis (Supplementary Figure 1), coupled with 100% bootstrap support by phylogeny (Figure 1) and pair-wise distance measurements (Table 1), clearly grouped 2 newly described genotype 4 sequences, G1248/PB66742 and G1249/PB65568, to near full-length genomes from the recently identified 4v provisional subtype [28], thereby confirming 4v as a distinct subtype of genotype 4.
Figure 1.
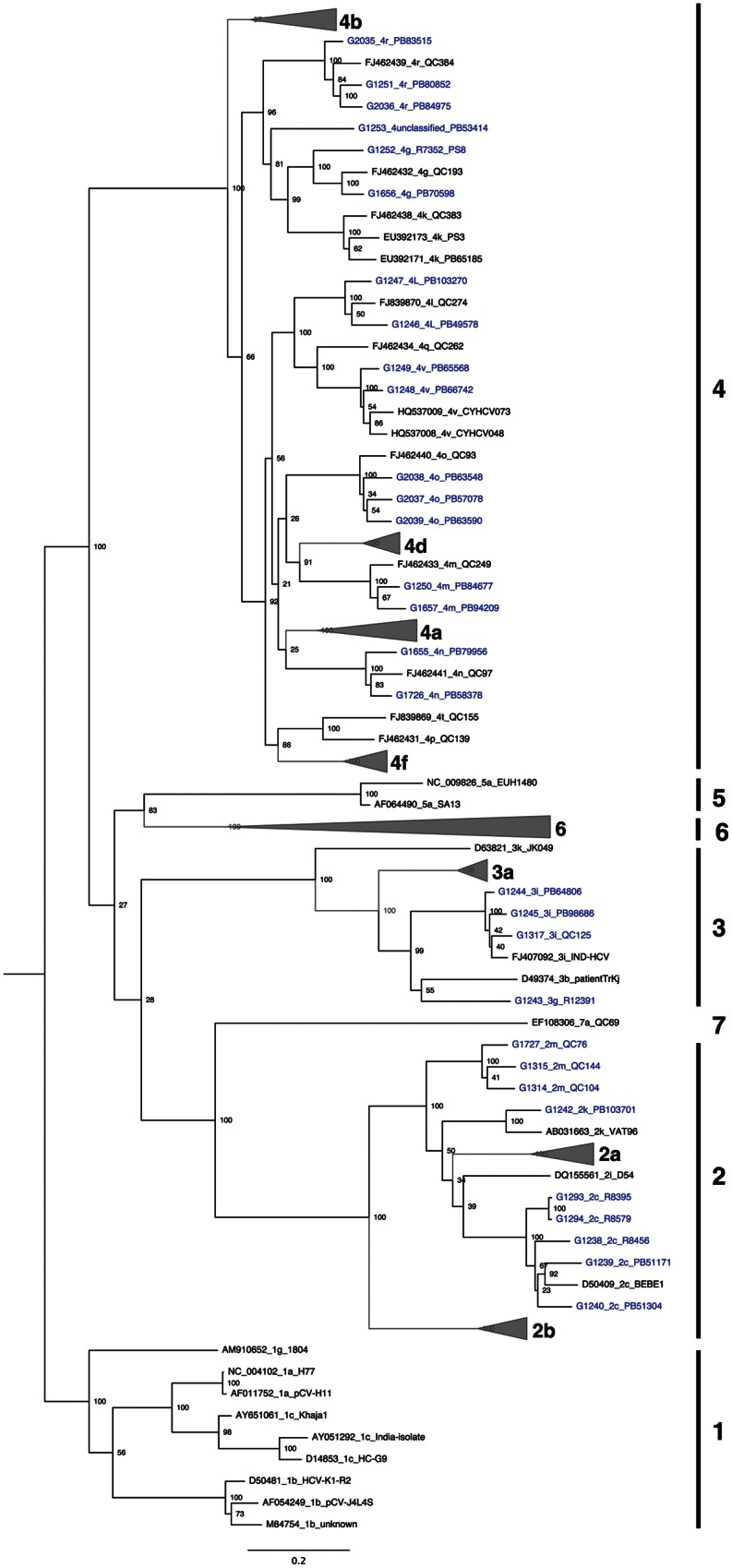
Maximum likelihood tree of 30 complete, nonrecombinant hepatitis C virus coding-region nucleotide sequences generated as part of this study (highlighted in blue), and 163 sequences from public sequence repositories. Bootstrap resampling (1000 replications) support values are shown at nodes. The tree is rooted using genotype 1 sequences, and all horizontal branch lengths are drawn to a scale of nucleotide substitutions per site.
Table 1.
Average Internucleotide and Intranucleotide Divergence Among Hepatitis C Virus Subtypes
| Genotype 1 | 1a (2) | 1b (3) | 1c (3) | 1g (1) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a (2) | 0.73 | 21.41 | 19.23 | 21.51 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 1b (3) | 21.41 | 8.36 | 22.26 | 22.02 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 1c (3) | 19.23 | 22.26 | 8.16 | 22.31 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 1g (1) | 21.51 | 22.02 | 22.31 | — | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| Genotype 2 | 2a (18) | 2b (14) | 2c (6) | 2i (1) | 2k (2) | 2m (3) | |||||||||||||||||
| 2a (18) | 7.96 | 23.08 | 19.75 | 19.37 | 20.37 | 19.44 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 2b (14) | 23.08 | 6.63 | 22.56 | 22.93 | 23.44 | 22.27 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 2c (6) | 19.75 | 22.56 | 9.39 | 18.86 | 19.82 | 19.12 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 2i (1) | 19.37 | 22.93 | 18.86 | — | 19.89 | 19.53 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 2k (2) | 20.37 | 23.44 | 19.82 | 19.89 | 9.67 | 19.04 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 2m (3) | 19.44 | 22.27 | 19.12 | 19.53 | 19.04 | 7.83 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| Genotype 3 | 3a (7) | 3b (1) | 3g (1) | 3i (4) | 3k (1) | ||||||||||||||||||
| 3a (7) | 6.20 | 21.75 | 20.78 | 21.04 | 24.78 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 3b (1) | 21.75 | — | 20.29 | 19.67 | 25.36 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 3g (1) | 20.78 | 20.29 | — | 19.41 | 25.11 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 3i (4) | 21.04 | 19.67 | 19.41 | 5.55 | 24.33 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 3k (1) | 24.78 | 25.36 | 25.11 | 24.33 | — | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| Genotype 4 | 4a (16) | 4b (4) | 4c (1) | 4d (4) | 4f (6) | 4g (3) | 4k (3) | 4l (3) | 4m (3) | 4n (3) | 4o (4) | 4p (1) | 4q (1) | 4r (4) | 4t (1) | 4v (4) | 4∼ (1) | ||||||
| 4a (16) | 7.45 | 20.61 | 14.61 | 17.99 | 18.54 | 20.00 | 19.99 | 18.01 | 18.72 | 18.67 | 18.43 | 18.78 | 17.87 | 20.39 | 19.32 | 18.19 | 20.26 | … | … | … | … | … | … |
| 4b (4) | 20.61 | 15.24 | 20.73 | 21.07 | 20.76 | 20.16 | 20.19 | 20.61 | 21.13 | 20.94 | 21.00 | 20.74 | 20.09 | 20.49 | 21.27 | 20.44 | 20.02 | … | … | … | … | … | … |
| 4c (1) | 14.61 | 20.73 | — | 18.05 | 18.65 | 20.19 | 20.11 | 17.79 | 18.27 | 18.31 | 18.54 | 18.35 | 17.53 | 20.78 | 18.79 | 18.04 | 19.79 | … | … | … | … | … | … |
| 4d (4) | 17.99 | 21.07 | 18.05 | 6.52 | 19.20 | 20.91 | 20.43 | 18.27 | 18.66 | 19.25 | 18.76 | 19.11 | 18.02 | 21.06 | 19.78 | 18.60 | 20.01 | … | … | … | … | … | … |
| 4f (6) | 18.54 | 20.76 | 18.65 | 19.20 | 8.16 | 20.31 | 20.40 | 19.00 | 19.38 | 19.39 | 19.30 | 18.93 | 18.56 | 20.55 | 19.39 | 19.15 | 20.07 | … | … | … | … | … | … |
| 4g (3) | 20.00 | 20.16 | 20.19 | 20.91 | 20.31 | 11.18 | 16.83 | 20.63 | 20.60 | 20.26 | 20.10 | 20.62 | 19.88 | 17.80 | 20.82 | 20.02 | 17.70 | … | … | … | … | … | … |
| 4k (3) | 19.99 | 20.19 | 20.11 | 20.43 | 20.40 | 16.83 | 8.19 | 20.56 | 20.70 | 20.28 | 20.55 | 20.78 | 20.19 | 18.88 | 20.95 | 20.24 | 18.25 | … | … | … | … | … | … |
| 4l (3) | 18.01 | 20.61 | 17.79 | 18.27 | 19.00 | 20.63 | 20.56 | 7.85 | 18.69 | 18.46 | 18.76 | 18.55 | 15.83 | 20.72 | 19.03 | 16.50 | 19.81 | … | … | … | … | … | … |
| 4m (3) | 18.72 | 21.13 | 18.27 | 18.66 | 19.38 | 20.60 | 20.70 | 18.69 | 7.79 | 18.91 | 19.11 | 18.76 | 18.22 | 20.61 | 19.37 | 18.57 | 20.41 | … | … | … | … | … | … |
| 4n (3) | 18.67 | 20.94 | 18.31 | 19.25 | 19.39 | 20.26 | 20.28 | 18.46 | 18.91 | 8.28 | 19.13 | 19.34 | 17.28 | 20.58 | 19.55 | 18.18 | 20.19 | … | … | … | … | … | … |
| 4o (4) | 18.43 | 21.00 | 18.54 | 18.76 | 19.30 | 20.10 | 20.55 | 18.76 | 19.11 | 19.13 | 8.02 | 19.27 | 18.16 | 20.42 | 19.76 | 18.67 | 20.02 | … | … | … | … | … | … |
| 4p (1) | 18.78 | 20.74 | 18.35 | 19.11 | 18.93 | 20.62 | 20.78 | 18.55 | 18.76 | 19.34 | 19.27 | — | 18.41 | 20.52 | 14.72 | 18.55 | 20.52 | … | … | … | … | … | … |
| 4q (1) | 17.87 | 20.09 | 17.53 | 18.02 | 18.56 | 19.88 | 20.19 | 15.83 | 18.22 | 17.28 | 18.16 | 18.41 | — | 20.52 | 18.37 | 14.30 | 19.62 | … | … | … | … | … | … |
| 4r (4) | 20.39 | 20.49 | 20.78 | 21.06 | 20.55 | 17.80 | 18.88 | 20.72 | 20.61 | 20.58 | 20.42 | 20.52 | 20.52 | 7.87 | 21.20 | 21.07 | 18.28 | … | … | … | … | … | … |
| 4t (1) | 19.32 | 21.27 | 18.79 | 19.78 | 19.39 | 20.82 | 20.95 | 19.03 | 19.37 | 19.55 | 19.76 | 14.72 | 18.37 | 21.20 | — | 19.04 | 21.12 | … | … | … | … | … | … |
| 4v (4) | 18.19 | 20.44 | 18.04 | 18.60 | 19.15 | 20.02 | 20.24 | 16.50 | 18.57 | 18.18 | 18.67 | 18.55 | 14.30 | 21.07 | 19.04 | 6.67 | 20.21 | … | … | … | … | … | … |
| 4∼ (1) | 20.26 | 20.02 | 19.79 | 20.01 | 20.07 | 17.70 | 18.25 | 19.81 | 20.41 | 20.19 | 20.02 | 20.52 | 19.62 | 18.28 | 21.12 | 20.21 | — | … | … | … | … | … | … |
| Genotype 5 | 5a (2) | ||||||||||||||||||||||
| 5a (2) | 9.43 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| Genotype 6 | 6a (16) | 6b (1) | 6c (1) | 6d (1) | 6e (4) | 6f (3) | 6g (2) | 6h (1) | 6i (3) | 6j (2) | 6k (4) | 6l (2) | 6m (4) | 6n (4) | 6o (2) | 6p (1) | 6q (1) | 6r (1) | 6s (1) | 6t (4) | 6u (1) | 6v (1) | 6w (1) |
| 6a (16) | 5.32 | 19.92 | 26.53 | 27.07 | 27.14 | 27.02 | 27.09 | 26.99 | 26.85 | 27.04 | 26.76 | 26.18 | 26.68 | 26.98 | 26.09 | 26.14 | 25.60 | 26.87 | 27.14 | 27.09 | 26.36 | 26.77 | 27.40 |
| 6b (1) | 19.92 | — | 26.54 | 26.85 | 26.98 | 26.96 | 26.85 | 27.03 | 27.40 | 26.93 | 26.89 | 26.71 | 26.56 | 26.83 | 26.41 | 26.22 | 25.90 | 26.63 | 27.10 | 26.84 | 26.64 | 26.80 | 27.31 |
| 6c (1) | 26.53 | 26.54 | — | 20.89 | 22.45 | 22.40 | 25.02 | 26.47 | 25.70 | 26.26 | 25.09 | 25.38 | 25.42 | 25.48 | 21.68 | 21.46 | 21.19 | 22.39 | 25.40 | 22.32 | 25.38 | 26.30 | 26.07 |
| 6d (1) | 27.07 | 26.85 | 20.89 | — | 22.59 | 22.79 | 25.43 | 26.47 | 26.32 | 26.31 | 26.11 | 25.78 | 25.61 | 25.50 | 22.39 | 22.32 | 21.75 | 22.36 | 24.83 | 22.42 | 25.16 | 26.64 | 26.54 |
| 6e (4) | 27.14 | 26.98 | 22.45 | 22.59 | 14.49 | 22.99 | 25.46 | 26.46 | 26.24 | 26.24 | 26.43 | 25.96 | 26.27 | 25.88 | 21.84 | 21.80 | 21.17 | 23.54 | 25.21 | 22.19 | 25.53 | 27.24 | 26.01 |
| 6f (3) | 27.02 | 26.96 | 22.40 | 22.79 | 22.99 | 4.14 | 25.15 | 26.50 | 26.44 | 26.09 | 25.91 | 25.61 | 25.72 | 25.75 | 22.43 | 22.86 | 21.64 | 17.26 | 25.25 | 22.61 | 25.59 | 26.36 | 25.93 |
| 6g (2) | 27.09 | 26.85 | 25.02 | 25.43 | 25.46 | 25.15 | 6.70 | 26.74 | 26.98 | 26.77 | 26.71 | 25.98 | 26.58 | 26.33 | 25.68 | 25.57 | 24.52 | 25.76 | 26.25 | 25.48 | 26.74 | 27.31 | 24.69 |
| 6h (1) | 26.99 | 27.03 | 26.47 | 26.47 | 26.46 | 26.50 | 26.74 | — | 20.99 | 20.47 | 23.00 | 22.59 | 22.63 | 22.38 | 26.08 | 26.12 | 25.07 | 26.59 | 26.96 | 26.48 | 26.55 | 26.52 | 27.29 |
| 6i (3) | 26.85 | 27.40 | 25.70 | 26.32 | 26.24 | 26.44 | 26.98 | 20.99 | 5.13 | 17.04 | 23.50 | 22.63 | 22.51 | 22.68 | 25.76 | 25.25 | 24.60 | 26.39 | 26.67 | 25.97 | 26.40 | 26.29 | 27.23 |
| 6j (2) | 27.04 | 26.93 | 26.26 | 26.31 | 26.24 | 26.09 | 26.77 | 20.47 | 17.04 | 3.26 | 23.25 | 22.44 | 22.79 | 22.58 | 26.30 | 26.05 | 24.75 | 26.46 | 26.70 | 26.14 | 25.89 | 26.45 | 27.00 |
| 6k (4) | 26.76 | 26.89 | 25.09 | 26.11 | 26.43 | 25.91 | 26.71 | 23.00 | 23.50 | 23.25 | 10.47 | 19.00 | 21.42 | 21.54 | 26.13 | 25.60 | 24.95 | 26.38 | 26.35 | 26.19 | 25.66 | 26.30 | 26.55 |
| 6l (2) | 26.18 | 26.71 | 25.38 | 25.78 | 25.96 | 25.61 | 25.98 | 22.59 | 22.63 | 22.44 | 19.00 | 4.94 | 21.26 | 20.93 | 25.74 | 25.33 | 24.26 | 25.78 | 25.96 | 25.53 | 25.46 | 25.74 | 26.72 |
| 6m (4) | 26.68 | 26.56 | 25.42 | 25.61 | 26.27 | 25.72 | 26.58 | 22.63 | 22.51 | 22.79 | 21.42 | 21.26 | 3.30 | 18.82 | 25.30 | 25.16 | 24.51 | 25.76 | 26.27 | 25.57 | 25.43 | 25.74 | 26.91 |
| 6n (4) | 26.98 | 26.83 | 25.48 | 25.50 | 25.88 | 25.75 | 26.33 | 22.38 | 22.68 | 22.58 | 21.54 | 20.93 | 18.82 | 5.03 | 25.08 | 24.86 | 23.93 | 25.47 | 26.27 | 25.61 | 25.47 | 26.18 | 26.85 |
| 6o (2) | 26.09 | 26.41 | 21.68 | 22.39 | 21.84 | 22.43 | 25.68 | 26.08 | 25.76 | 26.30 | 26.13 | 25.74 | 25.30 | 25.08 | 7.52 | 19.54 | 20.81 | 22.81 | 24.70 | 21.27 | 25.30 | 25.87 | 25.71 |
| 6p (1) | 26.14 | 26.22 | 21.46 | 22.32 | 21.80 | 22.86 | 25.57 | 26.12 | 25.25 | 26.05 | 25.60 | 25.33 | 25.16 | 24.86 | 19.54 | — | 20.94 | 22.70 | 25.09 | 21.39 | 25.56 | 26.47 | 26.00 |
| 6q (1) | 25.60 | 25.90 | 21.19 | 21.75 | 21.17 | 21.64 | 24.52 | 25.07 | 24.60 | 24.75 | 24.95 | 24.26 | 24.51 | 23.93 | 20.81 | 20.94 | — | 21.53 | 23.84 | 20.32 | 25.25 | 24.94 | 24.59 |
| 6r (1) | 26.87 | 26.63 | 22.39 | 22.36 | 23.54 | 17.26 | 25.76 | 26.59 | 26.39 | 26.46 | 26.38 | 25.78 | 25.76 | 25.47 | 22.81 | 22.70 | 21.53 | — | 24.76 | 22.79 | 25.86 | 26.57 | 25.97 |
| 6s (1) | 27.14 | 27.10 | 25.40 | 24.83 | 25.21 | 25.25 | 26.25 | 26.96 | 26.67 | 26.70 | 26.35 | 25.96 | 26.27 | 26.27 | 24.70 | 25.09 | 23.84 | 24.76 | — | 25.01 | 25.88 | 27.02 | 26.39 |
| 6t (4) | 27.09 | 26.84 | 22.32 | 22.42 | 22.19 | 22.61 | 25.48 | 26.48 | 25.97 | 26.14 | 26.19 | 25.53 | 25.57 | 25.61 | 21.27 | 21.39 | 20.32 | 22.79 | 25.01 | 7.51 | 25.71 | 26.02 | 25.75 |
| 6u (1) | 26.36 | 26.64 | 25.38 | 25.16 | 25.53 | 25.59 | 26.74 | 26.55 | 26.40 | 25.89 | 25.66 | 25.46 | 25.43 | 25.47 | 25.30 | 25.56 | 25.25 | 25.86 | 25.88 | 25.71 | — | 24.58 | 27.14 |
| 6v (1) | 26.77 | 26.80 | 26.30 | 26.64 | 27.24 | 26.36 | 27.31 | 26.52 | 26.29 | 26.45 | 26.30 | 25.74 | 25.74 | 26.18 | 25.87 | 26.47 | 24.94 | 26.57 | 27.02 | 26.02 | 24.58 | — | 27.20 |
| 6w (1) | 27.40 | 27.31 | 26.07 | 26.54 | 26.01 | 25.93 | 24.69 | 27.29 | 27.23 | 27.00 | 26.55 | 26.72 | 26.91 | 26.85 | 25.71 | 26.00 | 24.59 | 25.97 | 26.39 | 25.75 | 27.14 | 27.20 | — |
Data are no. of base differences per sequence (divided by total no. of positions [ie, 9314] in the master alignment) from between sequences. The analysis involved 193 nucleotide sequences. All ambiguous positions were removed for each sequence pair. Numbers in parentheses next to subtype names indicate no. of genomes of each subtype included in pair-wise analysis. Bold text shows distances within a given subtype. Subtypes with <2 sequences for comparison are denoted by —.
Figure 2.

Maximum likelihood tree based on concatenated core/E1 (869–1292 on H77 reference) and NS5B (8276–8615 on H77 reference) nucleotide sequences. Hepatitis C virus sequences are designated by accession number or sequence ID, followed by subtype designation and unique name. Sequences for the 30 nonrecombinant isolates described in this study are colored in blue. Bootstrap resampling (1000 replications) support values are shown at nodes. The tree is rooted using genotype 1 sequences, and all horizontal branch lengths are drawn to a scale of nucleotide substitutions per site.
In addition to validating previously identified HCV genetic subtypes, phylogenetic analysis also identified a novel genotype 4 variant. While isolate G1253/PB53414 grouped with genotype 4 sequences in the full-genome phylogenetic tree (Figure 1), bootstrap resampling strongly supported G1253/PB53414 as a unique subtype as compared to genotypes 4g and 4k (Figure 1). Similarly, G1253/PB53414 was genetically distinct from subtypes 4h, 4g, and 4k in the core/E1-NS5B tree (Figure 2). In addition, nucleotide sequence divergence of G1253/PB53414 from all other genotype 4 full genome sequences was >17.7% (Table 1). These lines of evidence strongly suggest that G1253/PB53414 represents a novel subtype of a genotype 4. This new genotype 4 variant could be designated as subtype 4w, but since only 1 example of this subtype has been sequenced, accepted convention for assigning provisional subtypes calls for designation of G1253/PB53414 as an unclassified genotype 4 variant.
One recombinant sequence was also identified. SimPlot analysis showed G1241/PB41880 to be a recombinant between subtypes 2k (K1-S2/AB031663) and 1b (Vat-96/D50485), sharing a recombination break point with 2k/1b recombinants previously identified in St. Petersburg, CRF_01_1b2k (Figure 3). Despite high sequence identity (93%–94%), G1241/PB41880 is not identical to previously reported 2k/1b recombinants. Nevertheless, separate phylogenetic analysis of 2k and 1b components of the new strain identified here along with other nonrecombinant 2k and 1b sequences confirm that there is 100% bootstrap support for G1241/PB41880 grouping with all other reported 2k/1b strains (Figure 4).
Figure 3.

Recombination similarity plot of the 2k/1b recombinant genome. Colored lines indicate the percentage identity (y-axis) of G1253 to each of 9 full-length nonrecombinant genomes across the entire genome (x-axis).
Figure 4.

Phylogenetic analysis of genotype 2k and genotype 1b sequences composing the G1241/PB41880 2k/1b recombinant. Hepatitis C virus sequences are designated by accession number, followed by subtype. A, Maximum likelihood tree of 2k nucleotide sequences corresponding to nucleotides 1–3291 of the G1241/PB41880 open reading frame (ORF). B, Maximum likelihood tree of 1b nucleotide sequences corresponding to nucleotides 3292–8926 of the G1241/PB41880 ORF. Genomes corresponding to other confirmed 2k/1b recombinants are colored in blue, while G1241/PB41880 is colored in red. Bars, 0.2 nucleotide substitutions per site. Black text denotes sequences from nonrecombinant genomes.
In the case of subtypes 2 m and 3 g, for which no full genome sequences were available in public databases, classification of these new sequences on the basis of genotyping regions alone was clearly supported (Figure 2). As expected, in instances where both full genome and partial genome data were available, bootstrap support for subtype classification was stronger in full-genome trees as compared to partial genome trees (Figures 1 and 2).
Prevalence of Dominant, Naturally Occurring Polymorphisms at Positions Associated with Drug Resistance
Alignments of these full genome sequences allowed us to investigate the prevalence of dominant amino acid substitutions among rare HCV subtypes and identify those known to be associated with resistance to NS3 protease and NS5B polymerase inhibitors (Table 2). In NS3, the V36L substitution that confers resistance to telaprevir [29] was found to be dominant in 100% of genotype 2–5 sequences analyzed, the single genotype 7 sequence analyzed, and 20% of genotype 6 sequences analyzed, while the A39V substitution responsible for resistance to ACH-806 [30], was dominant in 100% of genotype 2 sequences analyzed. Moreover, all genotype 3 sequences and the 1 available genotype 7 sequence analyzed contained a D to Q substitution at amino acid 168, a site at which other mutations conferring up to 10 000-fold resistance to TMC-435 have been described in HCV genotype 1 [31]. Additionally, a D168E substitution known to confer 40-fold resistance to TMC-435 was found in 1 of 2 genotype 5 sequences and in 2 of 62 genotype 6 sequences [31]. In rare instances, we also identified genomes with dominant drug resistance mutations at positions C16, T54, A156, or V170 (Table 2).
Table 2.
Dominant Mutations Associated With Resistance to NS3 Protease and NS5B Polymerase Inhibitors in Complete Genome Sequences of Hepatitis C Virus Genotypes 1–7
| Protein | Amino Acid Position | Dominant Amino Acid | Mutant Amino Acid | Drug | Substitution (% of Genomes), by Genotype |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype 1 (n = 9) | Genotype 2 (n = 43) | Genotype 3 (n = 14) | Genotype 4 (n = 62) | Genotype 5 (n = 2) | Genotype 6 (n = 61) | Genotype 7 (n = 1) | |||||
| NS3 | 16 | C | S | ACH806 | ▪ | A (77)/S (2)/T (21) | T (100) | A (100) | T (100) | T (100) | |
| NS3 | 36 | V | AMLG | Telaprevir, boceprevir, SCH900518 | ▪ | L (100) | L (100) | L (100) | L (100) | V (75)/L (20)/ I (5) | L (100) |
| NS3 | 39 | A | V | ACH806 | ▪ | V (98)/I (2) | A (65)/S (14)/T (21) | A (92)/T (8) | ▪ | A (97)/T (1.5)/ D (1.5) | S (100) |
| NS3 | 41 | Q | R | Boceprevir, ITMN-191 | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| NS3 | 43 | F | SC | ITMN-191, telaprevir, boceprevir | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| NS3 | 54 | T | AS | Telaprevir, boceprevir, SCH900518 | T (89)/S (11) | ▪ | ▪ | T (97)/S (3) | ▪ | ▪ | G (100) |
| NS3 | 109 | R | K | SCH6 | ▪ | ▪ | ▪ | R (100) | ▪ | ▪ | ▪ |
| NS3 | 138 | S | T | ITMN-191 | ▪ | ▪ | ▪ | S (98)/F (2) | ▪ | S (98.5)/F (1.5) | ▪ |
| NS3 | 155 | R | K,T,I,M, G,L,S,Q | Telaprevir, boceprevir, TMC 435, R7227, MK 7009, BI 201335, SCH900518, BILN-2061 | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| NS3 | 156 | A | STVI | Telaprevir, boceprevir, R7227, SCH900518, BILN-2061 | ▪ | ▪ | ▪ | ▪ | ▪ | A (98.5)/V (1.5) | ▪ |
| NS3 | 168 | D | QAVET | TMC 435, R7227, MK 7009, BI201335, BILN-2061 | ▪ | ▪ | Q (100) | ▪ | D (50)/E (50) | D (97)/E (3) | Q (100) |
| NS3 | 170 | V | AT | Telaprevir, boceprevir | V (56)/I (44) | I (95)/V (2.5) /unresolved (2.5) | I (71)/V (29) | V (97)/I (3) | I (50)/V (50) | V (65.5)/A (1.5)/I (33) | ▪ |
| NS3 | 489 | S | L | ITMN-191 | S (67)/V (33) | V (100) | V (100) | S (89)/A (9)/V (2) | V (100) | V (85)/I (15) | ▪ |
| NS5B | 95 | H | Q | A-782759 | ▪ | ▪ | ▪ | ▪ | ▪ | H (97)/C (1.5)/R (1.5) | ▪ |
| NS5B | 96 | S | T | R1479 | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| NS5B | 282 | S | T | 2'C-methyl- ribonucleosides | ▪ | ▪ | ▪ | S (98)/T (2) | ▪ | ▪ | ▪ |
| NS5B | 316 | C | Y | HCV-796, A-837093 | C (89)/N (11) | C (98)/W (2) | ▪ | C (87)/N (8)/H (5) | ▪ | ▪ | ▪ |
| NS5B | 365 | S | TA | HCV-796 | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ |
| NS5B | 411 | N | S | A-782759 | ▪ | ▪ | N (93)/S (7) | ▪ | ▪ | ▪ | ▪ |
| NS5B | 414 | M | LT | A-782759 | ▪ | Q (98)a | ▪ | L (53)/V (32)/I (13)/Q (2) | ▪ | M (98.5)/A (1.5) | ▪ |
| NS5B | 423 | M | TVI | AG-021541 | ▪ | ▪a | ▪ | ▪ | I (100) | ▪ | ▪ |
| NS5B | 448 | Y | HC | A-782759, A-837093 | ▪ | ▪a | ▪ | ▪ | ▪ | ▪ | ▪ |
| NS5B | 495 | P | LA | JTK-109 | ▪ | ▪a,b | ▪c | ▪e | ▪ | P (98.5)/L (1.5) | ▪ |
| NS5B | 554 | G | DS | A-837093 | ▪ | G (74)/S (21)a,b | ▪c,d | ▪e,f | ▪ | ▪ | C (100) |
| NS5B | 559 | D | GSN | A-837093 | ▪ | D (93)/H (2)a,b | ▪c,d | ▪e,f | ▪ | ▪ | ▪ |
Filled squares indicate that the dominant residue found in genotype 1 is found in 100% of the genomes analyzed for any given genotype. Substitutions known to confer drug resistance are in bold, and substitutions of unknown impact are in italics.
Abbreviation: AA, amino acid.
a No data at this position for G1727_2m_QC76.
b No data at this position for DQ430815_2b_TN9-0FL.
c No data at this position for AY956467_3a_32E and DQ430819_3a_TN78-0.
d No data at this position for G1245_3i_PB98686.
e No data at this position for DQ988079_4a_Eg12 and DQ988073_4a_Eg2.
f No data at this position for EF589160_4f_IFBT84, DQ988074_4a_Eg3, DQ988075_4a_Eg4, DQ988076_4a_Eg7, DQ988077_4a_Eg9, DQ988078_4a_Eg10, and G1726_4n_PB58378.
In NS5B, the M414L substitution conferring resistance to the polymerase inhibitor A-782759 was found in 53% of genotype 4 sequences spanning multiple subtypes. The S282T mutation associated with resistance to the nucleoside analog mericitabine and the nucleotide analog PSI-7977 [37] was found to be dominant in only one of the 191 sequences analyzed, a previously described genotype 4a isolate (ED43) from Egypt [32]. A small number of genomes harboring dominant drug resistance mutations at positions N411, M423, P495, and G554 were also identified (Table 2).
Combinations of dominant resistance mutations in NS3 and/or NS5B were found in genotype 2 (NS3:V36L and A39V), genotype 3 (NS3:V36L, and D168Q), and genotype 4 (NS3:V36L and M414L) sequences, suggesting possible resistance to multiple antiviral drugs (telaprevir/ACH806, telaprevir/TCM-435, and telaprevir/A-782759, respectively). Further examination of the highly polymorphic residue 414 of NS5B in genotype 4 sequences revealed a subtype-specific pattern whereby all subtype 4g, 4k-4r, and 4v genomes and 3 of 4 subtype b sequences had the M414L mutation reported to confer resistance to A-782759, while the remaining subtypes (a, c, d, f, and t) harbored multiple variants of unknown phenotype (M414I, M414Q, or M414V; Table 2 and Supplementary Table 2).
In addition to known resistance changes, other dominant substitutions of unknown resistance status were also common at key sites in NS3 and NS5B (Table 2). The dominant residue at amino acid 16 of NS3 was cysteine, in genotype 1, but was alanine, in genotypes 2 (74.4%) and 5 (100%), and threonine, in genotypes 4 and 6 (both 100%). The dominant NS3 V170 residue found in genotype 1 was an isoleucine, in 95% of genotype 2 and 71% of genotype 3 sequences. Isoleucine was also found at this position in genotypes 4–6, although at lower frequency (3%–50%). All genotype 2 and 3 sequences had a valine at residue 489 in NS3, rather than the serine found in genotype 1. Finally, the M414 in NS5B was replaced with glutamine, in 100% of genotype 2 sequences, and with isoleucine, in 32% of genotype 4 sequences.
Analysis of Viral Populations for Minor Variants at Drug Resistant Sites
The average depth of coverage generated by pyrosequencing across all 31 samples ranged from 2 to 3200 across the complete HCV genome and from 22 to 3041 at the drug resistance sites analyzed in NS3 and NS5B. This led to the identification of minor variants at drug resistance sites in NS3 in 7 of 31 samples. Minor variants ranged in frequency, from 0.10% to 7.78% (Table 3). Only the dominant amino acid was detected in the remaining 24 sequences, despite 500–900-fold sequence coverage. Even within genomes with extremely high sequence coverage, no minor variants were detected at known drug resistance sites in NS5B. It is possible that sequencing genomes to higher coverage could result in identification of additional low-frequency mutations at these drug resistance sites. Furthermore, we cannot exclude the possibility that some mutations were not detected because of primer selection bias.
Table 3.
Minor Variants at Amino Acids (AAs) Associated With Resistance to NS3 Protease Inhibitors in the Viral Population of Patients Infected With Hepatitis C Virus
| Position | Dominant AA in Genotype 1 | Mutant AA in Genotype 1 | Drug | Sample | Genotype | Dominant AA (Frequency) | Minor AA (Frequency) | Fold-Sequence Coverage |
|---|---|---|---|---|---|---|---|---|
| 16 | C | S | ACH806 | G2035/PB83515 | 4r | T (98.86) | A (1.14) | 2458 |
| 36 | V | AMLG | Telaprevir, boceprevir, SCH900518 | G2037/PB57078 | 4o | L (99.51) | P (0.2)/R (0.10) | 3030 |
| 39 | A | V | ACH806 | G2036/PB84975 | 4r | A (99.39) | V (0.61) | 2300 |
| 43 | F | SC | ITMN-191, telaprevir, boceprevir | G2038/PB63548 | 4o | F (98.21) | L (1.79) | 2345 |
| F | SC | ITMN-191, telaprevir, boceprevir | G2039/PB63590 | 4o | F (98.77) | L (1.23) | 2690 | |
| F | SC | ITMN-191, telaprevir, boceprevir | G2037/PB57078 | 4o | F (99.34) | L (0.66) | 3041 | |
| 54 | T | AS | Telaprevir, boceprevir, SCH900518 | G2036/PB84975 | 4r | T (99.55) | P (0.45)/A (0.11) | 2537 |
| 109 | R | K | SCH6 | G2036/PB84975 | 4r | R (99.89) | Q (0.11) | 2717 |
| 155 | R | K,T,I,M,G,L,S,Q | Telaprevir, boceprevir, TMC 435, R7227, MK 7009, BI 201335, SCH900518, BILN-2061 | G2036/PB84975 | 4r | R (99.89) | W (0.11) | 2846 |
| 156 | A | STVI | Telaprevir, boceprevir, R7227, SCH900518, BILN-2061 | G1314/QC104 | 2m | A (92.22) | V (7.78) | 90 |
| A | STVI | Telaprevir, boceprevir, R7227, SCH900518, BILN-2061 | G2036/PB84975 | 4r | A (99.86) | T (0.14) | 2066 | |
| 168 | D | QAVET | TMC 435, R7227, MK 7009, BI201335, BILN-2061 | G1251/PB80852 | 4r | D (94.64) | E (5.36) | 56 |
| D | QAVET | TMC 435, R7227, MK 7009, BI201335, BILN-2061 | G2035/PB83515 | 4r | D (99.7) | E (0.3) | 2109 | |
| 170 | V | AT | Telaprevir, boceprevir | G2036/PB84975 | 4r | V (99.61) | A (0.39) | 2818 |
Substitutions known to confer drug resistance are in bold, and substitutions of unknown impact are in italics.
The viral population of 3 individuals contained single low-frequency polymorphisms in NS3 that have been associated with resistance to BI-201335 or SCH-900518 (G1251/PB80852:D168E, G1314/QC104:A156V, and G2035/PB83515:D168E). One individual (G2036/PB84975) had multiple mutations (A29V, T54A, A156T, and V170A) associated with resistance to the protease inhibitors ACH806, telaprevir, boceprevir, SCH-900518, or R7227. The viral populations also contained variants of unknown phenotype at key sites in NS3 (Table 3). One individual, G2037/PB57078, harbored low-frequency HCV variants L36P/R altering the dominant leucine that is associated with drug resistance to mutations of unknown drug response status.
DISCUSSION
In this study, we report the use of pyrosequencing combined with a hemi-genome amplification approach to rapidly sequence, assemble, and classify 30 complete HCV genome sequences of poorly characterized HCV subtypes (2c, 2k, 2m, 3g, 3i, 4g, 4l, 4m, 4n, 4o, 4r, and 4v) and 1 intergenotypic recombinant (2k/1b) for a better view of global sequence diversity, enabling formal confirmation of the provisionally classified HCV subtypes 2c, 3g, and 4v and the identification of a novel variant of genotype 4. Together with sequences available from public databases, these new data enhance our knowledge of potential drug resistance mutations, especially for subtypes of genotypes 2 and 4 that are mainly found on the African continent [2]. Pyrosequencing of HCV hemi-genomes, without previous knowledge of target sequence or use of time-consuming and labor-intensive methods such as primer walking, allowed a high-resolution assessment of both high-frequency and low-frequency drug resistance mutations across the full viral genome in infected individuals.
The importance of accurate HCV genotyping, with at least 1 full genome sequence required to resolve discrepancies in HCV subtype classification, has been emphasized for the study of HCV evolution and potential associations with treatment response to DAAs [2, 12, 28, 33–37]. Unlike core-E1/NS5B genotyping, analysis of full genome sequences allows identification of divergence across all regions of the genome, thereby facilitating parsing of closely related, yet distinct subtype sequences. This is further evidenced in our phylogenetic analysis by the increase in bootstrap support for subtype assignments based on full genome sequences as compared to assignments based on core-E1/NS5B sequences. As a result, we are able to provide definitive confirmation of several provisionally classified subtypes and to identify a potential novel subtype of genotype 4. We expect that as more full genome sequences become available, new HCV subtypes will be teased apart from existing ones and that additional nomenclature criteria will be necessary.
The ability to robustly identify and classify genotype recombinants in full genome sequences is also critical in determining the prevalence and clinical relevance of such genetic events. The genotype 2k/1b recombinant sequenced here, G1241/PB41880, appears to be a member of the same circulating recombinant form as that identified in St. Petersburg [4, 5]. However, despite sharing the same mosaic structure as the St. Petersburg isolate, phylogenetic and break point site evidence suggests that G1241/PB41880 is likely to be an independently transmitted and diverged copy of the previously observed recombinant type, rather than a novel recombinant of similar parental strains.
Dominant drug resistance mutations in response to treatment with DAAs have been extensively studied in genotype 1 infection, but data on drug resistance mutations in other genotypes remain scarce [38–40]. Furthermore, very few studies explore sensitive techniques for the identification of minor drug resistance variants in viral populations [41]. The application of pyrosequencing to poorly characterized HCV genotypes allowed the identification of naturally occurring genotype and subtype-specific drug resistance mutations to mutations conferring resistance to DAAs, as well as a high-resolution view of intrapatient sequence diversity. In these poorly described HCV genotypes, we find a genotype and subtype-specific distribution of dominant changes in key resistance sites in NS3 and NS5B proteins, as well as minor variants in NS3. While mutations at drug resistance sites in NS3 in genotypes 2–5 have been previously reported [39, 40], this study extends the analysis to resistance mutations in NS5B and includes genotype 6 and 7 sequences in the analysis.
The availability of full genome sequences also allowed the identification of genotype 4 subtypes that harbor resistance mutations in both NS3 (V36L) and NS5B (M414L) that have been associated with increased resistance to treatment with telaprevir and A-782759. The presence of both of these dominant resistance mutations in subtype 4g, 4k-4r, and 4v virus isolated from infected patients suggests that these patients might respond poorly to treatment with both drugs, although this remains to be tested. Furthermore, the finding that these 2 changes are found only in a subset of genotype 4 subtypes reinforces the importance of subtype classification in helping to predict clinical response to treatment, as has recently been found for treatment response rates to DAAs for HCV subtypes 1a and 1b [15, 16].
It is important to note that for a significant proportion of amino acid changes identified here, their impact on drug resistance is unknown. Moreover, given the significant nucleotide differences between HCV genotypes and subtypes, it is unclear whether the drug resistance mutations that have been extensively characterized in genotype 1 would have the same impact on resistance in other genotypic backgrounds. A recent in vitro pilot study on the activity of the NS5A inhibitor BMS-790052 in genotype 4, however, found overlapping resistance profiles between HCV genotypes 1 and 4 [42], suggesting that mutations analyzed here may also be important for treatment-response rates in areas with a high HCV prevalence, such as Africa, where HCV genotype 4 subtypes are prevalent. It is also possible that previously uncharacterized mutations such as the D168Q mutation prevalent in 100% of HCV genotype 3 sequences may help explain the poor treatment response of this genotype to TMC-435 [43]. It will be important to study the consequence of these changes in the various genetic backgrounds on replication and resistance to antiviral drugs.
Taken together, our results indicate that deep sequencing of full genomes from HCV-infected patients can be an efficient method to classify virus subtypes and may help to inform treatment strategies. Appraisal of consensus full genome sequence from HCV-infected individuals can be used to identify the presence of dominant antiviral drug resistance mutations before initiation of a treatment regimen, while deep sequence data can be used to identify resistance mutations present at low frequency in the viral population of the same individual. Our approach identified minor drug-resistant variants in a subset of the treatment-naive patients tested, while a recent report describing the use of Illumina sequencing to identify DAA resistance mutations in treatment-naive genotype 1b–infected patients showed the presence of DAA resistant mutations in both NS3 and NS5B in up to 74.1% of the patients tested [44], suggesting that these low-level mutations may be highly prevalent in selected HCV subtypes. This information can be used to study the impact of dominant or subdominant mutations on treatment response rates for future interferon-free treatment regimens with DAAs and to eventually steer selection of appropriate single or combinatorial antiviral therapies in order to minimize treatment failure due to preexisting drug resistance mutations among viral subtypes.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the Broad Institute's Sequencing and Biological Samples Repository Platforms for their assistance in generation of genomic data and sample handling.
Disclaimer. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services (contract HHSN272200900018C to B. W. B., contract HHSN272200900006C to B. W. B., grant R01-AI067926 to T. M. A., and grant U19-AI082630 to T. M. A.; and the Deutsche Forschungsgemeinschaft (grant DFG KU2250/1-1 to T. K.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Murphy DG, Chamberland J, Dandavino R, Sablon E. A new genotype of hepatitis C virus originating from Central Africa. Hepatology. 2007;46(Suppl 1):623A. doi: 10.1128/JCM.02831-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simmonds P, Bukh J, Combet C, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology (Baltimore, Md). 2005;42:962–73. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 3.Murphy DG, Willems B, Deschênes M, Hilzenrat N, Mousseau R, Sabbah S. Use of sequence analysis of the NS5B region for routine genotyping of hepatitis C virus with reference to C/E1 and 5′ untranslated region sequences. Journal of Clinical Microbiology. 2007;45:1102–12. doi: 10.1128/JCM.02366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalinina O, Norder H, Magnius LO. Full-length open reading frame of a recombinant hepatitis C virus strain from St Petersburg: proposed mechanism for its formation. Journal of General Virology. 2004;85:1853–7. doi: 10.1099/vir.0.79984-0. [DOI] [PubMed] [Google Scholar]
- 5.Kalinina O, Norder H, Mukomolov S, Magnius LO. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. Journal of Virology. 2002;76:4034–43. doi: 10.1128/JVI.76.8.4034-4043.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raghwani J, Thomas XV, Koekkoek SM, et al. The origin and evolution of the unique HCV circulating recombinant form 2k/1b. Journal of Virology. 2011;86:2212–20. doi: 10.1128/JVI.06184-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreau I, Hegarty S, Levis J, et al. Serendipitous identification of natural intergenotypic recombinants of hepatitis C in Ireland. Virology Journal. 2006;3:95. doi: 10.1186/1743-422X-3-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharya D, Accola MA, Ansari IH, Striker R, Rehrauer WM. Naturally occurring genotype 2b/1a hepatitis C virus in the United States. Virology Journal. 2011;8:458. doi: 10.1186/1743-422X-8-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kageyama S, Agdamag DM, Alesna ET, et al. A natural inter-genotypic (2b/1b) recombinant of hepatitis C virus in the Philippines. Journal of Medical Virology. 2006;78:1423–8. doi: 10.1002/jmv.20714. [DOI] [PubMed] [Google Scholar]
- 10.Legrand-Abravanel F, Claudinon J, Nicot F, et al. New natural intergenotypic (2/5) recombinant of hepatitis C virus. Journal of Virology. 2007;81:4357–62. doi: 10.1128/JVI.02639-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noppornpanth S, Lien TX, Poovorawan Y, Smits SL, Osterhaus ADME, Haagmans BL. Identification of a naturally occurring recombinant genotype 2/6 hepatitis C virus. Journal of Virology. 2006;80:7569–77. doi: 10.1128/JVI.00312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuiken C, Simmonds P. Nomenclature and numbering of the hepatitis C virus. Methods in Molecular Biology. 2009;510:33–53. doi: 10.1007/978-1-59745-394-3_4. [DOI] [PubMed] [Google Scholar]
- 13.Morel V, Fournier C, François C, et al. Genetic recombination of the hepatitis C virus: clinical implications. Journal of Viral Hepatitis. 2011;18:77–83. doi: 10.1111/j.1365-2893.2010.01367.x. [DOI] [PubMed] [Google Scholar]
- 14.Foster GR, Hezode C, Bronowicki JP, et al. Telaprevir alone or with peginterferon and ribavirin reduces HCV RNA in patients with chronic genotype 2 but not genotype 3 infections. Gastroenterology. 2011;141:881–889 e1. doi: 10.1053/j.gastro.2011.05.046. [DOI] [PubMed] [Google Scholar]
- 15.Fridell RA, Wang C, Sun J-H, et al. Genotypic and phenotypic analysis of variants resistant to Hepatitis C Virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology. 2011;54:1924–35. doi: 10.1002/hep.24594. [DOI] [PubMed] [Google Scholar]
- 16.Hazuda D, Ogert R, Howe J, et al. Analysis of resistance associated variants by HCV genotype 1 subtype (1a/1b) in phase 3 (SPRINT-2/RESPOND-2) boceprevir plus standard of care clinical trials. 2011 Sixth International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention, Rome, Italy, [Google Scholar]
- 17.Kuntzen T, Timm J, Berical A, et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naive patients. Hepatology. 2008;48:1769–78. doi: 10.1002/hep.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. New England Journal of Medicine. 2012;366:216–24. doi: 10.1056/NEJMoa1104430. [DOI] [PubMed] [Google Scholar]
- 19.Gane EJ, Stedman CA, Hyland RH, et al. Once daily PSI-7977 plus RBV: pegylated interferon-ALFA not required for complete rapid viral response in treatment-naive patients with HCV GT2 or GT3. 2011 The Liver Meeting 2011: American Association for the Study of Liver Diseases (AASLD) 62nd Annual Meeting, Boston, MA. [Google Scholar]
- 20.Kuntzen T, Berical A, Ndjomou J, et al. A set of reference sequences for the hepatitis C genotypes 4d, 4f, and 4k covering the full open reading frame. Journal of Medical Virology. 2008;80:1370–8. doi: 10.1002/jmv.21240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henn MR, Boutwell CL, Charlebois P, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathogens. 2012;8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–3. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lole KS, Bollinger RC, Paranjape RS, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. Journal of Virology. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 26.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macalalad AR, Zody MC, Charlebois P, et al. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput Biol. 2012;8:e1002417. doi: 10.1371/journal.pcbi.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demetriou VL, Kostrikis LG. Near-full genome characterization of unclassified hepatitis c virus strains relating to genotypes 1 and 4. Journal of Medical Virology. 2011;83:2119–27. doi: 10.1002/jmv.22237. [DOI] [PubMed] [Google Scholar]
- 29.Bartels DJ, Zhou Y, Zhang EZ, et al. Natural prevalence of hepatitis C virus variants with decreased sensitivity to NS3.4A protease inhibitors in treatment-naive subjects. Journal of Infectious Diseases. 2008;198:800–7. doi: 10.1086/591141. [DOI] [PubMed] [Google Scholar]
- 30.Yang W, Zhao Y, Fabrycki J, et al. Selection of replicon variants resistant to ACH-806, a novel hepatitis C virus inhibitor with no cross-resistance to NS3 protease and NS5B polymerase inhibitors. Antimicrobial Agents and Chemotherapy. 2008;52:2043–52. doi: 10.1128/AAC.01548-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenz O, Verbinnen T, Lin TI, et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrobial Agents and Chemotherapy. 2010;54:1878–87. doi: 10.1128/AAC.01452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chamberlain RW, Adams N, Saeed AA, Simmonds P, Elliott RM. Complete nucleotide sequence of a type 4 hepatitis C virus variant, the predominant genotype in the Middle East. Journal of General Virology. 1997;78(Pt 6):1341–7. doi: 10.1099/0022-1317-78-6-1341. [DOI] [PubMed] [Google Scholar]
- 33.Cummings MD, Lindberg J, Lin TI, et al. Induced-fit binding of the macrocyclic noncovalent inhibitor TMC435 to its HCV NS3/NS4A protease target. Angew Chem Int Ed Engl. 2010;49:1652–5. doi: 10.1002/anie.200906696. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Lu L, Wu X, et al. Complete genomic sequences for hepatitis C virus subtypes 4b, 4c, 4d, 4g, 4k, 4l, 4m, 4n, 4o, 4p, 4q, 4r and 4t. Journal of General Virology. 2009;90:1820–6. doi: 10.1099/vir.0.010330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pang PS, Planet PJ, Glenn JS. The evolution of the major Hepatitis C genotypes correlates with clinical response to interferon therapy. PLoS ONE. 2009;4:e6579. doi: 10.1371/journal.pone.0006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reesink HW, Fanning GC, Farha KA, et al. Rapid HCV-RNA decline with once daily TMC435: a phase I study in healthy volunteers and hepatitis C patients. Gastroenterology. 2010;138:913–21. doi: 10.1053/j.gastro.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 37.Soriano V, Vispo E, Poveda E, et al. Directly acting antivirals against hepatitis C virus. J Antimicrob Chemother. 2011;66:1673–86. doi: 10.1093/jac/dkr215. [DOI] [PubMed] [Google Scholar]
- 38.Akhavan S, Schnuriger A, Lebray P, Benhamou Y, Poynard T, Thibault V. Natural variability of NS3 protease in patients infected with genotype 4 hepatitis C virus (HCV): implications for antiviral treatment using specifically targeted antiviral therapy for HCV. J Infect Dis. 2009;200:524–7. doi: 10.1086/600893. [DOI] [PubMed] [Google Scholar]
- 39.Lopez-Labrador FX, Moya A, Gonzalez-Candelas F. Mapping natural polymorphisms of hepatitis C virus NS3/4A protease and antiviral resistance to inhibitors in worldwide isolates. Antiviral Therapy. 2008;13:481–94. [PubMed] [Google Scholar]
- 40.Vallet S, Viron F, Henquell C, et al. NS3 protease polymorphism and natural resistance to protease inhibitors in French patients infected with HCV genotypes 1-5. Antiviral Therapy. 2011;16:1093–1102. doi: 10.3851/IMP1900. [DOI] [PubMed] [Google Scholar]
- 41.Lauck M, Alvarado-Mora MV, Becker EA, et al. Analysis of hepatitis C virus intra-host diversity Across The Coding Region by Ultra-Deep pyrosequencing. Journal of Virology. 2012;86:3952–60. doi: 10.1128/JVI.06627-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C, Jia L, Huang H, et al. In vitro activity of BMS-790052 on hepatitis C virus genotype 4 NS5A. Antimicrobial Agents and Chemotherapy. 2011;55:3795–802. doi: 10.1128/AAC.06169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreno C, Berg T, Tanwandee T, et al. Antiviral activity of TMC435 monotherapy in patients infected with HCV genotypes 2 to 6: TMC435-C202, a phase IIa, open-label study. Journal of Hepatology. 2012;56:1247–53. doi: 10.1016/j.jhep.2011.12.033. [DOI] [PubMed] [Google Scholar]
- 44.Nasu A, Marusawa H, Ueda Y, et al. Genetic heterogeneity of hepatitis C virus in association with antiviral therapy determined by ultra-deep sequencing. PLoS One. 2011;6:e24907. doi: 10.1371/journal.pone.0024907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.