

Abstract
Background. Acquired immunity to malaria develops with increasing age and repeated infections. Understanding immune correlates of protection from malaria would facilitate vaccine development and identification of biomarkers that reflect changes in susceptibility resulting from ongoing malaria control efforts.
Methods. The relationship between immunoglobulin G (IgG) antibody and both interferon γ (IFN-γ) and interleukin 10 (IL-10) responses to the 42-kD C-terminal fragment of Plasmodium falciparum merozoite surface protein 1 (MSP142) and the risk of (re)infection were examined following drug-mediated clearance of parasitemia in 94 adults and 95 children in an area of holoendemicity of western Kenya.
Results. Positive IFN-γ enzyme-linked immunosorbent assay (ELISA) and enzyme-linked immunosorbent spot assay (ELISPOT) responses to MSP142 3D7 were associated with delayed time to (re)infection, whereas high-titer IgG antibodies to MSP142 3D7 or FVO alleles were not independently predictive of the risk of (re)infection. When IFN-γ and IL-10 responses were both present, the protective effect of IFN-γ was abrogated. A Cox proportional hazard model including IFN-γ, IL-10, MSP142 3D7 IgG antibody responses, hemoglobin S genotype, age, and infection status at baseline showed that the time to blood-stage infection correlated positively with IFN-γ responses and negatively with IL-10 responses, younger age, and asymptomatic parasitemia.
Conclusions. Evaluating combined allele-specific cellular and humoral immunity elicited by malaria provides a more informative measure of protection relative to evaluation of either measure alone.
Keywords: human immunity, Merozoite Surface Protein 1, humoral immunity, cellular immunity, interferon-gamma, interleukin-10, sickle cell trait, longitudinal study, observational study
Globally, falciparum malaria remains a major cause of childhood mortality and morbidity. Considerable reduction in malaria transmission has been achieved through use of insecticidal bed nets and indoor spraying [1]. Protective immunity to blood-stage Plasmodium falciparum, the parasite stage underlying malaria pathogenesis, develops in children and is maintained in adults as a consequence of periodic boosting manifested as asymptomatic low-density parasitemia [2]. Anticipating future decreases in malaria transmission, understanding the mechanisms underlying protective immunity is pertinent to identifying markers predictive of age-related shifts in malaria susceptibility and developing vaccines to blood-stage P. falciparum.
Validating correlates of immune protection in malaria-endemic populations is challenging because both humoral and cellular immunity are induced and because only a limited number of candidate antigens have been studied to date. Most have described the association between the malarial immunoglobulin G (IgG) or IgG subclass antibodies and susceptibility to clinical malaria [3]. Antibodies are clearly important since transfer of IgG from immune African adults to children and nonimmune adults experiencing acute malaria rapidly reduces parasitemia and abrogates fever [4, 5]. These antibodies are directed against a breadth of blood-stage antigens, which, in combination and after achieving a critical threshold level, correlate with decreased susceptibility to high-density parasitemia and clinically mild malaria [6, 7]. The mechanisms by which such antibodies mediate protective immunity are incompletely resolved [2]. With respect to cellular immunity, studies of rodent malaria [8], malaria-naive humans exposed to sporozoites and liver-stage P. falciparum [9] or to a small number of infected erythrocytes followed by drug cure [10], and studies of residents of malaria endemic areas [11] indicate that T-cell cytokine responses, particularly interferon γ (IFN-γ) responses, correlate with protection from blood-stage infection and malaria illness [12, 13].
Merozoite surface protein 1 (MSP1) has been considered as a blood-stage vaccine candidate [14, 15]. Although a clinical trial of the C-terminal 42-kDa region of the MSP1 (MSP142) 3D7 allele showed no impact on the susceptibility to mild malaria in Kenyan children [16], improved immunogenicity and efficacy may be possible by using viral vectors [17] and different adjuvants [18, 19], allowing MSP1 to remain a candidate for a multistage vaccine [20]. IgG antibody and T-cell cytokine responses are directed to epitopes contained within fragments of MSP142, MSP119, and MSP133, that are generated by endogenous parasite proteases prior to invasion of the erythrocyte [15]. Studies of mouse malaria and malaria-naive human volunteers experimentally inoculated with blood-stage P. falciparum suggest that T-cell cytokine responses to epitopes contained within MSP133 mediate protection from parasitemia independent of antibodies [10, 21, 22]. T-helper cell responses that support anti-MSP1 antibody production by B cells may also be generated [23, 24]. Polymorphisms of MSP119 and MSP133 are common in areas of endemicity [25], raising concern that variant-specific immunity needs to be considered when studying MSP1 and other polymorphic blood-stage antigens as targets of acquired and vaccine-induced immunity [26]. We addressed these issues by conducting an observational study of adults and children living in a malaria-holoendemic area of Kenya in 2003, prior to widespread introduction of insectical bed nets and control strategies. IgG antibody, IFN-γ, and interleukin 10 (IL-10) responses specific to MSP142 were evaluated as correlates of delay in the time to (re)infection and association with clinical malaria following drug-mediated cure of blood-stage parasitemia.
METHODS
Study Participants and Design
Asymptomatic healthy adults (n = 101) and children (n = 100) from Kanyawegi in Nyanza Province, Kenya, were enrolled in July 2003 at the beginning of a seasonal peak in malaria transmission. Women were excluded if they self-reported to be pregnant. Venous blood was collected for immunologic studies prior to administration of a weight-adjusted 6-dose regimen of artemether-lumefantrine that all participants received at baseline (week 0) regardless of malaria status determined by blood smear.
Blood samples were collected weekly by finger stick for 11 consecutive weeks to determine the time to (re)infection. Giemsa-stained thick and thin smears were scored as negative when no asexual-stage P. falciparum or other malaria species was observed after microscopic inspection that included ≥200 leukocytes. Parasite density was expressed as the number of parasites per microliter of blood, on the assumption of a leukocyte count of 8000 per µL blood. Parasite clearance following artemether-lumefantrine administration at baseline was defined by a malaria-negative blood smear 2 weeks later. If the blood smear was positive at week 2, these individuals were excluded from the remaining analysis. Passive surveillance for clinical malaria was performed and defined by a positive blood smear, as well as an axillary temperature of ≥37.5°C and malaria symptoms [7]. Asymptomatic blood-stage infections during weeks 2–11 were not treated with antimalarial drugs, consistent with 2003 Kenya Ministry of Health guidelines. Malaria-naive adults (n = 20) from Cleveland, Ohio, were screened to evaluate nonspecific immune responses.
Ethical Approval
Ethical approval was obtained from the Institutional Review Board at the University Hospitals Case Medical Center and from the Kenya Medical Research Institute Ethical Review Committee. Adults signed a written informed consent form in the local language (ie, English or Duhluo). Parents or guardians signed for minors <18 years old.
Recombinant MSP142 and MSP133 Peptide T-Cell Epitopes
Recombinant MSP142 corresponding to the dimorphic allelic variants PNG-MAD20 (3D7 strain, E-TSR haplotype) and Wellcome-K1 (FVO strain, Q-KNG haplotype) were expressed in Escherichia coli and used for peripheral blood mononuclear (PBMC) cytokine recall assays and IgG antibody assays [20]. Synthetic M2 peptide (GISYYEKVLAKYKDDLE; MSP1 residues 1467–1483, SigmaGenosys, Gainsville, TX) corresponding to a region of MSP133 3D7 was selected as an immunodominant T-cell epitope [27].
Cytokine Recall Assays
Enzyme-linked immunosorbent spot assays (ELISPOT) and enzyme-linked immunosorbent assays (ELISAs) were performed as previously described [28, 29] by incubating PBMCs for 84 hours with 5 µg/mL MSP142 3D7, MSP142 FVO, or M2 3D7 peptide. IFN-γ and IL-10 ELISA responses were considered positive if the culture supernatant contained ≥20 pg/mL following stimulation with malaria antigen. An IFN-γ ELISPOT response was considered positive if the number of spot-forming units (SFU) in the stimulated well was significantly greater than that in the unstimulated background well, using a P value of < .05, calculated by χ2 analysis of two proportions, with adjustment for small sample size. The mean number of SFU per 106 PBMCs was <5 in unstimulated wells for all study participants. The magnitude of IFN-γ ELISPOT responses was determined by subtracting the number of SFU in the unstimulated well from that for the well containing malaria antigen. Malaria-naive controls (n = 20) showed no IFN-γ or IL-10 responses to recombinant MSP142 or M2 peptide by ELISA or ELISPOT.
IgG Antibody ELISA
IgG antibodies to MSP142 3D7 and FVO were measured by ELISA, as previously described [7]. Plasma from 9 malaria-naive North American adults was pooled and used as a negative control, and plasma from 20 Kenyan adults was pooled and used to create a standard curve. Individual antibody responses were expressed in arbitrary units (AU) and categorized into quartiles. Individuals with high-AU IgG responses (ie, those with an AU in the upper 2 quartiles; defined as high-titer antibodies) were considered positive, high-responders for the survival analysis.
Hemoglobin S Genotype
Genomic DNA was extracted from blood using the QIAamp DNA blood mini kit (Qiagen, Valencia, CA). The sickle hemoglobin mutation was detected by polymerase chain reaction amplification, followed by Bsu 361 digestion, as described elsewhere [30].
Statistical Analysis
Differences in the frequency of immunological variables were compared across age groups (ie, adult aged ≥18 years vs children aged 1–14 years), using McNemar χ2 tests. Median cytokine recall responses were compared across age groups for adults and subsets of children 10–14, 5–9, and 1–4 years old, using the Kruskal-Wallis nonparametric method.
To proceed from a stepwise analytical approach, we first conducted survival analyses (for 9 weeks, from weeks 2–11), using dichotomized variables for antibodies, cytokine responses, hemoglobin AA/AS genotype, and parasitemia prior to treatment one at a time, in keeping with similar studies [3]. To explore synergistic and antagonistic immune responses, survival analyses were then conducted with a combination of 2 variables, such as IFN-γ and IL-10 or IFN-γ and IgG antibody. The time to (re)infection was determined with a Kaplan-Meier log-rank test for survival analysis, using SAS 9.2 (SAS Institute, Cary, NC). Multiple factors that simultaneously might influence time to (re)infection were next evaluated. Cox regression models were used to estimate the association between these variables or combination of variables and the risk for malaria where proportional hazards assumptions were met. Covariates in the analysis included, IFN-γ, IL-10, IgG antibody, HbAS/AA genotype, malaria status prior to treatment and age (continuous). Finally, we correlated baseline antibody and cytokine responses with susceptibility to clinical malaria.
RESULTS
Study Population Characteristics
Entomologic inoculation rates in July 2003 were estimated to be >300 cases per person per year [31]. Thirty-four adults had asymptomatic parasitemia at baseline, and 40 remained blood smear negative during the 11-week follow-up period (Table 1). One adult failed to clear blood-stage infection 2 weeks after treatment because of noncompliance with directly observed antimalarial treatment. Six were lost to follow-up, resulting in 94 adults eligible for analysis. Eighty-two of 100 children had blood-stage P. falciparum at baseline, with higher average parasite densities than adults (Table 1). Only 8 children did not have a positive blood smear during the follow-up period, and the average time to (re)infection was significantly shorter, at 5.3 weeks, compared with 7.4 weeks for adults. When children were reinfected, they tended to have 2.5-fold higher parasite densities as compared to reinfected adults. Five children were lost to follow-up, leaving 95 children included in the analysis. Twenty-three children developed symptomatic malaria during the 12-week observation period. The frequency of sickle cell trait was 30% for both adults and children. No individuals were homozygous for hemoglobin S.
Table 1.
Study Population Characteristics
| Characteristic | Adults (n = 101) | Children (n = 100) |
|---|---|---|
| Age, y, mean (range) | 39.6 (18–79) | 7.7 (1–14) |
| Parasitemic yet asymptomatic prior to treatmenta | 34 | 82 |
| Hemoglobin level, g/dL, mean | 12.6 | 11.4 |
| Parasite density, parasites/μL blood, mean (range)a | 148 (80–5120) | 3740 (80–48 000) |
| Cleared blood-stage parasites 2 weeks after treatment, by blood smear | 100 | 100 |
| Remained aparasitemic during 12-wk follow-up perioda | 40 | 8 |
| Time to reinfection, wk, mediana | 7.4 | 5.3 |
| Lost to follow-up | 6 | 5 |
| Required treatment for symptomatic malariaa | 3 | 23 |
| Frequency of hemoglobin AS genotype, % | 30 | 30 |
| Total included in analysis of immune correlates | 94 | 95 |
a Significant differences were observed when comparing adults to children for median time to (re)infection, parasite prevalence, mean parasite density at baseline, number who remained aparasitemic during the follow-up period and for those who had symptomatic malaria infections (P < .0001).
MSP142 IFN-γ and IL-10 Responses
To appreciate differences in cellular immunity to MSP142 between adults and children, we compared the baseline prevalence of IFN-γ and IL-10 responses (Table 2). Higher frequencies of IFN-γ ELISA responses among adults, compared with children, were observed for M2 3D7 peptide (P = .007), MSP142 FVO (P = .003), and MSP142 3D7 (P < .001), with responses to 3D7 more common than those to FVO. Analysis of children grouped according to age 10–14, 5–9, and 1–4 years revealed that the frequency of IFN-γ ELISA responses had a positive, age-related trend for MSP142 FVO and 3D7, but neither was statistically significant. IFN-γ ELISPOT results showed a lower proportion of responders than IFN-γ recall measured by ELISA (eg, 9.5% vs 47.7% for MSP142 FVO; P < .0001, by the Fisher exact test). In general, adults had more positive responses to malaria antigens, as measured by IFN-γ ELISPOT; however, only responses to MSP142 FVO (P = .036) were significantly higher in adults as compared to children. As was the case for IFN-γ ELISA, the frequency of positive ELISPOT responses was greater when PBMCs were stimulated with MSP142 3D7 than with MSP142 FVO (Table 2). With respect to IL-10, responses to both MSP142 FVO and 3D7 showed no significant differences according to age; however, IL-10 responses were more common to 3D7 as compared to the FVO variant (59.1%–44.6% and 21.6%–16.9%, respectively).
Table 2.
Frequency of Naturally Acquired Immune Responses to Plasmodium falciparum Merozoite Surface Protein 1 (MSP1) in Adults and Children
| Assay, Variable | Adults Aged ≥18 y | Children Aged 1–14 y | Pa | Children Aged 10–14 y | Children Aged 5–9 y | Children Aged 1–4 y |
|---|---|---|---|---|---|---|
| IFN-γ ELISA | ||||||
| Subjects, no. | 90 | 88 | 22 | 44 | 22 | |
| Stimulus, % | ||||||
| PHA | 100.0 | 98.0 | .500 | 100 | 95 | 100 |
| M2 peptide | 30.7 | 13.6 | .007 | 13.4 | 18.2 | 12.5 |
| MSP142 3D7 | 62.5 | 34.1 | .001 | 45.4 | 27.3 | 36.4 |
| MSP142 FVO | 47.7 | 26.1 | .003 | 36.4 | 25.0 | 18.2 |
| IFN-γ ELISPOT | ||||||
| Subjects, no. | 94 | 91 | 19 | 46 | 26 | |
| Stimulus, % | ||||||
| PHA | 95.0 | 91.0 | .447 | 86.4 | 93.9 | 89.7 |
| M2 peptide | 9.5 | 4.4 | .152 | 5.3 | 3.8 | 7.7 |
| MSP142 3D7 | 37.9 | 26.4 | .126 | 21.1 | 21.7 | 38.5 |
| MSP142 FVO | 9.5 | 2.2 | .036 | 0.0 | 2.2 | 3.8 |
| IL-10 ELISA | ||||||
| Subjects, no. | 90 | 88 | 22 | 44 | 22 | |
| Stimulus, % | ||||||
| PHA | 100.0 | 100.0 | NS | 100.0 | 100.0 | 100.0 |
| MSP142 3D7 | 59.1 | 44.6 | .058 | 42.9 | 42.9 | 55.0 |
| MSP142 FVO | 21.6 | 16.9 | .434 | 4.8 | 16.7 | 30.0 |
| IgG antibody ELISA | ||||||
| Subjects, no. | 90 | 88 | 22 | 44 | 22 | |
| Stimulus, % | ||||||
| MSP142 3D7 | 49.5 | 25.0 | .003 | 22.7 | 28.6 | 20.7 |
| MSP142 FVO | 36.6 | 28.0 | .190 | 22.7 | 34.7 | 20.7 |
Bold values denote significant results.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; IFN-γ, interferon γ; IgG, immunoglobulin G; IL-10, interleukin 10; MSP142, 42-kD C-terminal fragment of Plasmodium falciparum MSP1; NS, not significant.
a Adults vs children.
We next compared the magnitude of IFN-γ responses among adults and children to address whether a threshold level is important for immune protection against malaria. The amount of IFN-γ produced by adults versus children for MSP142 3D7 was similar (median level, 150 and 110 pg/mL, respectively). However, IFN-γ concentrations were negatively related to age, with the youngest children secreting relatively more IFN-γ in response to MSP142 3D7 ex vivo stimulation (median levels for children aged 10–14, 5–9, and 1–4 years, 49, 123 and 160 pg/mL, respectively; P = .006, by the Kruskal-Wallis test; Supplementary Figure 1). MSP142 FVO–driven IFN-γ production was uniformly low across all age groups (Supplementary Figure 1). With respect to IFN-γ ELISPOT, the level was similar among children and adults who responded to MSP142 3D7 (median level, 60 SFU per 106 PBMC). The median value for IFN-γ ELISPOT responses to MSP142 FVO was one-half that of MSP142 3D7 (data not shown).
MSP142 IgG Antibody Responses
A greater proportion of adults than children had high-titer IgG antibodies to MSP142 3D7 (49.5% vs 25.0%; P = .003), whereas there was no difference for anti-MSP142 FVO antibodies (36.6% and 28.0%, respectively; Table 2). Antibody levels to MSP142 3D7 and FVO did not differ significantly by age group among the children (Table 2).
MSP142 Allele-Specific Immunity and Risk of (Re)infection
Kaplan-Meier survival analyses were performed to determine which variables were independently associated with risk of (re)infection. M2 3D7 peptide IFN-γ ELISPOT responses (Figure 1A) and MSP142 3D7 IFN-γ ELISA responses (Figure 1B) were significantly associated with a decreased risk of (re)infection (hazard ratio [HR], 0.35 [95% confidence interval {CI}, .13–.98]; P = .046) and a delayed time to (re)infection (HR, 0.65 [95% CI, .44–.94]; P = .023). IFN-γ recall to MSP142 FVO was present (Figure 2C) but not associated with a decreased risk of (re)infection (HR, 0.91 [95% CI, .062–1.34]; P = .663). IL-10 ELISA responses were not an independent predictor of the risk of (re)infection (data not shown). Individuals with high-titer IgG antibody responses to MSP142 3D7 and FVO were not associated with a delayed time to (re)infection relative to that for individuals with responses in the lower 2 quartiles (Figure 2), even after adjustment for age.
Figure 1.

Survival analyses for interferon γ (IFN-γ) responses to the Plasmodium falciparum merozoite surface protein 1 (MSP1). The solid black line represents nonresponders and the dashed line represents responders. A) Significant delay in the time to (re)infection was observed for those with IFN-γ ELISPOT responses to the M2 3D7 peptide (hazard ratio [HR], 0.35 [95% confidence interval {CI}, .13–.98]; P = .046; and B) IFN-γ enzyme-linked immunosorbent assay (ELISA) responses to recombinant MSP142 3D7 (HR, 0.65 [95% CI, .44–.94]; P = .023; but not for those with C) IFN-γ ELISA responses to recombinant MSP142 FVO (HR, 0.91 [95% CI, .62–1.34]; P = .663.
Figure 2.

Survival analyses for immunoglobulin G (IgG) antibody responses to the 42-kD C-terminal fragment of Plasmodium falciparum merozoite surface protein 1 (MSP142). The solid black line represents nonresponders, and the dashed line represents responders. Study participants were considered negative for IgG antibody responses if levels were in the lower 2 quartiles, whereas those with responses in the upper 2 quartiles were considered antibody positive. A, A slight delay in the time to (re)infection was observed for those with high IgG antibody responses to MSP142 3D7, but this did not achieve statistical significance (hazard ratio, 1.02 [95% confidence interval {CI}, .71–1.49]; P = .897). B, Antibody responses to MSP142 FVO were also not associated with a risk of (re)infection (HR, 1.0 [95% CI, .70–1.16]; P = .969).
We next evaluated combined measurements of immunity to determine whether this was a better fit for the model. IFN-γ and IL-10 responses were evaluated first because IL-10 has been associated with protection from malaria but may have an antagonistic effect on IFN-γ responses [32]. Cytokine responses to MSP142 3D7 were categorized into 4 groups: IFN-γ positive and IL-10 negative, IFN-γ positive and IL-10 positive, IFN-γ negative and IL-10 positive, IFN-g negative and IL-10 negative. Figure 3 demonstrates that individuals who produced only IFN-γ had a significantly reduced risk of (re)infection as compared to IFN-γ negative and IL-10 negative (HR, 0.65 [95% CI, .49–.88]) or the other 3 groups combined (HR, 0.48 [95% CI, .28–.82]; P = .008). Similar analyses were done for MSP142 FVO, confirming that cytokine responses to this allele were not associated with a reduced risk of (re)infection (data not shown).
Figure 3.
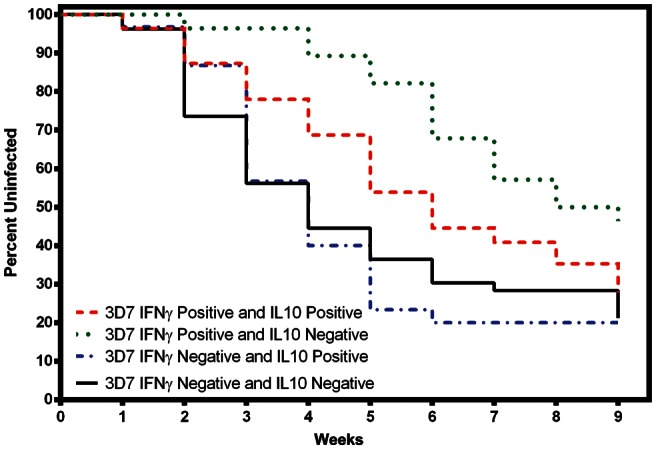
Survival analyses for interferon γ (IFN-γ) and interleukin 10 (IL-10) responses to 42-kD C-terminal fragment of Plasmodium falciparum merozoite surface protein 1 (MSP142) 3D7. Cytokine responses to MSP142 3D7 were categorized into 4 groups: IFN-γ positive and IL-10 positive (red), IFN-γ positive and IL-10 negative (green), IFN-γ negative and IL-10 positive (blue), IFN-γ negative and IL-10 negative (black). Study participants with IFN-γ positive and IL-10 negative responses (n = 55) had a significantly reduced risk of (re)infection as compared to those with IFN-γ negative and IL-10 negative responses (hazard ratio [HR], 0.65 [95% confidence interval {CI}, .49–.88]) or the other 3 groups combined (HR, 0.48 [95% CI, .28–.82]; P = .008). By using study participants with no cytokine responses (ie, IFN-γ negative and IL-10 negative) as the referent group (n = 53), those who were double positive for IFN-γ and IL-10 (n = 28; HR, 0.82 [95% CI, .53–1.27]; P = .382) or IFN-γ negative and IL-10 positive (n = 31; HR, 1.03 [95% CI, .62–1.72]; P = .897) did not have a significantly delayed time to (re)infection. Cytokine responses to MSP142 FVO were too infrequent for meaningful analysis (data not shown).
Combined MSP142 3D7 and FVO IFN-γ and antibody responses were evaluated next to determine whether there was an additive effect for individuals with both responses. Responses were classified into 4 groups, similar to the description above: IFN-γ positive and antibody positive, IFN-γ positive and antibody negative, etc. For MSP142 3D7, any positive response was associated with significantly reduced risk of (re)infection, compared with the double-negative group (Figure 4A). Antibody responses to MSP142 3D7 and FVO were highly correlated (Spearman r = 0.78; P < .0001). Yet improved survival estimates were observed for MSP142 FVO only when IFN-γ and antibody responses were included in the analysis (HR, 0.80 [95% CI, .64–.98]; P = .042; Figure 4B).
Figure 4.
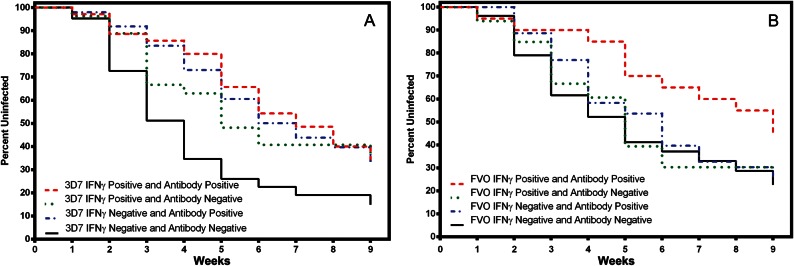
Survival analyses for combined interferon γ (IFN-γ) and immunoglobulin G (IgG) antibody responses to 42-kD C-terminal fragment of Plasmodium falciparum merozoite surface protein 1 (MSP142) 3D7 and FVO. Responses were divided into 4 categories: IFN-γ positive and IL-10 positive (red), IFN-γ positive and IL-10 negative (green), IFN-γ negative and IL-10 positive (blue), IFN-γ negative and IL-10 negative (black). A, For MSP142 3D7, IFN-γ positive and antibody positive (hazard ratio [HR], 0.78 [95% confidence interval {CI}, .66–.92]; P = .0037), IFN-γ positive and antibody negative (HR, 0.50 [95% CI, .32–.78]; P = .0022), and IFN-γ negative and antibody positive (HR, 0.74 [95% CI, .56–.98]; P = .036) responses were associated with a reduced risk of (re)infection, compared with those who were negative for both measurements. The combined effect for any positive IFN-γ or antibody response relative to IFN-γ negative or antibody negative response was an HR of 0.79 (95% CI, .7–.89; P = .0001). B, For MSP142 FVO, those who were IFN-γ positive and antibody positive were the only group who experienced a significant delay in the time to (re)infection (HR, 0.80 [95% CI, .64–.98]; P = .042), compared with those who were IFN-γ negative and antibody negative. IFN-γ positive and antibody positive (HR, 0.93 [95% CI, .73–1.19]; P = .5672) and IFN-γ negative and antibody positive (HR, 0.85 [95% CI, .55–1.32]; P = .472) had nonsignificant delays to reinfection, relative to IFN-γ negative and antibody negative individuals.
Finally, Cox proportional hazard models were created for all MSP142 3D7 and FVO immunologic measurements (ie, IFN-γ, IL-10, and antibodies), with adjustment for HbAS genotype, parasitemia status at baseline, and age (continuous). Table 3 demonstrates that the 3 immune responses to MSP142 FVO, the interactions among them, and HbAS genotype were not significant or informative. Colinearity was evaluated and found not to significantly influence the parameter estimates. However, age (continuous), and malaria status at baseline (ie, asymptomatic parasitemia and parasite density; Supplementary Materials) were significantly associated with a risk for (re)infection. The full model confirmed that IL-10 responders to MSP142 3D7 had a 62% increase in the risk of (re)infection, compared with IL-10 nonresponders (P = .019). In contrast, IFN-γ responders to MSP142 3D7 had a 46% decrease in the risk of (re)infection, compared with IFN-γ nonresponders (P = .002). In the full model, MSP142 3D7 IgG antibody responses were not significantly associated with the risk of malaria but increasing age was associated with a lower risk of infection (P < .0001). Finally, there were no significant differences in antibody and cytokine responses revealed by a comparison of the 23 children with febrile (re)infections to the 72 children who remained uninfected or asymptomatic when (re)infected.
Table 3.
Hazard Ratio Estimates for Cox Regression Survival Analyses
| Antigen | Predictor Variablea | HR (95% CI) | P |
|---|---|---|---|
| 3D7 | IFN-γ ELISA | 0.54 (.36–.80) | .002 |
| 3D7 | IgG Ab ELISA | 1.00 (.66–1.53) | .988 |
| 3D7 | IL-10 ELISA | 1.62 (1.08–2.43) | .019 |
| 3D7 | Age (continuous) | 0.97 (.95–.98) | <.0001 |
| 3D7 | Baseline Pf infection | 1.75 (1.13–2.74) | .013 |
| 3D7 | HbAS genotype | 0.95 (.61–1.47) | .820 |
| FVO | IFN-γ ELISA | 0.93 (.62–1.38) | .703 |
| FVO | IgG Ab ELISA | 0.86 (.56–1.32) | .495 |
| FVO | IL-10 ELISA | 1.44 (.91–2.28) | .116 |
| FVO | Age (continuous) | 0.97 (.95–.98) | <.0001 |
| FVO | Baseline Pf infection | 1.56 (1.01–2.41) | .044 |
| FVO | HbAS genotype | 0.81 (.53–1.24) | .335 |
Survival analysis for the full model, including both cellular and humoral immunity to MSP142 Bold values denote significant results.
Abbreviations: Ab, antibody; CI, confidence interval; ELISA, enzyme-linked immunosorbent assay; ELISPOT, enzyme-linked immunosorbent spot assay; HR, hazard ratio; IFN-γ, interferon γ; IgG, immunoglobulin G; IL-10, interleukin 10; MSP142, 42-kD C-terminal fragment of Plasmodium falciparum merozoite surface protein 1; NS, not significant; Pf, Plasmodium falciparum.
a Adjusted for HbAS genotype, parasitemia status at baseline, and age.
DISCUSSION
We sought to more clearly define cellular and humoral immune correlates of protection from P. falciparum blood-stage infection in a malaria-holoendemic setting. We focused on immunity to MSP142, the most abundant of the merozoite proteins, a ligand essential to erythrocyte invasion, and a malaria vaccine candidate. Taking into account various polymorphic MSP142 variants circulating in western Kenya [33], we also evaluated whether allele-specific immunity is relevant to these immune correlates. IgG antibodies to MSP142 3D7 and FVO were abundant but not predictive of protection. In contrast, MSP142 3D7–driven IFN-γ responses measured by ELISA and ELISPOT correlated, respectively, with a 41% and 73% lower risk of (re)infection during the 3-month follow-up period, whereas IFN-γ responses to the FVO variant were infrequent and not independently predictive of protection, suggesting that immunity to polymorphic T-cell epitopes within MSP133 is important to protection from parasitemia. In addition, IL-10 responses did not correlate with time to (re)infection; however, when IL-10 responses were observed to coexist with IFN-γ, they were associated with abrogation of protection. This finding is consistent with the counterregulatory influence of IL-10 on IFN-γ [32] and indicates that the cytokine milieu induced by malaria antigens should be considered during vaccine design and evaluation. Covariate analyses of the risk for (re)infection that include IFN-γ, IgG antibody, and human genetic and demographic factors showed that only IFN-γ responses to MSP142 3D7 were protective and not additive or synergistic, that HbAS genotype was not relevant to the risk of (re)infection (contrary to results of a study from an area with seasonal malaria transmission [34]), and that asymptomatic parasitemia at baseline was associated with increased risk of (re)infection. A limitation of this study was an insufficient sample size to make firm conclusions regarding the relevance of these immune end points to the susceptibility to clinical malaria.
Protective immunity to asexual-stage P. falciparum is an age-related process signified by antiparasite immunity marked by control of parasite density and clinical immunity marked by reduced susceptibility to fever with parasitemia [2]. Since both phenotypes appear by late childhood and are maintained during adulthood, it is intuitive that asymptomatic low-density parasitemia is a critical factor to the development and maintenance of this immunity. Age, independent of malaria exposure history, may also be an important variable in the development of immunologic memory [35]. With respect to antibody responses, higher antimerozoite antigen IgG titers are found in asymptomatic infected children, compared with noninfected children, and rapidly wane following clinical malaria [36], suggesting that serologic responses are not stable [37, 38] and that memory B cells are slow to develop and, when considered alone, are a poor correlate of protection from parasitemia. An earlier study we conducted in a region of Kenya with low malaria transmission reported that functional antibodies to MSP119, as measured by invasion inhibitory activity, may be more informative than standard serologic analysis [39]. However, MSP119 invasion inhibitory activity did not correlate with protection from malaria in an area of considerably higher transmission [33]. It is clear that more research is needed to understand the significance of antimerozoite antibodies to the control of parasitemia and susceptibility to clinical malaria, including the breadth of merozoite antigens to which memory B cells and antibodies are generated [37, 40].
Studies of cellular immunity in regulating malaria susceptibility are challenging given the low frequency of T-cell subsets in peripheral blood and the many cytokines produced by antigen-specific T-cells, B-cells, and monocytes. Although we did not address the cellular source of IFN-γ or IL-10, recent observations show that IL-10 is primarily secreted by activated effector CD4+ T-cells that also secrete IFN-γ [32]. This regulatory IL-10 may diminish the protective effect of IFN-γ during blood-stage malaria while limiting immunopathology. Nevertheless, enhancing long-lived T-cell memory is a desired end point for human malaria vaccines, including those that include merozoite antigens such as MSP1 [41, 42]. Inadequate T-cell immunity may also result from elicitation of short-lived, effector T cells [43] and active interference [44]. Longitudinal studies of naturally infected humans have demonstrated instability of cytokine responses over time [28, 45] and qualitative differences in IFN-γ–secreting MSP1-specific T-cells associated with age [43]. Further studies are underway to determine which T-cell subsets are associated with fewer subsequent malaria episodes and to provide a more expansive picture for the role of IL-10, in addition to transforming growth factor β, in inhibiting protective immunity.
Natural malaria infections in endemic areas display significant genetic heterogeneity and antigenic diversity [46], which complicate the correlation of antigen-specific immunity with strain-specific protection from infection. Studies of strain-specific immunity to MSP142 are further challenged by the fact that major T-cell and B-cell epitopes are encoded in segments of msp1 that have undergone recombination, leading to different haplotypes that are not easily sequenced from mixed-strain infections [29, 33]. Using disparate yet region-specific genotyping methods for msp1, we observed allele-specific immunologic correlations of protection for antibodies to MSP119 [33] but apparently only for children, not adults. This may be due to strain-specific immunity that clears individual infections until more general immunity (including T-cell help) has been acquired [47]. Our past studies in this population show higher temporal stability of IgG to the C-terminal MSP1 FVO as compared to 3D7 [48], even though Uganda-PA and FVO, which share downstream B-cell epitopes but differ upstream where the T-cell epitopes are located, tended to be the dominant allele [33]. Combined with results presented here, this suggests that antibodies to FVO do not confer protection from infection unless IFN-γ is also present. Immunity to 3D7 MSP142 was dominated by IFN-γ responses, which would also cover the Uganda-PA predominant genotype. Studies in animal models demonstrate that immunity to MSP1 is strain specific [49, 50]; however, naturally acquired immunity in humans who experience mixed-strain infections are more difficult to deconvolute.
Additional studies that monitor the cumulative effect of malaria on the development and maintenance of B-cell and T-cell immunity should increase our understanding of the key elements underlying protective immunity to malaria. This information is pertinent to vaccine development and to understanding how decreasing malaria transmission intensity impacts the strength and quality of protective immunity.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the parents and guardians, for enrolling their children in this study; our team of community health workers, without whom this study would not have been possible; Garret S. Bailey; and the Malaria Vaccine Development Branch, National Institutes of Health and Infectious Diseases. This work is published with the permission of the Director of the Kenya Medical Research Institute.
Financial support. This work was supported by National Institutes of Health RO1 AI43906 (J. W. K.), R01 CA134051 (A. M. M.), Fogarty International Center 1D43TW006576 (P. O. S., K. C.), Burroughs Wellcome Fund 1006818 (A. E. D.), UL1RR031982, National Center for Research Resources (H. J. F.) and the intramural research program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (C. L.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Moonen B, Cohen JM, Snow RW, et al. Operational strategies to achieve and maintain malaria elimination. Lancet. 2010;376:1592–603. doi: 10.1016/S0140-6736(10)61269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doolan DL, Dobano C, Baird JK. Acquired immunity to malaria. Clin Microbiol Rev. 2009;22:13–36. doi: 10.1128/CMR.00025-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fowkes FJ, Richards JS, Simpson JA, Beeson JG. The relationship between anti-merozoite antibodies and incidence of Plasmodium falciparum malaria: a systematic review and meta-analysis. PLoS Med. 2010;7:e1000218. doi: 10.1371/journal.pmed.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen S, McGregor IA, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. 1961;192:733–7. doi: 10.1038/192733a0. [DOI] [PubMed] [Google Scholar]
- 5.Sabchareon A, Burnouf T, Ouattara D, et al. Parasitologic and clinical human response to immunoglobulin administration in falciparum malaria. Am J Trop Med Hyg. 1991;45:297–308. doi: 10.4269/ajtmh.1991.45.297. [DOI] [PubMed] [Google Scholar]
- 6.Greenhouse B, Slater M, Njama-Meya D, et al. Decreasing efficacy of antimalarial combination therapy in Uganda is explained by decreasing host immunity rather than increasing drug resistance. J Infect Dis. 2009;199:758–65. doi: 10.1086/596741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.John CC, Tande AJ, Moormann AM, et al. Antibodies to pre-erythrocytic Plasmodium falciparum antigens and risk of clinical malaria in Kenyan children. J Infect Dis. 2008;197:519–26. doi: 10.1086/526787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langhorne J, Ndungu FM, Sponaas AM, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. 2008;9:725–32. doi: 10.1038/ni.f.205. [DOI] [PubMed] [Google Scholar]
- 9.Roestenberg M, McCall M, Hopman J, et al. Protection against a malaria challenge by sporozoite inoculation. N Engl J Med. 2009;361:468–77. doi: 10.1056/NEJMoa0805832. [DOI] [PubMed] [Google Scholar]
- 10.Pombo DJ, Lawrence G, Hirunpetcharat C, et al. Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet. 2002;360:610–7. doi: 10.1016/S0140-6736(02)09784-2. [DOI] [PubMed] [Google Scholar]
- 11.Bejon P, Mwacharo J, Kai O, et al. The induction and persistence of T cell IFN-gamma responses after vaccination or natural exposure is suppressed by Plasmodium falciparum. J Immunol. 2007;179:4193–201. doi: 10.4049/jimmunol.179.6.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCall MB, Sauerwein RW. Interferon-gamma–central mediator of protective immune responses against the pre-erythrocytic and blood stage of malaria. J Leukoc Biol. 2010;88:1131–43. doi: 10.1189/jlb.0310137. [DOI] [PubMed] [Google Scholar]
- 13.Struik SS, Riley EM. Does malaria suffer from lack of memory? Immunol Rev. 2004;201:268–90. doi: 10.1111/j.0105-2896.2004.00181.x. [DOI] [PubMed] [Google Scholar]
- 14.Genton B. Malaria vaccines: a toy for travelers or a tool for eradication? Expert Rev Vaccines. 2008;7:597–611. doi: 10.1586/14760584.7.5.597. [DOI] [PubMed] [Google Scholar]
- 15.Holder AA. The carboxy-terminus of merozoite surface protein 1: structure, specific antibodies and immunity to malaria. Parasitology. 2009;136:1445–56. doi: 10.1017/S0031182009990515. [DOI] [PubMed] [Google Scholar]
- 16.Ogutu BR, Apollo OJ, McKinney D, et al. Blood stage malaria vaccine eliciting high antigen-specific antibody concentrations confers no protection to young children in Western Kenya. PLoS One. 2009;4:e4708. doi: 10.1371/journal.pone.0004708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheehy SH, Duncan CJ, Elias SC, et al. Phase Ia clinical evaluation of the Plasmodium falciparum blood-stage antigen MSP1 in ChAd63 and MVA vaccine vectors. Mol Ther. 2011;19:2269–76. doi: 10.1038/mt.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Cassan SC, Forbes EK, Douglas AD, et al. The requirement for potent adjuvants to enhance the immunogenicity and protective efficacy of protein vaccines can be overcome by prior immunization with a recombinant adenovirus. J Immunol. 2011;187:2602–16. doi: 10.4049/jimmunol.1101004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian F, Reiter K, Zhang Y, et al. Immunogenicity of self-associated aggregates and chemically cross-linked conjugates of the 42 kDa Plasmodium falciparum merozoite surface protein-1. PLoS One. 2012;7:e36996. doi: 10.1371/journal.pone.0036996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh S, Miura K, Zhou H, et al. Immunity to recombinant plasmodium falciparum merozoite surface protein 1 (MSP1): protection in Aotus nancymai monkeys strongly correlates with anti-MSP1 antibody titer and in vitro parasite-inhibitory activity. Infect Immun. 2006;74:4573–80. doi: 10.1128/IAI.01679-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawabata Y, Udono H, Honma K, et al. Merozoite surface protein 1-specific immune response is protective against exoerythrocytic forms of Plasmodium yoelii. Infect Immun. 2002;70:6075–82. doi: 10.1128/IAI.70.11.6075-6082.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wipasa J, Hirunpetcharat C, Mahakunkijcharoen Y, Xu H, Elliott S, Good MF. Identification of T cell epitopes on the 33-kDa fragment of Plasmodium yoelii merozoite surface protein 1 and their antibody-independent protective role in immunity to blood stage malaria. J Immunol. 2002;169:944–51. doi: 10.4049/jimmunol.169.2.944. [DOI] [PubMed] [Google Scholar]
- 23.Keitel WA, Kester KE, Atmar RL, et al. Phase I trial of two recombinant vaccines containing the 19kd carboxy terminal fragment of Plasmodium falciparum merozoite surface protein 1 (msp-1(19)) and T helper epitopes of tetanus toxoid. Vaccine. 1999;18:531–9. doi: 10.1016/s0264-410x(99)00221-2. [DOI] [PubMed] [Google Scholar]
- 24.Pusic KM, Hashimoto CN, Lehrer A, Aniya C, Clements DE, Hui GS. T cell epitope regions of the P. falciparum MSP1-33 critically influence immune responses and in vitro efficacy of MSP1-42 vaccines. PLoS One. 2011;6:e24782. doi: 10.1371/journal.pone.0024782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiwanuka GN. Genetic diversity in Plasmodium falciparum merozoite surface protein 1 and 2 coding genes and its implications in malaria epidemiology: a review of published studies from 1997–2007. J Vector Borne Dis. 2009;46:1–12. [PubMed] [Google Scholar]
- 26.Thera MA, Doumbo OK, Coulibaly D, et al. A field trial to assess a blood-stage malaria vaccine. N Engl J Med. 2011;365:1004–13. doi: 10.1056/NEJMoa1008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qari SH, Shi YP, Goldman IF, Nahlen BL, Tibayrenc M, Lal AA. Predicted and observed alleles of Plasmodium falciparum merozoite surface protein-1 (MSP-1), a potential malaria vaccine antigen. Mol Biochem Parasitol. 1998;92:241–52. doi: 10.1016/s0166-6851(98)00010-3. [DOI] [PubMed] [Google Scholar]
- 28.Moormann AM, John CC, Sumba PO, Tisch D, Embury P, Kazura JW. Stability of interferon-gamma and interleukin-10 responses to Plasmodium falciparum liver stage antigen-1 and thrombospondin-related adhesive protein in residents of a malaria holoendemic area. Am J Trop Med Hyg. 2006;74:585–90. [PubMed] [Google Scholar]
- 29.Spring MD, Chelimo K, Tisch DJ, et al. Allele specificity of gamma interferon responses to the carboxyl-terminal region of Plasmodium falciparum merozoite surface protein 1 by Kenyan adults with naturally acquired immunity to malaria. Infect Immun. 2010;78:4431–41. doi: 10.1128/IAI.00415-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Husain SM, Kalavathi P, Anandaraj MP. Analysis of sickle cell gene using polymerase chain reaction & restriction enzyme Bsu 361. Indian J Med Res. 1995;101:273–6. [PubMed] [Google Scholar]
- 31.Bloland PB, Boriga DA, Ruebush TK, et al. Longitudinal cohort study of the epidemiology of malaria infections in an area of intense malaria transmission II. Descriptive epidemiology of malaria infection and disease among children. Am J Trop Med Hyg. 1999;60:641–8. doi: 10.4269/ajtmh.1999.60.641. [DOI] [PubMed] [Google Scholar]
- 32.Freitas do Rosario AP, Langhorne J. T cell-derived IL-10 and its impact on the regulation of host responses during malaria. Int J Parasitol. 2012;42:549–55. doi: 10.1016/j.ijpara.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Dent AE, Moormann AM, Yohn CT, et al. Broadly reactive antibodies specific for Plasmodium falciparum MSP-119 are associated with the protection of naturally exposed children against infection. Malar J. 2012;11:287. doi: 10.1186/1475-2875-11-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crompton PD, Traore B, Kayentao K, et al. Sickle cell trait is associated with a delayed onset of malaria: implications for time-to-event analysis in clinical studies of malaria. J Infect Dis. 2008;198:1265–75. doi: 10.1086/592224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moormann AM. How might infant and paediatric immune responses influence malaria vaccine efficacy? Parasite Immunol. 2009;31:547–59. doi: 10.1111/j.1365-3024.2009.01137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kinyanjui SM, Bejon P, Osier FH, Bull PC, Marsh K. What you see is not what you get: implications of the brevity of antibody responses to malaria antigens and transmission heterogeneity in longitudinal studies of malaria immunity. Malar J. 2009;8:242. doi: 10.1186/1475-2875-8-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crompton PD, Kayala MA, Traore B, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci U S A. 2010;107:6958–63. doi: 10.1073/pnas.1001323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanisic DI, Richards JS, McCallum FJ, et al. Immunoglobulin G subclass-specific responses against Plasmodium falciparum merozoite antigens are associated with control of parasitemia and protection from symptomatic illness. Infect Immun. 2009;77:1165–74. doi: 10.1128/IAI.01129-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.John CC, O'Donnell RA, Sumba PO, et al. Evidence that invasion-inhibitory antibodies specific for the 19-kDa fragment of merozoite surface protein-1 (MSP-1 19) can play a protective role against blood-stage Plasmodium falciparum infection in individuals in a malaria endemic area of Africa. J Immunol. 2004;173:666–72. doi: 10.4049/jimmunol.173.1.666. [DOI] [PubMed] [Google Scholar]
- 40.Barry AE, Trieu A, Fowkes FJ, et al. The stability and complexity of antibody responses to the major surface antigen of Plasmodium falciparum are associated with age in a malaria endemic area. Mol Cell Proteomics. 2011;10:M111 008326. doi: 10.1074/mcp.M111.008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huaman MC, Martin LB, Malkin E, et al. Ex vivo cytokine and memory T cell responses to the 42-kDa fragment of Plasmodium falciparum merozoite surface protein-1 in vaccinated volunteers. J Immunol. 2008;180:1451–61. doi: 10.4049/jimmunol.180.3.1451. [DOI] [PubMed] [Google Scholar]
- 42.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–58. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 43.Chelimo K, Embury PB, Sumba PO, et al. Age-related differences in naturally acquired T cell memory to Plasmodium falciparum merozoite surface protein 1. PLoS One. 2011;6:e24852. doi: 10.1371/journal.pone.0024852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun T, Holowka T, Song Y, et al. A Plasmodium-encoded cytokine suppresses T-cell immunity during malaria. Proc Natl Acad Sci U S A. 2012;109:E2117–26. doi: 10.1073/pnas.1206573109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riley EM, Morris-Jones S, Blackman MJ, Greenwood BM, Holder AA. A longitudinal study of naturally acquired cellular and humoral immune responses to a merozoite surface protein (MSP1) of Plasmodium falciparum in an area of seasonal malaria transmission. Parasite Immunol. 1993;15:513–24. doi: 10.1111/j.1365-3024.1993.tb00639.x. [DOI] [PubMed] [Google Scholar]
- 46.Kemp DJ. Antigenic diversity and variation in blood stages of Plasmodium falciparum. Immunol Cell Biol. 1992;70(Pt 3):201–7. doi: 10.1038/icb.1992.25. [DOI] [PubMed] [Google Scholar]
- 47.Pinkevych M, Petravic J, Chelimo K, Kazura JW, Moormann AM, Davenport MP. The dynamics of naturally acquired immunity to Plasmodium falciparum infection. PLoS Comput Biol. 2012;8:e1002729. doi: 10.1371/journal.pcbi.1002729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dent AE, Chelimo K, Sumba PO, et al. Temporal stability of naturally acquired immunity to merozoite surface protein-1 in Kenyan adults. Malar J. 2009;8:162. doi: 10.1186/1475-2875-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hui G, Hashimoto C. Plasmodium falciparum anti-MSP1–19 antibodies induced by MSP1-42 and MSP1-19 based vaccines differed in specificity and parasite growth inhibition in terms of recognition of conserved versus variant epitopes. Vaccine. 2007;25:948–56. doi: 10.1016/j.vaccine.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 50.Lyon JA, Angov E, Fay MP, et al. Protection induced by Plasmodium falciparum MSP1(42) is strain-specific, antigen and adjuvant dependent, and correlates with antibody responses. PLoS One. 2008;3:e2830. doi: 10.1371/journal.pone.0002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.