

Abstract
The mechanisms by which articular surface impact causes post-traumatic osteoarthritis are not well understood, but studies of cartilage explants implicate the mitochondrial electron transport chain as a source of oxidants that cause chondrocyte death from mechanical injury. The linkage of mitochondria to the cytoskeleton suggests that they might release oxidants in response to mechanical strain, an effect that disrupting the cytoskeleton would prevent. To test this we investigated the effects of agents that promote the dissolution of microfilaments (cytochalasin B) or microtubules (nocodazole) on oxidant production and chondrocyte death following impact injury. Osteochondral explants treated with cytochalasin B or nocodazole for 4 hours were impacted (7J/cm2) and stained for oxidant production directly after impact and for cell viability 24 hours after impact. Surfaces within and outside impact sites were then imaged by confocal microscopy. Both agents significantly reduced impact-induced oxidant release (p < 0.05); however, cytochalasin B was more effective than nocodazole (> 60% reduction versus 40% reduction respectively). Both agents also prevented impact induced cell death. Dissolution of the cytoskeleton by both drugs was confirmed by phalloidin staining and confocal microscopy. These findings show that chondrocyte mortality from impact injury depends substantially on mitochondrial-cytoskeletal linkage, suggesting new approaches to stem mechanically-induced cartilage degeneration.
Keywords: Cartilage injury, cytoskeleton, chondrocyte death, mitochondria, oxidants
Introduction
Impact injury sustained by cartilage, typically resulting from an intraarticular fracture, leads to chondrocyte metabolic dysfunction and death.1, 2 It is hypothesized that injury-related chondrocyte death plays a primary role in progression to posttraumatic osteoarthritis (PTOA).3–5 Therefore, deciphering the pathologic mechanobiologic link between impact injury and chondrocyte death will provide foundational information to develop new treatments to prevent PTOA.
Chondrocyte death after an articular surface impact occurs by both necrosis and apoptosis.4–8 Recent evidence suggests that mitochondria play a central pathoetiologic role in both necrotic and apoptotic chondrocyte death pathways.4, 5 Martin and colleagues demonstrated that in bovine osteochondral explants subjected to a high energy impact injury, most chondrocyte death was necrotic and occurred in the first 12 hours after impact. Subsequently, Goodwin and colleagues showed that the injury-associated reactive oxygen species (ROS) were mitochondrial in origin, and by blocking mitochondrial electron transport (ET), the injury-related production of ROS was reduced and chondrocyte viability was increased.6 Furthermore, chondrocyte death was mitigated by treating impacted specimens with N-acetylcysteine (NAC), a potent scavenger of ROS.5 Others have shown that chondrocyte death continues for days after an impact injury, presumably as a result of secondary apoptosis. Factors that promote apoptosis, including caspases, are associated with mitochondria and caspase inhibition reduces death in injured cartilage.4, 7 Current evidence strongly suggests that impact injury to cartilage causes mitochondrial dysfunction which leads to chondrocyte necrosis, apoptosis, or both. Therefore, understanding how impact injury induces mitochondrial dysfunction leading to chondrocyte death would provide important foundational information to prevent impact-related PTOA.
The physical structure of the chondrocyte is maintained by the intracellular cytoskeleton consisting of microfilaments, intermediate filaments, and microtubules.8–10 Microfilaments or filamentous actin, f-actin, primarily resist compressive forces applied to the cell.11 Furthermore, filamentous actin has been shown to be highly concentrated around the cell periphery, thereby maintaining the chondrocyte’s shape and structure, and facilitating attachment of the chondrocyte to the surrounding extracellular matrix.10 F-actin assembly is a highly dynamic process. A readily available pool of globular actin is rapidly assembled into structural f-actin in response to changes in the local mechanical environment.9, 12, 13 Microtubules have been shown to provide secondary structural support to the f-actin cytoskeleton which is significantly stiffer and less brittle under loading with an intact microtubular lattice.11, 17
F-actin also serves as an important scaffold for mitochondrial attachment.14 Studies have depicted an elaborate mechanism that binds mitochondria to the f-actin cytoskeleton facilitating optimal mitochondrial distribution within the cell and also allowing local intracellular mitochondrial movement.14, 15 This physical attachment of the mitochondria to the cytoskeleton potentially subjects the mitochondria to external loads encountered by the chondrocyte. Tissue level strains are transduced into cellular level deformations, including deformation of the intracellular mitochondria, primarily via the actin cytoskeleton with support from the microtubules.
Following on this concept, transient high-magnitude pathologic strains encountered during an impact injury would be transmitted to the mitochondria via the intracellular cytoskeleton. Therefore, it is possible that impact-related mitochondrial dysfunction is enacted by the physical connection between the mitochondria and the intracellular cytoskeleton.
In this study, we tested the hypothesis that impact-related mitochondrial dysfunction leading to chondrocyte death could be mitigated by dissociating the intracellular cytoskeleton prior to an impact injury. Experimental specimens were treated with cytochalasin B, a compound that disrupts f-actin, or nocodazole, a compound that dissociates the microtubule cytoskeleton, prior to impact. Specimens were then subjected to injury using a drop tower apparatus. Subsequently, chondrocyte production of ROS was measured half an hour after impact, and chondrocyte viability was determined 24 hours post-impact using confocal microscopy.
Methods
Mature bovine stifle joints were obtained after slaughter from a local abattoir. Osteochondral explants (2.5 cm by 2.5 cm by 1.0 cm) were cut from the center of the lateral tibial plateau, primarily encompassing the area uncovered by the meniscus. Explants were maintained overnight at 37°C in culture medium containing 45% DMEM (Dulbecco Modified Eagle Medium), 45% Ham’s F-12, and 10% fetal bovine serum (FBS) (Invitrogen) under low oxygen conditions (5% O2, 5% CO2). The next day, explants were transferred to low O2 equilibrated Phenol Red-Free culture medium (10% FBS, 1:1 DMEM/F12 (Ham) 1x) and incubated overnight under the same low O2 conditions.
Prior to impact testing, experimental explants were treated in Phenol Red-Free culture medium with 20 μM cytochalasin B (Calbiochem) (n = 8) or 10 μM nocodazole (Sigma) (n = 8) for four hours under low O2 conditions. Untreated explants (n = 7) were maintained in low O2 media until impact. Explants were secured in a custom testing fixture for impact loading and covered with low O2 equilibrated culture medium. Explants were then subjected to a 7 J/cm2 impact using a drop tower apparatus as previously described5, 6. In brief, a 2 kg mass was dropped from a height of 7 cm onto a brass rod (5.0 mm diameter) that was in contact with the explant surface, resulting in nominal impact energy of 7J/cm2 (2kg × 9.8m/s2 × 0.07m/0.2cm2). The mass was allowed to bounce and come to rest before removal. This consistently caused superficial-transitional zone matrix cracks and superficial chondrocyte mortality (64% +/− 7% standard deviation). Immediately after impact, explants were fixed securely to a plastic imaging plate using polycaprolactone (PCL) (Aldrich) to facilitate image analysis.
To determine how pre-impact cytoskeletal dissolution affected ROS production, experimental and untreated explants were placed in Phenol Red-Free culture medium containing 5 μM dihydroethidium (DHE) (Invitrogen), a superoxide probe, and 1 mM Calcein AM, a live cell probe, at the previously specified low O2 conditions for 30 minutes immediately after impact and placement on the plastic plate. After staining, explants were secured to a custom x-y imaging table specifically designed to position explants under our confocal microscope. The custom imaging table allows specimens to remain submerged in low O2 equilibrated DMEM/F12 (Invitrogen) media. Explants were imaged on a BioRad 1024 Confocal Microscope equipped with a Krypton/Argon laser. We analyzed three sites, 0.5 mm in diameter, within the impact region and three similar control sites 5.0 mm outside of the impact zone. Images at each site were captured using wavelengths of 568 nm and 488 nm, starting at the cartilage surface and imaging to a depth of about 140 μm at intervals of 20 μm. Numbers of DHE positive cells and Calcein AM positive cells in each slice of the z-axis stack were analyzed using ImageJ (rsb.info.nih.gov/ij) to determine percentage positive values for ROS producing cells. After imaging for ROS production, explants were placed in fresh, low O2 equilibrated Phenol Red-Free culture medium and incubated overnight in low O2 conditions. The data are reported as means and standard deviations.
To determine the effects of pre-impact cytoskeletal dissolution on chondrocyte viability, untreated and experimental specimens were stained with Calcein AM and ethidium homodimer-2 (EHD), a dead cell probe, in low O2 equilibrated Phenol Red-Free culture medium for 30 minutes prior to imaging. Explants were imaged on our x-y imaging table with the confocal microscope as described above. Identical z-axis stacks of live and dead cells were generated at the same three impact and control sites. The data are reported as means and standard deviations.
Three medial osteochondral explants were obtained and cultured as reported above. Explants were either treated with cytochalasin B for four hours, nocodazole for four hours, or were not treated. Immediately after treatment, explants were cryo-imbedded. Ten micron thick sections were cut for imaging f-actin. Sections were fixed with 4% paraformaldehyde in 1x PBS, 0.1% Triton-X-100 for five minutes. They were then rinsed two times with 1x PBS. Sections were stained with a 1:40 dilution of phalloidin, an f-actin stain, in 1% bovine serum albumin in PBS and incubated at 37°C for 30 minutes. Sections were then stained with DAPI and a coverslip was placed on the slides. Images were taken using a Zeiss 710 confocal microscope at 20x and 63x magnification.
ROS production and viability data were pooled for the impact and control sites within their respective experimental groups. The effects of treatments on ROS production and on viability were evaluated using Kruskal-Wallis One Way ANOVA on Ranks and Dunn’s method for post-hoc testing.
Results
Confocal images of chondrocytes within impact sites and control sites away from the impact injury were analyzed using ImageJ software to determine the percent of DHE positive cells present directly after impact and the percent of viable cells 24 hours after impact injury. In general, DHE stained cells were more evident in the impact sites of untreated specimens than in the impact sites of the two treatment groups (Figure 1). Also, confocal images showed that viability was greater in the impact sites of treated specimens than in the impact sites of untreated specimens.
Figure 1.
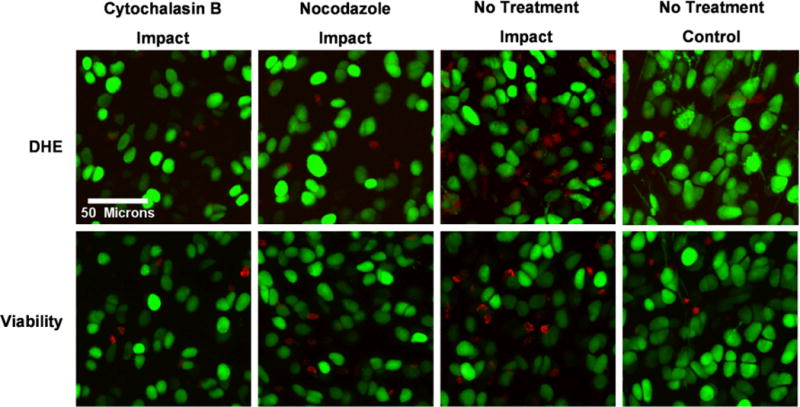
Representative confocal images of ROS production and chondrocyte viability in specimens treated with cytoskeletal disruptors prior to impact. ROS producing chondrocytes (cells fluorescing red in the upper row of images) are detected in the greatest concentration in untreated impacted cartilage compared to impacted cartilage that was treated with cytoskeletal disruptors prior to impact. Chondrocyte viability (cells fluorescing green in the bottom row of images) was greater in impacted cartilage in treated specimens compared to impacted cartilage in untreated specimens.
After blunt impact, impact sites of the cytochalasin B and nocodazole treated explants produced significantly less ROS (19.7% +/− 13.2% and 24.9 +/− 14.9% positive, respectively) than impact sites of untreated explants (42.4% +/− 21.5% positive) (p < 0.05) (Figure 2). The only significant difference between impact and control sites within groups was in the untreated group (p < 0.05). Also, there was no significant difference in ROS production between the control groups (p = 0.237).
Figure 2.
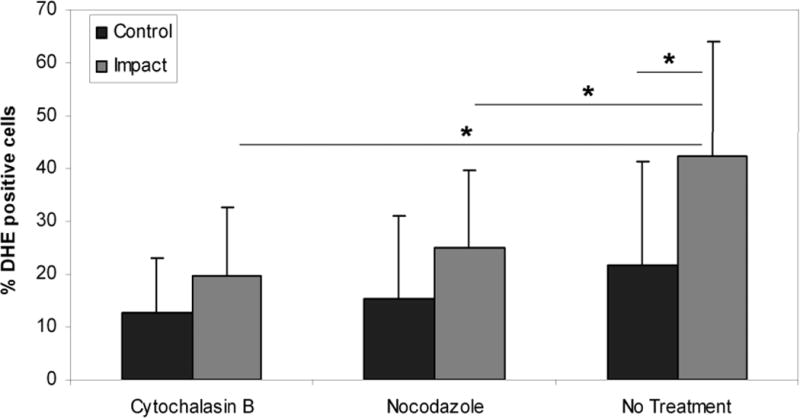
Percentages of ROS-producing chondrocytes in impacted and peripheral cartilage immediately after impact in control specimens and specimens treated with cytoskeletal disruptors prior to impact. Cytoskeletal dissolution significantly decreased ROS production in the impacted cartilage in experimental specimens compared to untreated control specimens. Columns and error bars represent means and standard deviations. Asterisks indicate significant differences (p < 0.05)
Viability 24 hours after impact injury showed significantly higher viability in the impact sites of the cytochalasin B (80.0% +/− 11.6%) and nocodazole (79.9% +/− 13.5%) treatment groups than in the untreated specimens (60.8% +/− 17.3%) (p < 0.001 and p < 0.001, respectively) (Figure 3). There was no significant difference between the impact and control sites within the two treatment groups (p > 0.05 for both groups) but there was a difference within the untreated group (p < 0.05). Twenty-four hour viability in the control sites between groups ranged from 86.9% to 92.5% and showed no significant difference between groups (p = 0.387).
Figure 3.
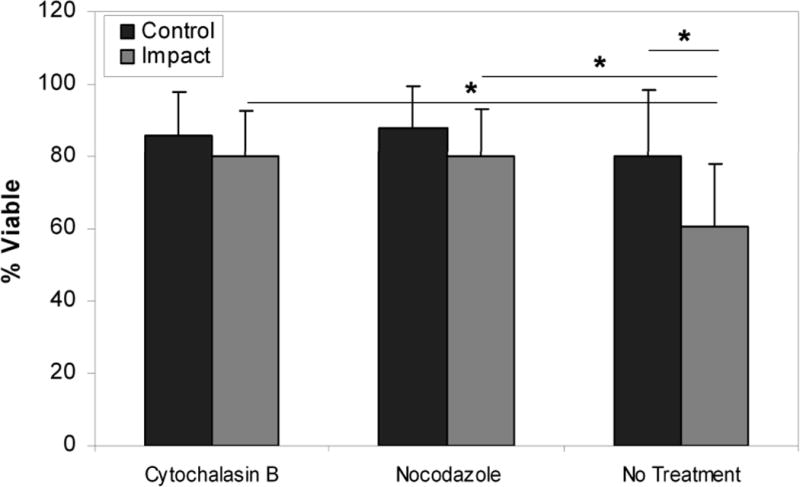
Chondrocyte viability in impacted and peripheral cartilage 24 hours after impact. Chondrocyte viability decreased significantly in impacted cartilage in untreated specimens compared to impacted cartilage treated with cytoskeletal dissolution prior to impact, and compared to peripheral unimpacted cartilage in all groups. Minimal differences in viability were detected in impacted cartilage compared to peripheral unimpacted cartilage in experimental groups. Columns and error bars represent means and standard deviations. Asterisks indicate significant differences (p < 0.05)
Treatment of explants with either cytochalasin B or nocodazole altered phalloidin staining patterns in superficial zone chondrocytes (Figure 4). Staining was much less intense in cells at the cartilage surface in treated explants versus an untreated control. Moreover, treatment caused a change in the intracellular distribution of the stain, which was diffuse in appearance in control cells, but highly granular in treated explants.
Figure 4.
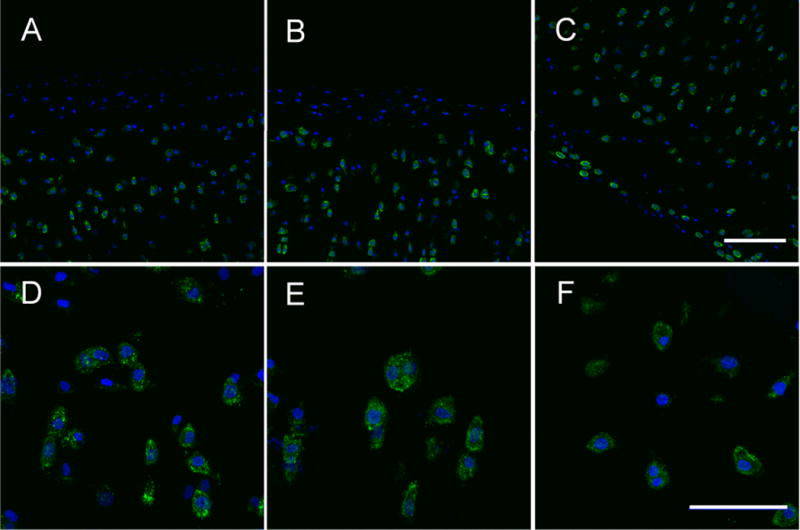
Effects of cytochalasinB and nocodazole on chondrocyte cytoskeleton. Confocal images showing phalloidin stained f-actin (green) and DAPI stained (blue) bovine cartilage of cytochalasinB (A,D), nocodazole (B,E), and untreated (C,F) specimens. CytochalasinB and nocodazole groups show dissociated f-actin in the superficial zone whereas the untreated group has intact f-actin. Scale bars in C and F are 100 and 50 microns, respectively.
Discussion
The results of this study show that dissolution of the chondrocyte cytoskeleton prior to an impact injury significantly reduced impact-related release of ROS and chondrocyte death. Excessive impact-related ROS is likely to be causally related to chondrocyte death. Previous studies have shown that scavenging excessive ROS after an impact injury to bovine osteochondral specimens reduced impact-related chondrocyte death.5 Furthermore, it was shown in this same line of investigation that the chondrocyte mitochondria were the source of impact-related release of ROS.6 These previous observations, coupled with the current experimental findings, suggest that dissolution of the chondrocyte cytoskeleton prior to an impact injury prevented excessive mitochondrial production of ROS which in turn prevented necrotic chondrocyte death. These data also strongly suggest that mitochondria play a central mechanobiologic role in the initiation of chondrocyte death after an impact injury.
Treatment of osteochondral explants with either cytochalasin B or nocodazole clearly dissociated chondrocytes’ cytoskeletons as evidenced by the confocal images in Figure 4. Even though nocodazole does not depolymerize f-actin, the nocodazole treated specimen showed dissociated f-actin in the superficial zone because the microtubule network is depolymerized; therefore, the tensegrity of the chondrocytes’ entire cytoskeleton is compromised. Also, images show a definite boundary between intact and compromised f-actin networks which may be a result of the drug penetration into the explants. The apparent depth of penetration was ~150 microns, which was comparable depth to confocal scans (~150–200 microns).
The chondrocyte cytoskeleton performs multiple functions. Actin microfilaments, which are found throughout the entire chondrocyte, are the stiffest component of the cytoskeleton and dissolution of the actin cytoskeleton has been shown to have the greatest effect on the determination of cell stiffness in both chondrocytes and endothelial cells.10, 16 Actin microfilaments are particularly concentrated in the chondrocyte periphery, playing a primary role in anchoring the chondrocyte to the surrounding cartilage matrix via integrin proteins.4, 10, 13, 16 Therefore, f-actin plays an important, potentially dominant, mechanical role in transducing tissue-level forces applied to the cartilage matrix to those encountered by intracellular organelles and the nucleus at the cellular-level. The f-actin of the cytoskeleton is in dynamic flux and responds rapidly to changes in the mechanical environment by both synthesis and dissolution within minutes of changes in stress.9, 10, 12, 13, 16 Mitochondria are firmly attached to f-actin throughout the cell.14 These cytoskeletal attachments afford the mitochondria mobility to move within the cytoplasm and stability to anchor in regions of high energy demands. It has been shown that dissolution of actin microfilaments by cytochalasin A stops mitochondrial movement, detaches mitochondria from actin, and results in substantial changes in mitochondrial shape.15 It has also been shown that cytoskeletal dissolution with both cytochalasin D and nocodazole eliminated mitochondrial deformation under load showing that mitochondrial deformation is linked to an intact cytoskeleton.17 Taken together, there is strong rationale to support the hypothesis that tissue-level stresses in cartilage are transduced to intracellular mitochondrial strain via the cytoskeleton, and disruption of microfilaments and microtubules affects mitochondrial deformation under loading. It is plausible that pre-impact cytoskeletal dissolution prevented excessive intracellular ROS by an unknown extra-mitochondrial mechanism we did not measure. However, it is more likely that pre-impact cytoskeletal dissolution physically uncoupled the mitochondria from the cytoskeleton, shielding the mitochondria from pathologic impact strains, thereby reducing the expected mitochondrial ROS hyperproduction associated with impact injury. The decreases in ROS production led to increases in chondrocyte viability 24 hours after impact.
The findings in our study compare favorably to a similar study conducted using cultured human umbilical vein endothelial cells.18 In this study, endothelial cells cultured on plates were subjected to cyclical strain and were treated with inhibitors of electron transport, inhibitors of extra-mitochondrial sources of ROS, and with cytochalasinD and nocodazole. In this study, ROS production increased by greater than 100% in strained cells with an intact cytoskeleton. In cells treated with rotenone which blocked mitochondrial electron transport, this strain-induced increase in ROS production was eliminated. In contrast, in cells treated with apocynin and allopurinol, inhibitors of extra-mitochondrial sources of ROS, the strain-induced ROS increases were unaffected. The investigators concluded that strain-induced increases in ROS production were primarily mitochondrial in origin. Subsequently, cells that were pretreated with cytochalasin D did not increase strain-induced ROS production. The investigators concluded that repetitive strain led to increase production of ROS that came from the mitochondria. Furthermore, severing mitochondrial attachments to the actin cytoskeleton prevented strain from inciting mitochondrial ROS production.
The central role that mitochondria play in causing cell death secondary to injury has been studied in multiple organ systems including traumatic brain injury, brain anoxia, cardiac injury, polytrauma, sepsis, and endothelial cellular injury.19–22 Consistent evidence has shown that mitochondrial depolarization leads to increased ROS concentrations and cell death. Conversely, blocking mitochondrial depolarization can prevent apoptotic and necrotic cell death.23, 24 The role of mitochondria in chondrocyte function is just beginning to be understood. In two separate experiments, chondrocyte cultures were stimulated to increase mitochondrial production of ROS. In both experiments, the elevated mitochondrial ROS concentrations decreased chondrocyte viability.25, 26 In a series of experiments, it was determined that impact injury to cartilage explants caused immediate increase in production of ROS and scavenging the impact-related ROS significantly reduced chondrocyte death.5 The impact-associated increases in ROS were eliminated by blocking mitochondrial electron transport at Complex I which led to increased chondrocyte viability after impact.6 Evidence is clearly mounting that mitochondria play a central role in chondrocyte death after cartilage impact injury. This experiment demonstrates a plausible physical link between injurious impact stresses and the intracellular biochemical response leading to cell death. The findings are potentially important for conceiving therapeutic interventions to preserve chondrocyte viability after an impact injury.
The confocal analyses can effectively image cells in the superficial 150 to 200 microns of cartilage, but cannot image cells in the deep zone of the cartilage. Therefore, the analyses in this experiment are limited to the superficial layer and the upper transitional layer of the cartilage. However, several investigators have shown that the majority of cell death occurs in the superficial layer and adjacent to matrix cracks after an impact injury, so it is likely that the techniques used in this study represent a valid population of chondrocytes at risk.4, 27, 28 Local chondrocyte density within specimens varies enough that it can obscure experimental effects when making comparisons between different sites. Thus, we wanted to keep our images in the same place so that the density of cells in the DHE imaging session was not different from that of the viability imaging session. This was achieved with ~10 micron precision using the x-y imaging table and the plastic imaging plates, which kept the explants in a constant orientation for spatial registration.
Several investigators have shown that in injured cartilage, apoptotic chondrocyte death likely begins one to two days after injury and can progress over seven to fourteen days.4, 7, 29, 30 Therefore, it is unknown if cytochalasin B treatment would have prevented, or potentially caused, apoptotic cell death. It is possible that cytochalasin B had a more direct effect on mitochondrial function which was independent of disrupting the actin cytoskeleton. Mitochondrial morphology was significantly altered and mitochondrial motility ceased in fungi cells treated with cytochalasin A which the authors thought was most likely secondary to disruption of the actin cytoskeleton but could not exclude a direct effect of cytochalasin B on mitochondrial function.15 We did not specifically exclude the potential that extra-mitochondrial sources of ROS were produced by the impact injury. However, our previous investigations demonstrated that impact-related increases in ROS were blocked by stopping mitochondrial electron transport, making it likely that extra-mitochondrial sources of ROS production are minimal.
In conclusion, cartilage explants treated with agents that promote cytoskeletal dissolution prior to impact had greater chondrocyte viability 24 hours after injury compared to impacted specimens that were untreated. It is likely that cytoskeletal dissolution reduces impact-related chondrocyte death by decreasing ROS production by chondrocyte mitochondria. These findings support further investigation into the molecular pathways of impact-related chondrocyte death which could be important in developing treatment strategies that could prevent PTOA.
Acknowledgments
This work was supported by the National Institutes of Health (CORT NIH P50 AR055533), and by a Merit Review Award from the Department of Veterans Affaires.
References
- 1.Borrelli J, Jr, Silva MJ, Zaegel MA, et al. Single high-energy impact load causes posttraumatic OA in young rabbits via a decrease in cellular metabolism. J Orthop Res. 2009;27:347–352. doi: 10.1002/jor.20760. [DOI] [PubMed] [Google Scholar]
- 2.Phillips DM, Haut RC. The use of a non-ionic surfactant (P188) to save chondrocytes from necrosis following impact loading of chondral explants. J Orthop Res. 2004;22:1135–1142. doi: 10.1016/j.orthres.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Clements KM, Burton-Wurster N, Lust G. The spread of cell death from impact damaged cartilage: lack of evidence for the role of nitric oxide and caspases. Osteoarthritis Cartilage. 2004;12:577–585. doi: 10.1016/j.joca.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 4.D’Lima DD, Hashimoto S, Chen PC, et al. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthritis Cartilage. 2001;9:712–719. doi: 10.1053/joca.2001.0468. [DOI] [PubMed] [Google Scholar]
- 5.Martin JA, McCabe D, Walter M, et al. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. J Bone Joint Surg Am. 2009;91:1890–1897. doi: 10.2106/JBJS.H.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodwin W, McCabe D, Sauter E, et al. Rotenone prevents impact-induced chondrocyte death. J Orthop Res. 2010;28:1057–1063. doi: 10.1002/jor.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.PascualGarrido C, Hakimiyan AA, Rappoport L, et al. Anti-apoptotic treatments prevent cartilage degradation after acute trauma to human ankle cartilage. Osteoarthritis Cartilage. 2009;17:1244–1251. doi: 10.1016/j.joca.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guilak F. Compression-induced changes in the shape and volume of the chondrocyte nucleus. J Biomech. 1995;28:1529–1541. doi: 10.1016/0021-9290(95)00100-x. [DOI] [PubMed] [Google Scholar]
- 9.Knight MM, Toyoda T, Lee DA, et al. Mechanical compression and hydrostatic pressure induce reversible changes in actin cytoskeletal organisation in chondrocytes in agarose. J Biomech. 2006;39:1547–1551. doi: 10.1016/j.jbiomech.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Trickey WR, Vail TP, Guilak F. The role of the cytoskeleton in the viscoelastic properties of human articular chondrocytes. J Orthop Res. 2004;22:131–139. doi: 10.1016/S0736-0266(03)00150-5. [DOI] [PubMed] [Google Scholar]
- 11.Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A. 1997;94:849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durrant LA, Archer CW, Benjamin M, et al. Organisation of the chondrocyte cytoskeleton and its response to changing mechanical conditions in organ culture. J Anat. 1999;194(Pt 3):343–353. doi: 10.1046/j.1469-7580.1999.19430343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haudenschild DR, D’Lima DD, Lotz MK. Dynamic compression of chondrocytes induces a Rho kinase-dependent reorganization of the actin cytoskeleton. Biorheology. 2008;45:219–228. [PubMed] [Google Scholar]
- 14.Boldogh IR, Nowakowski DW, Yang HC, et al. A protein complex containing Mdm10p, Mdm12p, and Mmm1p links mitochondrial membranes and DNA to the cytoskeleton-based segregation machinery. Mol Biol Cell. 2003;14:4618–4627. doi: 10.1091/mbc.E03-04-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suelmann R, Fischer R. Mitochondrial movement and morphology depend on an intact actin cytoskeleton in Aspergillus nidulans. Cell Motil Cytoskeleton. 2000;45:42–50. doi: 10.1002/(SICI)1097-0169(200001)45:1<42::AID-CM4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 16.Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 17.Ohashi T, Hagiwara M, Bader DL, et al. Intracellular mechanics and mechanotransduction associated with chondrocyte deformation during pipette aspiration. Biorheology. 2006;43:201–214. [PubMed] [Google Scholar]
- 18.Ali MH, Pearlstein DP, Mathieu CE, et al. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L486–496. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- 19.Gilmer LK, Roberts KN, Joy K, et al. Early mitochondrial dysfunction after cortical contusion injury. J Neurotrauma. 2009;26:1271–1280. doi: 10.1089/neu.2008.0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazzeo AT, Beat A, Singh A, et al. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol. 2009;218:363–370. doi: 10.1016/j.expneurol.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 24.Lee I, Bender E, Arnold S, et al. New control of mitochondrial membrane potential and ROS formation–a hypothesis. Biol Chem. 2001;382:1629–1636. doi: 10.1515/BC.2001.198. [DOI] [PubMed] [Google Scholar]
- 25.Grishko V, Xu M, Ho R, et al. Effects of hyaluronic acid on mitochondrial function and mitochondria-driven apoptosis following oxidative stress in human chondrocytes. J Biol Chem. 2009;284:9132–9139. doi: 10.1074/jbc.M804178200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu GJ, Chen TG, Chang HC, et al. Nitric oxide from both exogenous and endogenous sources activates mitochondria-dependent events and induces insults to human chondrocytes. J Cell Biochem. 2007;101:1520–1531. doi: 10.1002/jcb.21268. [DOI] [PubMed] [Google Scholar]
- 27.Lewis JL, Deloria LB, Oyen-Tiesma M, et al. Cell death after cartilage impact occurs around matrix cracks. J Orthop Res. 2003;21:881–887. doi: 10.1016/S0736-0266(03)00039-1. [DOI] [PubMed] [Google Scholar]
- 28.Milentijevic D, Helfet DL, Torzilli PA. Influence of stress magnitude on water loss and chondrocyte viability in impacted articular cartilage. J Biomech Eng. 2003;125:594–601. doi: 10.1115/1.1610021. [DOI] [PubMed] [Google Scholar]
- 29.Baars DC, Rundell SA, Haut RC. Treatment with the non-ionic surfactant poloxamer P188 reduces DNA fragmentation in cells from bovine chondral explants exposed to injurious unconfined compression. Biomech Model Mechanobiol. 2006;5:133–139. doi: 10.1007/s10237-006-0024-3. [DOI] [PubMed] [Google Scholar]
- 30.Hurtig M, Chubinskaya S, Dickey J, et al. BMP-7 protects against progression of cartilage degeneration after impact injury. J Orthop Res. 2009;27:602–611. doi: 10.1002/jor.20787. [DOI] [PubMed] [Google Scholar]