

Abstract

X-ray structures of several ternary substrate and product complexes of the catalytic subunit of cAMP-dependent protein kinase (PKAc) have been determined with different bound metal ions. In the PKAc complexes, Mg2+, Ca2+, Sr2+, and Ba2+ metal ions could bind to the active site and facilitate the phosphoryl transfer reaction. ATP and a substrate peptide (SP20) were modified, and the reaction products ADP and the phosphorylated peptide were found trapped in the enzyme active site. Finally, we determined the structure of a pseudo-Michaelis complex containing Mg2+, nonhydrolyzable AMP-PCP (β,γ-methyleneadenosine 5′-triphosphate) and SP20. The product structures together with the pseudo-Michaelis complex provide snapshots of different stages of the phosphorylation reaction. Comparison of these structures reveals conformational, coordination, and hydrogen bonding changes that might occur during the reaction and shed new light on its mechanism, roles of metals, and active site residues.
Phosphorylation of proteins is an essential regulatory process in biology. Activating kinase enzymes deliver the γ-phosphoryl group from an ATP cofactor to the hydroxyl group of a substrate protein’s serine, threonine, or tyrosine side chain to produce a phosphomonoester. Because kinases affect a variety of physiological responses and are associated with many diseases, their structures have been studied extensively. Among the diverse Ser/Thr protein kinase family, protein kinase A (PKA) is most studied and has become a paradigm for the whole class of kinase enzymes.1,2 When inactive, PKA is an R2C2 holoenzyme tetramer of two catalytic (C) and two regulatory (R) subunits. When the cAMP concentration is increased following β-adrenergic stimulation, PKA undergoes activation through binding of four cAMP molecules to the R subunits, and subsequent dissociation of the holoenzyme and release of the active C subunits (PKAc) for phosphorylation.3 This activation process and the subsequent phosphorylation process are highly regulated and are still not completely understood. PKAc folds into a bean-shaped structure, with two (small and large) lobes producing a cleft at the base between the lobes for ATP and a ledge for the protein substrate binding. The small lobe also contains the glycine-rich loop, a hairpin structure that spans residues 49–57, that comprises three Gly residues and acts as a lid for the catalytic site cleft.
Experimental and theoretical studies of the binding and dynamic events involved in the phosphoryl transfer reaction catalyzed by PKAc point to a precisely controlled dynamic switching process. Kinetic studies revealed that large conformational changes in PKAc are responsible for the slow rate of turnover,4−6 while the chemical step is at least an order of magnitude faster.7 The apo form (without ATP or substrate bound) shows an open conformation that partially closes when ATP binds to give a binary PKAc–ATP complex with an active site that is primed for substrate binding. It has been reported that one or two divalent metals are required for ATP binding and catalysis, with Mg2+ believed to be the physiologically relevant metal.8,9 The chemical reaction happens only in the fully closed conformation of PKAc after the protein or peptide substrate binds to produce a ternary complex.10,11 At physiological Mg2+ concentrations, the slow PKAc domain movements associated with binding of ATP and release of ADP each partially determine the catalytic rate. At higher metal concentrations, the removal of ADP is rate-limiting.5
At the atomic level of chemical catalysis, exact details of the phosphoryl group transfer remain uncertain. Theoretical, crystallographic, nuclear magnetic resonance, and solution stereochemistry studies suggest that the transfer proceeds through a concerted SN2 mechanism12−16 with a loose transition state (which is, however, mistakenly termed the dissociative mechanism in the literature!). However, a dissociative SN1 mechanism has also been proposed on the basis of a quantum mechanics/molecular mechanics study,17 and this possibility cannot be ruled out without observation of a stable metaphosphate intermediate. Further uncertainty surrounds the roles of active site residues and also the details of hydrogen transfer pathways among Ser/Thr substrate side chain groups, ATP phosphates, and PKAc. In particular, different functional roles have been proposed for the catalytically important and invariant residues Asp166 and Lys168.18−24 Our recent crystallographic studies of PKAc in complex with ATP and the 20-residue peptidic inhibitor (IP20) at high Mg2+ concentrations, called PKAc–Mg2ATP–IP20 in the text, have also challenged assumptions about the exact role of metal ions in catalysis.25
We present here crystallographic structures of various complexes of PKAc that provide new information about the detailed chemistry of the catalytic mechanism. The structures were determined at 100 K (LT) to maximize resolution, but representative structures were also determined at room temperature (RT, 293 K) to verify that conformational differences were not caused by temperature effects and that the structures are physiologically relevant. First, we determined the structure of a pseudo-Michaelis complex containing Mg2+, nonhydrolyzable AMP-PCP (β,γ-methyleneadenosine 5′-triphosphate), and a 20-residue pseudo-substrate peptide SP20 (PKAc–Mg2AMP-PCP–SP20) at 100 K. Next, we determined the structures of PKAc in complex with different divalent metals, ATP, and SP20. We found that all Mg2+, Ca2+, Sr2+, and Ba2+ alkaline earth metals bound in the active site along with trapped products of the phosphoryl transfer reaction, ADP, and the SP20 peptide phosphorylated at Ser21SP20 (pSP20). These structures are denoted in the text as PKAc–M2ADP–pSP20 (where M is either Mg2+, Ca2+, Sr2+, or Ba2+). These structures suggest that the currently held notion that most metals (apart from Mg2+) do not support phosphoryl transfer and do not allow ATP to bind in the active site requires revision. However, establishing the physiological relevance of metals other than Mg2+ will require solution studies under physiological conditions. Finally, the two pseudo-Michaelis complexes PKAc–Mg2AMP-PCP–SP20, newly determined here, and PKAc–Mg2ATP–IP20, previously determined,25 together with the PKAc–Mg2ADP–pSP20 product complex, provide snapshots of the PKAc active site at different stages of the phosphoryl transfer reaction. Comparison of these structures reveals conformational, coordination, and hydrogen bonding changes that might occur during the chemical reaction and might shed new light on the mechanism of the chemical step, in particular the roles of metals and active site residues Asp166 and Lys168 during the reaction, and the conformation of the Ser21SP20 side chain in the pseudo-Michaelis and product complexes.
Materials and Methods
General Information
Pseudo-substrate peptide SP20 (TTYADFIASGRTGRRASIHD; residues 5–24 of the heat-stable PKAc inhibitor PKI, where positions 20 and 21 have been mutated to Ala and Ser, respectively) was custom-synthesized by and purchased from Biomatik (Wilmington, DE). ATP as the magnesium or disodium salt and AMP-PCP as the disodium salt were purchased from Sigma-Aldrich (St. Louis, MO). Protein purification supplies were purchased from GE Healthcare (Piscataway, NJ). Crystallization reagents were purchased from Hampton Research (Aliso Viejo, CA).
Protein Expression and Purification
His6-tagged recombinant mouse PKAc was expressed in Escherichia coli using LB or minimal medium at 24 °C for 16–18 h. The recombinant enzyme was purified by affinity chromatography using HisTrap fast-flow chromatography columns supplied by GE Healthcare. The enzyme was then buffer-exchanged with 50 mM MES, 250 mM NaCl, and 2 mM DTT (pH 6.5) on a desalting column. Isoforms of PKAc that represent different phosphorylated states of the enzyme were not separated, without any obvious effect on crystallization of the ternary complexes.
Crystallization and Data Collection
For crystallization trials, PKAc was concentrated to 8–12 mg/mL. The ternary complexes with different metals, ATP (or AMP-PCP), and SP20 were made before crystallizations were set up. First, the concentrated PKAc solution was mixed with a solution of metal chloride salt to achieve the final metal concentration of ∼20 mM. Second, the nucleoside was added and the peptide substrate introduced into the mixture last. The PKAc:ATP:SP20 molar ratio was kept at 1:10:10. Crystals were grown in sitting drop microbridges or in nine-well glass plates using well solutions consisting of 100 mM MES (pH 6.5), 5 mM DTT, and 15–20% PEG 4000 at 4–14 °C. In addition, for complexes with different metal ions the corresponding metal chloride salts were introduced into the well solutions at concentrations of 50 mM prior to crystallization drops being set up.
Structure Determination and Refinement
X-ray crystallographic data were collected from frozen crystals at 100 K for all complexes. In the case of PKAc–Mg2ADP–pSP20 and PKAc–Sr2ADP–pSP20, equivalent diffraction data were also measured at room temperature. The data sets were collected on a Rigaku HomeFlux system, equipped with a MicroMax-007 HF generator and Osmic VariMax optics. The diffraction images were obtained using an RAXIS-IV++ image plate detector. Diffraction data were collected, integrated, and scaled using the HKL3000 software suite.26 The structures were refined using SHELX-9727 for resolutions equal to or better than 2.0 Å, and using CNS28 for resolutions of <2.0 Å. A summary of the crystallographic data and refinement is given in Table 1 of the Supporting Information. Similar to our previous observations,25 all the structures were of isoform 2 and contained three post-translationally phosphorylated residues, Ser139, Thr197, and Ser338. The structure of the ternary complex of PKAc with two Mg2+ atoms, ATP, and peptide inhibitor IP20 [Protein Data Bank (PDB) entry 4DH3]25 was used as a starting model to determine all the structures described here. The structures were built and manipulated with Coot,29 whereas the figures were generated using PyMol version 1.5.0.3 (Schrödinger, LLC). The presence of metal ions was established by ensuring that the strongest peaks in difference FO – FC maps with metals omitted and 2FO – FC maps with metals present corresponded to positions M1 and M2, and by checking that the coordination spheres of six to eight ligands were present around the metal ions. Also, differences in coordination geometries were used to distinguish among metal ions. Mg2+, Ca2+, Sr2+, and Ba2+ have increasing numbers of electrons; therefore, they were identified by electron density peaks that were larger than those for the smaller metals. The representative electron densities in the active sites are shown in Figures 1 and 5 and Figures 1 and 2 of the Supporting Information, while the coordination of metals in Ca, Sr, and Ba complexes is given in Table 2 and Figure 3 of the Supporting Information.
Figure 1.

(a) Close-up view of the enzyme active site in the PKAc–Mg2AMP-PCP–SP20 pseudo-Michaelis complex showing metals Mg1 and Mg2 bound at sites M1 and M2, respectively, AMP-PCP, Ser21SP20, and the residues of the enzyme that are important for metal binding or catalysis. Metal coordination is shown as black solid lines, whereas possible hydrogen bonds are represented as red dashed lines. (b) Electron density for the active site components AMP-PCP, Mg2+, water molecules, and Ser21SP20 of the substrate peptide SP20 contoured at the 2.0σ level.
Figure 5.
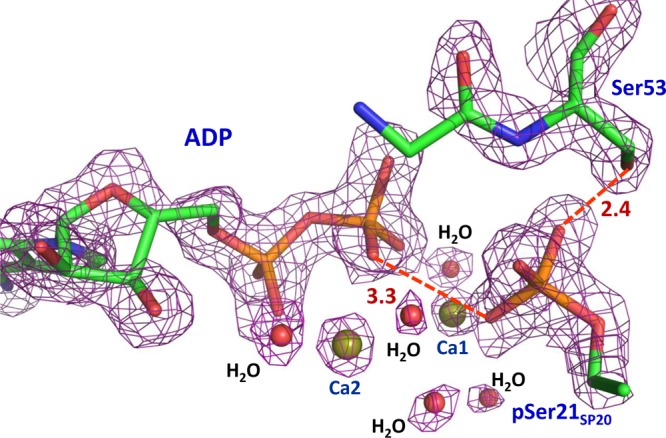
Electron density for the active site components ADP, pSer21SP20, and Ser53 contoured at the 2.0σ level (4σ for calcium cations) in PKAc–Ca2ADP–pSP20. Distances are in angstroms.
The Ramachandran statistics for the structures reported here are as follows. PKAc–Mg2AMP-PCP–SP20: 91.6% of residues in most favored regions, 8.4% of residues in additional allowed regions. PKAc–Mg2ADP–pSP20 at a low temperature: 90.5% of residues in most favored regions, 9.5% of residues in additional allowed regions. PKAc–Mg2ADP–pSP20 at room temperature: 90.8% of residues in most favored regions, 9.2% of residues in additional allowed regions. PKAc–Ca2ADP–pSP20: 91.8% of residues in most favored regions, 8.2% of residues in additional allowed regions. PKAc–Sr2ADP–pSP20 at a low temperature: 92.5% of residues in most favored regions, 7.5% of residues in additional allowed regions. PKAc–Sr2ADP–pSP20 at room temperature: 91.1% of residues in most favored regions, 8.9% of residues in additional allowed regions. PKAc–Ba2ADP–pSP20: 91.8% of residues in most favored regions, 8.2% of residues in additional allowed regions.
The structures have been deposited in the PDB and were assigned the following codes: 4IAC for PKAc–Mg2AMP-PCP–SP20, 4IAD for low-temperature PKAc–Mg2ADP–pSP20, 4IAF for room-temperature PKAc–Mg2ADP–pSP20, 4IAI for PKAc–Ca2ADP–pSP20, 4IAK for low-temperature PKAc–Sr2ADP–pSP20, 4IAY for room-temperature PKAc–Sr2ADP–pSP20, and 4IAZ for PKAc–Ba2ADP–pSP20.
Results
Pseudo-Michaelis Complex PKAc–Mg2AMP-PCP–SP20
The nonhydrolyzable ATP analogue AMP-PCP and the Ser21SP20 substrate are found to be chemically unchanged in the active site of PKAc–Mg2AMP-PCP–SP20 at LT, and thus, this complex represents a pseudo-Michaelis complex (Figure 1a,b). Our previously reported structure of PKAc–Mg2ATP–IP20 at LT25 is also a pseudo-Michaelis complex because IP20 contains Asn20IP20 and Ala21IP20 residues, instead of Ala20SP20 and Ser21SP20 in the substrate peptide, and thus is an inhibitor of PKAc activity. Comparison of these two structures reveals that they are similar. In particular, the metal ions coordinate to AMP-PCP in PKAc–Mg2AMP-PCP–SP20 in much the same way as they do to ATP in PKAc–Mg2ATP–IP20. The importance of the similarity of these two structures is that it tells us that the conformations of ATP, SP20, and PKAc in the two different structures, when combined, are representative of the actual Michaelis complex. We can thus compare the position of ATP in PKAc–Mg2ATP–IP20 and the position of SP20 in PKAc–Mg2AMP-PCP–SP20 to the position in the product complex to obtain meaningful mechanistic insights.
However, there are some differences between PKAc–Mg2AMP-PCP–SP20 and PKAc–Mg2ATP–IP20. M2 coordination is octahedral in PKAc–Mg2ATP–IP20, but because of the substitution of the β,γ-bridging CH2 group for an oxygen atom, the M2 site lacks a sixth coordination bond and consequently has trigonal bipyramidal coordination in PKAc–Mg2AMP-PCP–SP20. In PKAc–Mg2ATP–IP20, two of the γ-phosphate oxygen atoms that are bound to the metals form hydrogen bonds with side chains of Lys168 and Asp166, but in PKAc–Mg2AMP-PCP–SP20, the hydrogen bond with Asp166 is lost (Figure 1a). The larger steric volume of CH2 compared to that of an oxygen atom and the former’s inherent inability to bind to a metal ion pushes the γ-phosphate group farther from M2 and Asp166 in PKAc–Mg2AMP-PCP–SP20. Another notable difference is a 2.6–2.8 Å displacement of the glycine-rich loop, with the loop being closer to the metals in PKAc–Mg2ATP–IP20 and in a more open conformation in PKAc–Mg2AMP-PCP–SP20 (Figure 2), again because of the stronger steric effects of the CH2 group.
Figure 2.
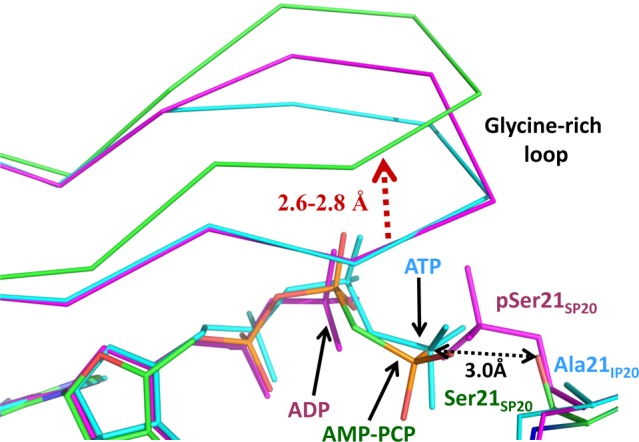
Superposition of the active sites of PKAc–Mg2AMP-PCP–SP20 (colored by atom type, carbon colored green), PKAc–Mg2ADP–pSP20 (magenta), and PKAc–Mg2ATP–IP20 (cyan). The distance between the γ-P atom of ATP and the nucleophilic oxygen of Ser21SP20 is only 3.0 Å, indicating correct positioning of the reactants in the studied complexes. Distances are in angstroms.
Product Complex PKAc–Mg2ADP–pSP20
Both Mg2+ ions have an octahedral configuration with M1 coordinated to phosphoryl oxygen atoms of pSer21SP20 and the β-phosphate of ADP, two water molecules, and chelated by the carboxylate group of Asp184 at LT (Figure 3). ADP’s β-phosphate oxygen is located in the axial position, while that of the phosphorylated serine is in the equatorial position. M2 is chelated by the α- and β-phosphates of ADP and has interactions with Asn171, Asp184, and two water molecules. The coordination of M2 has changed significantly from its coordination in PKAc–Mg2ATP–IP20 (Figure 4; see also Figure 1b of ref (25)). In the PKAc–Mg2ATP–IP20 complex, Mg2+ at site M2 is coordinated to oxygen atoms of the α- and γ-phosphates, to the β,γ-bridging oxygen, and to a single water molecule.25 In PKAc–Mg2ADP–pSP20, M2 has lost the interaction with the γ-phosphate, which has moved ∼2 Å onto Ser21SP20, and its M2 coordination has been replaced with a new water molecule. In PKAc–Mg2ATP–IP20 and PKAc–Mg2ADP–pSP20, the α- and β-phosphates are stabilized by bonding to the metal ions and by hydrogen bonding with Lys72. The γ-phosphate oxygens of ATP hydrogen bond with side chain atoms of Asp166 and Lys168 in PKAc–Mg2ATP–IP20 (Figure 4). However, surprisingly, the phosphate group on pSer21SP20 is too far (∼4 Å or more) from Asp166 and Lys168 to form hydrogen bonds in PKAc–Mg2ADP–pSP20. On the other hand, the phosphate group on pSer21SP20 gains a new 2.8 Å hydrogen bond with the OH group of Ser53 in the glycine-rich loop in PKAc–Mg2ADP–pSP20 compared to that in PKAc–Mg2ATP–IP20. Thus, the reactive γ-phosphate of ATP is transferred to the substrate with a net loss of one metal interaction, and one hydrogen bond. This occurs as a result of the Cβ–Oγ bond of pSer21SP20 being rotated away from the metals and Asp166 toward the bulk solvent. Importantly, a similar orientation of this phosphate group was found in the structure of the holoenzyme–product complex,30 indicating that our structure with a small peptide substrate is, in fact, physiologically relevant.
Figure 3.

Close-up view of the enzyme active site in the PKAc–Mg2ADP–pSP20 product complex showing metals Mg1 and Mg2 bound at sites M1 and M2, respectively, ADP, phosphorylated pSer21SP20, and the residues of the enzyme that are important for metal binding or catalysis. Metal coordination is shown as black solid lines, whereas possible hydrogen bonds are represented as red dashed lines.
Figure 4.
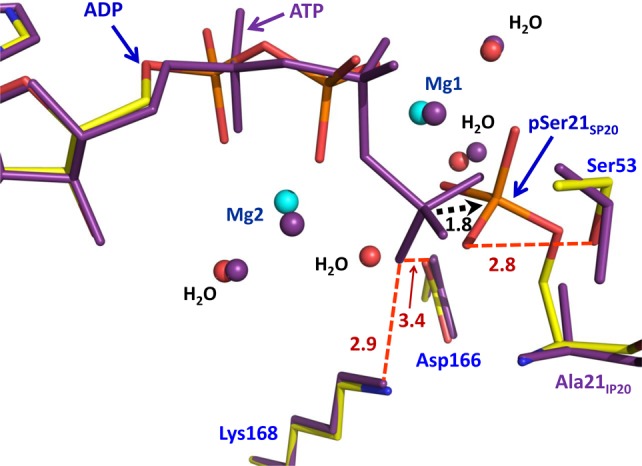
Superposition of the active sites of PKAc–Mg2ADP–pSP20 and PKAc–Mg2ATP–IP20. The distance between the γ-P atom of ATP and the phosphorus in pSer21SP20 is shown as a black dashed arrow, indicating that the phosphate has to move <2 Å during the phosphoryl transfer reaction to its new position in pSer21SP20.
Product Complexes with Other Alkali Earth Metals
In complexes of PKAc with Ca2+, Sr2+, and Ba2+, we found that the metals, when in excess, bind in the active site in both M1 and M2 sites and furthermore that the phosphotransfer products, ADP and pSP20, were also present. The ionic radii of these metals are significantly longer than that of Mg2+ (0.72 Å). Ba2+ represents the largest cation, with an ionic radius of 1.42 Å. Sr2+ is slightly smaller, with a radius of 1.26 Å, whereas Ca2+ has a radius of ∼1.1 Å. The active sites in all the structures of the metal complexes are very similar to each other and to that of PKAc–Mg2ADP–pSP20. In particular, the phosphoryl groups on pSer21SP20 are similarly oriented away from Asp166 and Lys168, keeping the length of the hydrogen bond to the OH group of Ser53 in the glycine-rich loop in the range of 2.4–2.7 Å (see Figure 5 for the highest-resolution PKAc–Ca2ADP–pSP20 complex), which compares well with the distance of 2.8 Å in PKAc–Mg2ADP–pSP20. However, because of differences in the metal size and electronic structure, there are differences in coordination spheres, metal–ligand distances, and metal hydration between complexes (for examples of electron density maps, see Figures 1 and 2 of the Supporting Information; for metal–ligand distances, see Figure 3 and Table 2 of the Supporting Information).
The octahedral geometry seen around ions in PKAc–Mg2ADP–pSP20 is not present in any of the other complexes. Both metal sites are surrounded by seven ligands in PKAc–Ca2ADP–pSP20, whereas in PKAc–Sr2ADP–pSP20 and PKAc–Ba2ADP–pSP20, M1 and M2 metals have coordination numbers of 8 and 7, respectively. As the protein residues cannot provide additional coordination to these metals, the extra ligands are water molecules. The sizes of the metal cations appear to have little effect on the positions of the side chains to which they are bound, but the terminal phosphoryl groups on ADP and pSer21SP20 are pushed away from each other compared to their locations in PKAc–Mg2ADP–pSP20. The increase in the βPADP···P21SP20 separation from 4.3 Å in PKAc–Mg2ADP–pSP20 is negligible (0.3–0.4 Å) in the calcium product complex but increases substantially to ∼1 Å for complexes with strontium and barium. These changes in the geometry of the active sites of different metal complexes can be partially explained by the variations in the metal ionic radii. Also, they can be linked to the increased coordination number of 8 for Sr2+ and Ba2+ cations at site M1. The larger the coordination sphere, the longer the metal–ligand distances, and consequently the phosphoryl groups are pushed apart.
Superposition of our product structures reveals that the main chain of the glycine-rich loop adopts two major positions related by a more than 2 Å sliding shift relative to its location in PKAc–Mg2ADP–SP20, which nonetheless does not affect the loop’s closed conformation (Figure 4 of the Supporting Information). The shift is observed in PKAc–Sr2ADP–pSP20 and PKAc–Ba2ADP–pSP20, where the ionic radii of the metals are longest. The side chain OH group of Ser53 has various conformations in the complexes studied, related by the Cβ–Oγ bond rotation, but keeps its hydrogen bond to the phosphate group of pSer21SP20 in all the structures. This observation is in contrast to the 3.1 Å structure of the tetrameric PKA complex,30 where the Ser53 side chain was not found to interact with the phosphate group on serine of the regulatory subunit. In addition, the position of the side chain phenyl of Phe54 changes dramatically when PKAc–Mg2ATP–IP20 and the product structures are compared (Figure 4 of the Supporting Information). In PKAc–Mg2ATP–IP20, when there is no phosphate at position 21 of the bound peptide, the phenyl is located within 3.3 Å of the oxygens of the γ-phosphate of ATP and is only 4.2 Å from the Cβ atom of Ala21IP20. In the product complexes, Phe54 is pushed away by the presence of the bulky side chain of pSer21SP20 toward a hydrophobic pocket lined by Leu74, Val79, and Leu82.
Discussion
The first PKA structure of a physiological product complex was reported recently by Zhang et al.30 for the tetrameric holoenzyme at 3.1 Å resolution (PDB entry 3TNQ), which was obtained by soaking in MgATP. That structure raised important questions about the role of autophosphorylation of the regulatory subunits in cells. However, the resolution of the holoezyme–product structure prevented a detailed analysis of the PKAc active site after the phosphoryl transfer had occurred, including accurate positions of products and enzyme residues, and the hydration of metal cations. In this work, the low-temperature structures of the PKAc–product complexes ranged between 1.55 and 1.90 Å resolution, whereas the two room-temperature product structures were obtained at 2.0 and 2.2 Å. These resolutions are sufficient to perform a detailed investigation of the active site, containing products, ADP, and phosphorylated SP20.
A previous report by Bhatnagar et al.8 on solution kinetics measurements of PKAc activity suggested that only a handful of divalent metal species promotes binding of ATP to the PKAc active site and is capable of facilitating the transfer of a phosphoryl group to a substrate. Most notably, Ca2+ and Sr2+ were found to allow ATP to bind, but no activity was detected. The heavier alkaline earth metal Ba2+ failed to support ATP binding or enzymatic activity. An exciting finding in this study is that all the alkaline earth metals, when in excess, appear to promote both ATP binding and phosphotransferase activity. The physiological relevance of our observation of the products in the reported structures has to be interpreted with great care. The SP20 pseudosubstrate peptide has been generated from the inhibitor peptide IP20 that represents a portion of the heat-stable protein kinase inhibitor, PKI, by mutation of residues 20 and 21. PKI, and hence IP20 and SP20, are able to bind to PKAc with high affinity.31,32 Possibly, in solution SP20 binds to PKAc first and facilitates the binding of ATP and metal ions by increasing the nucleoside affinity for PKAc, as has been seen for the PKI.32 In fact, a binary complex of PKAc with bound SP20 was reported previously.13 The phosphoryl transfer occurs, and the stable product complex is trapped in the crystals. Our ability to trap the products in all the reported structures can, thus, be attributed to the unique binding properties of the substrate peptide used. We are pursuing solution kinetics studies, including steady-state33 and single-turnover7 measurements, to clarify which metals do indeed support phosphoryl transfer. Our preliminary enzyme kinetics measurements of the RIIβ subunit and Kemptide acting as substrates have shown that alkaline earth divalent metals do in fact support phosphoryl transfer catalyzed by PKAc. Conversely, the phosphorylated RIIβ subunit could not be detected in the steady-state kinetics when calcium was used, perhaps because the phosphorylated product does not dissociate from PKAc, suggesting a low off rate of the product complex decomposition. However, in the single-turnover (“burst”) kinetics measurements, we were able to detect phosphorylation of the RIIβ subunit using both magnesium and calcium. A full kinetic study will be published separately.
In principle, the transfer of a phosphoryl group from ATP to a substrate’s Ser/Thr can proceed according to dissociative, associative, and concerted mechanisms.34 The dissociative mechanism, designated DN + AN or SN1 by IUPAC, necessitates the production of intermediate metaphosphate ion PO3– that is attacked by an incoming nucleophile. In the associative mechanism, termed AN + DN, a detectable pentacoordinated phosphorane intermediate is formed along the reaction path. In the concerted mechanism, ANDN or SN2, the reaction proceeds through a trigonal bipyramidal pentacoordinated transition state, in which fission of the bond of the leaving ADP group and formation of a bond to the nucleophile’s hydroxyl group (OH) both occur at the same time. This transition state can be loose or tight depending on the distances from the central phosphorus atom to the apical positions in the trigonal bipyramid and on how synchronous the nucleophilic attack and leaving group departure are. None of these mechanisms has been ruled out definitively for PKAc.
Recently, Montenegro and co-authors have reported a comprehensive molecular dynamics study on the Michaelis complexes containing Kemptide or SP20 as the substrate.35 Notably, they found that the OH group of Kemptide’s serine is rotated toward Asp166, forming hydrogen bonds with this aspartate and Lys168. On the other hand, the OH group of Ser21SP20 adopts a conformation very similar to that observed in our PKAc–Mg2AMP-PCP–SP20 structure. Furthermore, these authors argued that the chemical mechanism of the phosphoryl transfer may depend on the substrate bound to the enzyme, suggesting that with Kemptide the reaction proceeds through a loose transition state, resulting in the protonation of Asp166 in the product. With SP20, however, the transition state is tight and Ser21SP20 protonates the incoming phosphate. Importantly, the loose transition state in the concerted mechanism has been verified for the transfer of the phosphoryl group to alcohols in solution for small molecules.36 We, therefore, want to emphasize that our current structures of the pseudo-Michaelis and product complexes provide some insight only into the possible mechanism with substrate SP20. The detailed atomic rearrangements may be different when the substrate is an actual physiological protein. Nonetheless, our findings with respect to the chemical mechanism of the phosphoryl transfer step with SP20 as a substrate are summarized as follows. Before the reaction, (a) the OH group of Ser21SP20 is rotated away from metals and lacks a hydrogen bond to Asp166, with an Oγ–Cβ–Cα–N torsion angle of 64°, (b) the OH group of Ser21SP20 forms a 2.7 Å hydrogen bond with a γ-phosphate oxygen of ATP and is 3.5 Å from the γ-phosphorus atom, and (c) the γ-phosphate forms a hydrogen bond with Lys168 and is coordinated to both metals. After the phosphoryl transfer has taken place, (a) the orientation of the Cβ–Oγ bond in pSer21SP20 is identical to that in the pseudo-Michaelis complex, (b) the transferred PO3 maintains coordination to M1 but loses its bond to M2 while the sixth position in the coordination sphere of M2 is replaced by a new water molecule, and (c) the transferred PO3 lacks a hydrogen bond to either Asp166 or Lys168 but gains an interaction with the side chain of Ser53 in the glycine-rich loop. Ser53 is not a conserved residue in the kinase family, and previous solution studies showed that mutations of this serine did not affect the steady-state phosphorylation kinetics of a small peptide.37 However, it is possible that Ser53 may have a certain effect on the pre-steady-state kinetics, stabilizing the transition state or the metaphosphate intermediate by providing additional hydrogen bonding for the moving γ-PO3.
These results can also be placed in context with the previous structural reports of PKAc complexes obtained by cocrystallizing presynthesized MgADP, SP20, phosphorylated SP20, and AlF3,13,20 in which OH and PO3 groups of Ser21SP20 were rotated toward metals, with an Oγ–Cβ–Cα–N torsion angle of −65°, and formed hydrogen bonds with Asp166 and Lys168. Assuming our current structures represent stable intermediates before and after the reaction with SP20, while the previous structure with AlF3 truly mimics the transition state, the substrate OH group should rotate ∼110° from its position in PKAc–Mg2-AMP–PCP–SP20 to reach the conformation found in the transition-state mimic, and once the phosphate is transferred, it should rotate back 110° to assume its position in PKAc–Mg2ADP–pSP20. On the basis of this perspective, we can hypothesize that the initial conformational change of the flexible Ser21SP20 side chain may initiate the chemistry and play the role of the reaction driving force. Later, after the phosphoryl transfer, the transferred phosphoryl group rotates toward the bulk solvent so that the product can be released from the enzyme. The γ-P atom is ∼3–3.5 Å from the substrate’s OH group depending on its location in the pseudo-Michaelis PKAc–Mg2AMP-PCP–SP20 or PKAc–Mg2ATP–IP2025 structure (Figure 2). Theoretical calculations for the mechanism that accommodates our current enzyme–substrate and enzyme–product and the earlier transition-state mimic structures could resolve this issue for PKAc, although the calculations would be successful in mapping the correct reaction pathway only if the locations of hydrogen atoms before and after the reaction were established for the starting theoretical models. Future neutron crystallographic studies of PKAc may be the only direct structural technique that will provide us with this information and therefore a definitive answer.
Conclusions
In summary, our X-ray structure of a pseudo-Michaelis complex with AMP-PCP and substrate SP20 showed the conformation of the Ser21SP20 side chain rotated away from the metals and Asp166, in agreement with recent molecular dynamics simulations. γ-PO3 has been transferred, and the product’s side chain was observed similarly rotated away toward the bulk solvent, perhaps representing a state just before the product is released from the enzyme. We have observed that alkaline earth divalent metal cations can indeed bind and facilitate the phosphoryl transfer chemical reaction in the active site of PKAc. Therefore, our results provide new insights into the chemistry of the phophoryl transfer reaction catalyzed by PKAc when the SP20 high-affinity peptide substrate is used and challenge the conventional views about the roles metals play in it.
Glossary
Abbreviations
- PKAc
catalytic subunit of protein kinase A
- PKI
heat-stable protein kinase inhibitor
- SP20
20-residue peptide substrate
- IP20
20-residue peptide inhibitor.
Supporting Information Available
Crystallographic data collection, refinement, and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
O.G., S.T., and A.K. were partly supported by a UCOP grant. A.K., M.J.W., and O.G. were partly supported by a DOE-OBER grant to the neutron Protein Crystallography Station at LANSCE. A.Y.K. and P.L. were partly supported by DOE-BES. P.L. was partly supported by a National Institute of General Medical Sciences-funded consortium (1R01GM071939-01) between Oak Ridge National Laboratory and Lawrence Berkeley National Laboratory to develop computational tools for neutron protein crystallography. S.T. was also supported by National Institutes of Health Grant GM19301.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Adams J. A. (2001) Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 101, 2271–2290. [DOI] [PubMed] [Google Scholar]
- Johnson D. A.; Akamine P.; Radzio-Andzelm E.; Madhusudan M.; Taylor S. S. (2001) Dynamics of cAMP-dependent protein kinase. Chem. Rev. 101, 2243–2270. [DOI] [PubMed] [Google Scholar]
- Taylor S. S., Buechler J. A., and Knighton D. R. (1990) Peptides and protein phosphorylation (Kemp B. E., Ed.) pp 1–41, CRC Press, Boca Raton, FL. [Google Scholar]
- Shaffer J.; Adams J. A. (1999) An ATP-linked structural change in protein kinase A precedes phosphoryl transfer under physiological magnesium concentrations. Biochemistry 38, 5572–5581. [DOI] [PubMed] [Google Scholar]
- Lew J.; Taylor S. S.; Adams J. A. (1997) Identification of a partially rate-determining step in the catalytic mechanism of cAMP-dependent protein kinase: A transient kinetic study using stopped-flow fluorescence spectroscopy. Biochemistry 36, 6717–6724. [DOI] [PubMed] [Google Scholar]
- Shaffer J.; Adams J. A. (1999) Detection of conformational changes along the kinetic pathway of protein kinase A using a catalytic trapping technique. Biochemistry 38, 12072–12079. [DOI] [PubMed] [Google Scholar]
- Grant B.; Adams J. A. (1996) Pre-steady-state kinetic analysis of cAMP-dependent protein kinase using rapid quench flow techniques. Biochemistry 35, 2022–2029. [DOI] [PubMed] [Google Scholar]
- Bhatnagar D.; Roskoski R. Jr.; Rosendahl M. S.; Leonard N. J. (1983) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: A new fluorescence displacement titration technique for characterizing the nucleotide binding site on the catalytic subunit. Biochemistry 22, 6310–6317. [DOI] [PubMed] [Google Scholar]
- Adams J. A.; Taylor S. S. (1993) Divalent metal ions influence catalysis and active site accessibility in the cAMP-dependent protein kinase. Protein Sci. 2, 2177–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox S.; Radzio-Andzelm E.; Taylor S. S. (1994) Domain movements in protein kinases. Curr. Opin. Struct. Biol. 4, 893–901. [DOI] [PubMed] [Google Scholar]
- Masterson L. R.; et al. (2011) Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 6969–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M.-F.; Bramson H. N.; Hansen D. E.; Knowles J. R.; Kaiser E. T. (1988) Stereochemical course of the phosphor group transfer catalyzed by cAMP-dependent protein kinase. J. Am. Chem. Soc. 110, 2680–2681. [Google Scholar]
- Madhusudan M.; et al. (1994) cAMP-dependent protein kinase: Crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 3, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granot J.; Mildvan A. S.; Bramson H. N.; Kaiser E. T. (1980) Magnetic resonance measurements of intersubstrate distances at the active site of protein kinase using substitution inert cobalt(III) and chromium(III) complexes for adenosine 5′-(β,γ-methylenetriphosphate). Biochemistry 19, 3537–3543. [DOI] [PubMed] [Google Scholar]
- Valiev M.; Kawai R.; Adams J. A.; Weare J. H. (2003) The role of the putative catalytic base in the phosphoryl transfer reaction in a protein kinase: First-principles calculations. J. Am. Chem. Soc. 125, 9926–9927. [DOI] [PubMed] [Google Scholar]
- Montenegro M.; Garcia-Viloca M.; Lluch J. M.; Gonzalez-Lafont A. (2011) A QM/MM study of the phosphoryl transfer to the Kemptide substrate catalyzed by protein kinase A. The effect of the phosphorylation state of the protein on the mechanism. Phys. Chem. Chem. Phys. 13, 530–539. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Zhang Y.; McCammon J. A. (2005) How does the cAMP-dependent protein kinase catalyze the phosphorylation reaction: An ab initio QM/MM study. J. Am. Chem. Soc. 127, 1553–1562. [DOI] [PubMed] [Google Scholar]
- Yoon M.-Y.; Cook P. F. (1987) Chemical mechanism of the adenosine cyclic 3′,5′-monophosphate dependent protein kinase from pH studies. Biochemistry 26, 4118–4125. [DOI] [PubMed] [Google Scholar]
- Gibbs C. S.; Zoller M. J. (1991) Rational scanning mutagenesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. J. Biol. Chem. 266, 8923–8931. [PubMed] [Google Scholar]
- Madhusudan M.; Akamine P.; Xuong N.-H.; Taylor S. S. (2002) Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat. Struct. Biol. 9, 273–277. [DOI] [PubMed] [Google Scholar]
- Blachut-Okrasinska E.; Lesyng B.; Briggs J. M.; McCammon J. A.; Antosiewicz J. M. (1999) Poisson-Boltzmann model studies of molecular electrostatic properties of the cAMP-dependent protein kinase. Eur. Biophys. J. 28, 457–467. [DOI] [PubMed] [Google Scholar]
- Diaz N.; Field M. J. (2004) Insights into the phosphoryl-transfer mechanism of cAMP-dependent protein kinase from quantum chemical calculations and molecular dynamics simulations. J. Am. Chem. Soc. 126, 529–542. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Adams J. A. (1977) Is there a catalytic base in the active site of cAMP-dependent protein kinase?. Biochemistry 36, 2977–2984. [DOI] [PubMed] [Google Scholar]
- Kenyon C. P.; Roth R. L.; van der Westhuyzen C. W.; Parkinson C. J. (2012) Conserved phosphoryl transfer mechanisms within kinase families and the role of the C8 proton of ATP in the activation of phosphoryl transfer. BMC Res. Notes 5, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalevsky A. Y.; Johson H.; Hanson B. L.; Waltman M. J.; Fisher S. Z.; Taylor S.; Langan P. (2012) Low- and room-temperature X-ray structures of protein kinase A ternary complexes shed new light on its activity. Acta Crystallogr. D68, 854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinovski Z.; Chruszcz M. (2006) HKL3000: The integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D62, 859–866. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Brunger A. T. (2007) Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; et al. (2012) Structure and allostery of the PKA RIIβ tetrameric holoenzyme. Science 335, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herberg F. W.; Taylor S. S. (1993) Physiological inhibitors of the catalytic subunit of cAMP-dependent protein kinase: Effect of MgATP on protein-protein interactions. Biochemistry 32, 14015–14022. [DOI] [PubMed] [Google Scholar]
- Herberg F. W.; Doyle M. L.; Cox S.; Taylor S. S. (1999) Dissection of the nucleotide and metal-phosphate binding sites in cAMP-dependent protein kinase. Biochemistry 38, 6352–6360. [DOI] [PubMed] [Google Scholar]
- Cook P. F.; Neville M. E. Jr.; Vrana K. E.; Hartl F. T.; Roskoski R. Jr. (1982) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: Kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21, 5794–5799. [DOI] [PubMed] [Google Scholar]
- Hengge A. C. (2005) Mechanistic studies on enzyme-catalyzed phosphoryl transfer. Adv. Phys. Org. Chem. 40, 49–108. [Google Scholar]
- Montenegro M.; Masgrau L.; Gonzalez-Lafont A.; Lluch J. M.; Garcia-Viloca M. (2012) Influence of the enzyme phosphorylation state and the substrate on PKA enzyme dynamics. Biophys. Chem. 161, 17–28. [DOI] [PubMed] [Google Scholar]
- Admiraal S. J.; Herschlag D. (1995) Mapping the transition state for ATP hydrolysis: Implications for enzymatic catalysis. Chem. Biol. 2, 729–739. [DOI] [PubMed] [Google Scholar]
- Aimes R. T.; Hemmer W.; Taylor S. S. (2000) Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: Role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry 39, 8325–8332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.