

Abstract
Structural and functional remodelling of cardiomyocytes, capillaries and cardiac innervation occurs in left ventricular hypertrophy (LVH) and heart failure (HF) in response to pressure-induced overload. However, the onset, time course and the extent of these morphological alterations remain controversial. In the present study, we tested the hypothesis that the progression from hypertrophy to HF is accompanied by changes in the innervation (hyper- or hypoinnervation). Left ventricles of wild-type murine hearts subjected to pressure overload-induced hypertrophy by transverse aortic constriction (TAC) were investigated by morphometric and design-based stereological methods at 1 and 4 weeks after TAC and compared with sham-operated mice. Mice developed compensated LVH at 1 week and typical signs of HF, such as left ventricular dilation, reduced ejection fraction and increased relative lung weight at 4 weeks post-TAC. At the (sub-)cellular level, cardiomyocyte myofibrillar and mitochondrial volume increased progressively in response to mechanical overload. The total length of capillaries was not significantly increased after TAC, indicating a misrelationship between the cardiomyocyte and the capillary compartment. The myocardial innervation decreased already during the development of LVH and did not significantly decrease further during the progression to HF. In conclusion, our study suggests that early loss of myocardial innervation density and increased heterogeneity occur during pressure overload-induced hypertrophy, and that these changes appear to be independent of cardiomyocyte and capillary remodelling.
Keywords: cardiac innervation, design-based stereology, left ventricle, pressure overload, transverse aortic constriction
Introduction
Cardiac hypertrophic growth occurs in response to hypertension and increased afterload, and frequently progresses to heart failure (HF). The transition from a compensatory state of hypertrophy to clinically manifested HF is characterized by alterations in neural regulation and adverse myocardial remodelling, which causes contractile failure, arrhythmias and sudden cardiac death (Chien, 1999; Frey & Olson, 2003).
Recent studies have suggested that concomitant alterations in cardiac innervation activity and density due to mechanical stress may contribute to the progression of HF (Kaye et al. 2000; Kimura et al. 2007; Kanazawa et al. 2010). Right ventricular hypertrophy induced by pressure overload is accompanied by upregulated expression of nerve growth factor in the myocardium, leading to increased myocardial innervation, i.e. more nerve fibres, but a functional downregulation (Kimura et al. 2007). Immunohistochemical analysis of failing human myocardium has demonstrated a close correlation between regional hyperinnervation and the occurrence of ventricular arrhythmias, including sudden cardiac death (Cao et al. 2000). In addition to hyperinnervation, HF causes transdifferentiation of some noradrenergic neurons to a cholinergic phenotype in human hearts and animal models (Kanazawa et al. 2010). In contrast, other studies have reported that cardiac innervation is reduced in animal models and in humans with HF (Himura et al. 1993; Kaye et al. 2000). These seemingly conflicting reports raise an important, yet unanswered, question about the time course and onset of hyper- and/or hypoinnervation during the development and progression of hypertrophy and HF.
In addition to the changes in innervation, a number of morphological alterations of cardiomyocytes and the microcirculation have been described in hypertrophy and HF. Cardiomyocytes from hypertrophic or failing hearts show increased volume, convoluted nuclear membrane, variety in the size of mitochondria, disarray of myofibrils and remodelling of t-tubules, among others (Leyton & Sonnenblick, 1969; Maron et al. 1975; Rakusan & Tomanek, 1986; Wei et al. 2010). Quantitatively, in the setting of pressure overload the ratio between mitochondria and myofibrils is first increased, but then reduced when the growth of myofibrils exceeds that of mitochondria (Anversa et al. 1986). In mild or concentric hypertrophy, the capillary network is increased in proportion to the increase in muscle mass to meet the higher metabolic demands (Tomanek et al. 1982; Gruber et al. 2012a). In volume overload or eccentric hypertrophy, however, the capillary network seems to drop behind the increasing muscle mass, thus impairing microcirculatory tissue blood supply (Anversa et al. 1982, 1986). In humans, the progression from a compensated state of left ventricular hypertrophy (LVH) induced by pressure overload to a decompensated state of failure is associated with a decline of capillary density if the pressure stimulus sets on at adulthood (Pearlman et al. 1981; Rakusan et al. 1992). It is argued that the failure to increase the capillary network in proportion to the increase in muscle mass causes an increase in oxygen diffusion distance and a reduction of blood volume, a phenomenon that worsens during increasing hypertrophy and dilation (Anversa et al. 1986). However, it is still not fully understood which of the morphological changes within cardiomyocytes and capillaries occurs during the development of pressure-induced hypertrophy, or later during the transition to HF. Furthermore, it has not been investigated if any of these changes are correlated with quantitative alterations of the innervation.
Here we provide a comprehensive analysis of the mouse left ventricle performed by design-based stereology, and light (LM) and electron microscopy (EM). The present study delineates the quantitative morphological changes of cardiomyocytes, capillaries and nerve fibres during the development and progression of LVH and HF in a wild-type mouse model of pressure overload induced by transverse aortic constriction (TAC). In addition, the distribution of nerve fibres ramifying between cardiomyocytes was analysed qualitatively and quantitatively.
Materials and methods
Minimally invasive TAC
Hypertrophy of the left ventricle was induced by pressure overload by constricting the transverse aorta as described elsewhere (Hu et al. 2003; Toischer et al. 2010). C57BL/6 mice (10- to 14-week-old females) of 21 ± 1 g body weight were anaesthetized by ketamine/xylazine intraperitoneal administration (80 mg kg−1 and 5 mg kg−1 body weight, respectively). The aorta was then approached via a horizontal incision at the jugulum, and a 5-0 ligature was placed around the transversal part of the aorta using a 27-gauge needle to ensure a consistent occlusion. After removal of the 27-gauge needle, the skin was closed. Spontaneously breathing mice were then kept on a warming pad at 37 °C in a cage supplied with oxygen-enriched atmosphere until complete recovery from anaesthesia. Sham-operated mice underwent the same surgery without constriction of the aorta. All mice received the analgesic metamizol-Na (1 mL dissolved in 100 mL drinking water) for 1 week. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal procedures were approved by the Austrian Committee on the Ethics of Animal Experiments (Bundesministerium für Wissenschaft und Forschung; BMWF-66.010/0062-II/10b/2010).
Echocardiography
In vivo cardiac morphology and function were assessed by transthoracic echocardiography 1 and 4 weeks post-surgery using a high-resolution micro-imaging system Vevo 770® (VisualSonics, Canada) equipped with a 30-MHz linear array transducer (VisualSonics). Mice were maintained lightly sedated with 0.5% isoflurane during the experiment. Mice were positioned on a warming pad with the body temperature kept constant at 37 °C. M-mode tracings of the left ventricle (short axis) were recorded. The thickness of the posterior wall (PW) and the interventricular septum (IVS) as well as the left ventricular end-diastolic (LVEDD) and the left ventricular end-systolic diameters (LVESD) were averaged from three consecutive cardiac cycles. Left ventricular fractional shortening and left ventricular mass were calculated as described previously (Manning et al. 1994; Kiatchoosakun et al. 2002). The investigator (A.S.) was blinded to the surgical intervention.
Morphometric analysis
One or 4 weeks after surgery, mice were anaesthetized with isoflurane and killed by cervical dislocation. Hearts were excised, weighed and immediately fixed by perfusion in a retrograde manner with 4% paraformaldehyde in phosphate-buffered saline (PBS) through the aorta, left ventricle and coronary vasculature followed by immersion fixation in buffered 4% paraformaldehyde. Hearts were kept at 4 °C before the tissue was processed. Lungs were extracted, blotted dry and weighed. Hypertrophy was assessed by comparing the ratios between heart weight-to-body weight and heart weight-to-tibia length. Pulmonary congestion was assessed by comparing lung weight-to-body weight and lung weight-to-tibia length coefficients.
Tissue processing and stereology
Using state-of-the-art stereology (reviewed in Mühlfeld et al. 2010a), we quantified the total length of capillaries and nerve fibre axons as well as the volume and the subcellular composition of cardiomyocytes. After isolation from the rest of the heart, the left ventricle was weighed and its mass was divided by the density of muscle tissue (1.06 g cm−3) to obtain the volume of the left ventricular myocardium (Mendez & Keys, 1961). The left ventricle was then once cut longitudinally and three times transversely (Eisele et al. 2008). The resulting eight tissue samples were either allocated to paraffin embedding (immunohistochemistry) or epoxy resin embedding (transmission EM). One tissue block was chosen randomly for the estimation of tissue deformation/volume shrinkage due to embedding (Mühlfeld et al. 2010b). Apart from the latter, all tissue blocks were disorientated either by use of the isector (paraffin; Nyengaard & Gundersen, 1992) or the orientator (epoxy resin; Mattfeldt et al. 1990). Epon embedding included post-fixation in 1% osmium tetroxide, en bloc staining in half-saturated uranyl acetate, dehydration in an ascending ethanol series, and embedding in Epon.
Left ventricular tissue sections of 5-μm thickness were generated from the paraffin-embedded samples, mounted on glass slides and used for immunohistochemistry to visualize nerve fibres using the pan-neuronal marker protein gene product (PGP) 9.5 (Gulbenkian et al. 1987). Briefly, sections were deparaffinized using xylol and incubated with 1% H2O2 in methanol for 10 min. Demasking of epitopes was performed by microwaving the specimens in 10 mm citric acid (pH 6.0). Sections were then incubated with the primary antibody (rabbit polyclonal anti-PGP 9.5 antibody, dilution 1 : 5000; Biotrend, Köln, Germany) for 18 h. After washing, the sections were incubated with the secondary antibody (peroxidase-linked donkey-anti-rabbit IgG, dilution 1 : 100; Amersham Int. Biotech, Little Chalfont, Buckinghamshire, UK) for 1 h. After additional washes, visualization of the immunoreaction was performed by the diaminobenzidine reaction enhanced by 1.5% nickel ammonium sulphate. Sections were washed again, dehydrated, embedded in Eukitt (Sigma-Aldrich, Steinheim, Germany) and sealed with a cover slip.
Myocardial tissue blocks for estimation of volume shrinkage were carefully weighed, and the block volume was calculated from the mass and density. Blocks were then embedded in paraffin and exhaustively cut in 7-μm-thick sections. Using the Cavalieri method, the volume of the tissue block after embedding was estimated and further used for the estimation of volume shrinkage due to embedding, as described previously (Mühlfeld et al. 2010b). This estimate was used to correct stereological data from paraffin sections for shrinkage.
Tissue blocks embedded in Epon were cut using an ultramicrotome to obtain semi- and ultrathin sections, which were stained with toluidine blue or uranyl acetate and lead citrate, respectively.
At the LM level, stereology was performed using an Olympus BX51 microscope (Olympus, Hamburg, Germany), equipped with a digital camera (Olympus DP72) and the newCAST stereology software (Visiopharm, Horsholm, Denmark). Transmission EM was performed using a LEO 902 EM (Zeiss, Oberkochen, Germany). For the stereological analysis, all fields of view were obtained by systematic uniform random sampling (Gundersen & Jensen, 1987).
The total length of axons in the left ventricle was estimated as described elsewhere (Mühlfeld et al. 2010b). This parameter provides the length of all axons contained in the left ventricle as if they were put in a row. Its estimation – according to basic stereological principles – is based on nerve fibre profile counting and relies on a random orientation of nerve fibres in 3D. Briefly, immunostained nerve fibre profiles were counted in unbiased counting frames at a magnification of × 40, and the length density of nerve fibres was calculated by LV = 2*Q/A, where Q is the number of counted profiles and A is the total area of the counting frames hitting the reference volume. This parameter was multiplied by the mean number of axons per nerve fibre profile as estimated by EM (× 20 000) to obtain the length density of axons in the left ventricle. The length density was multiplied by the volume of the left ventricle to obtain the total length of axons, which was finally corrected for volume shrinkage due to embedding.
The total length of capillaries was estimated by counting the number of capillary profiles in unbiased counting frames on EM sections (× 3000), as described previously (Gruber et al. 2012b). Length density and total length were calculated as described above for the estimation of axon length. The reciprocal of the length density provides an estimate of the mean cross-sectional area of tissue surrounding an average capillary and helps to evaluate the diffusion distance from the capillaries to the tissue. Given that it includes the capillary cross-sectional area of the capillary itself, the volume of the capillaries (either including the capillary endothelium or not) was estimated using the point counting method on the same images used for length estimation. From these data, the mean cross-sectional area of capillaries (including the endothelium) or of the capillary lumen was calculated by dividing the respective volume by the length of capillaries. The arithmetic mean thickness of the capillaries was estimated by point and intersection counting according to Weibel (1979).
On semi-thin sections from Epon-embedded heart tissue, the volume of cardiomyocytes and interstitium was estimated using the point counting method (Weibel, 1979). At the EM level, point counting was used to estimate the composition of cardiomyocytes with respect to the compartments, such as myofibrils, mitochondria, nuclei and sarcoplasm.
Immunohistochemistry
Heart sections of 5 μm thickness were obtained from samples embedded in paraffin, mounted on glass slides and deparaffinized in xylol. Antigen retrieval was performed by microwaving the sections at 800 W for 20 min using the Dako Retrieval buffer pH 6.0 (Dako, Glostrup, Denmark). After washing, unspecific antibody interactions were blocked with 10% donkey serum (Dianova, Hamburg, Germany) and 0.15% TX-100 (Sigma-Aldrich, Steinheim, Germany) in PBS for 45 min.
For analysis of the distribution of nerve fibres, sections were incubated with the primary antibody rabbit polyclonal anti-PGP 9.5 antibody (diluted 1 : 1000; Biotrend, Cologne, Germany) diluted in PBS containing 1% bovine serum albumin (BSA) and 0.15% Tx-100 for 20 h. After washing, sections were incubated with the secondary antibody Cy3-linked goat-anti-rabbit antibody (diluted 1 : 1000; Jackson ImmunoResearch Laboratories, Suffolk, UK) and FITC-conjugated lectin (10 ng/mL; Sigma-Aldrich) for 1 h.
For analysis of the phenotype of nerve fibres, combined stainings for PGP 9.5 and tyrosine hydroxylase were performed. Sections were incubated with the primary antibodies rabbit polyclonal anti-PGP 9.5 antibody (diluted 1 : 1000; Biotrend) and sheep polyclonal anti-tyrosine hydroxylase antibody (diluted 1 : 50; Merck Millipore, Billerica, MA, USA), both diluted in PBS containing 1% BSA and 0.15% Tx-100 for 48 h. The sections were further incubated with Alexa546-linked goat-anti-rabbit antibody and Alexa488-linked donkey-anti-sheep antibody (both diluted 1 : 1000; both Life Technologies, Carlsbad, CA, USA), and To-Pro 3 Iodide (diluted 1 : 1000; Life Technologies) for 24 h. Finally, sections were embedded in Dako Fluorescence Mounting Medium and sealed with a cover slip.
Statistics
Echocardiographic data are shown as mean (SEM); gravimetric and stereological data are presented as mean (SD). Inter-group differences were analysed using one-way analysis of variance (anova) and Bonferroni's post hoc test, when an overall significance was detected. Sample sizes are provided in the figures and tables. Statistical differences between means were considered significant at P < 0.05.
Results
Echocardiography and organ dimensions
Assessment of LVEDD and LVESD by echocardiography demonstrated that the mice effectively compensated pressure overload 1 week post-TAC (Table 1). Four weeks after TAC, however, the mice underwent a significant left ventricular chamber dilatation. Thickening of the left ventricular walls induced by pressure overload, measured as the sum of IVS and PW thickness, was similarly increased at both 1 and 4 weeks after TAC compared with sham (Fig. 1A and B; P < 0.05). Ventricular systolic function progressively declined in the mice as indicated by significantly reduced ejection fraction 4 weeks after TAC with respect to 1 week after TAC (Fig. 1C). The heart rate was not different among the groups.
Table 1.
Left ventricular echocardiographic parameters in mice after 1 and 4 weeks sham/TAC
| Sham (N = 8) | 1-week TAC (N = 6) | 4-weeks TAC (N = 11) | |
|---|---|---|---|
| HR (bpm) | 541 (31) | 517 (26) | 518 (16) |
| LVEDD (mm) | 3.77 (0.03) | 3.54 (0.14) | 4.17 (0.11)*§ |
| LVESD (mm) | 2.34 (0.14) | 2.39 (0.20) | 3.21 (0.15)*§ |
| IVS (mm) | 0.71 (0.02) | 1.02 (0.04)* | 0.97 (0.06)* |
| PW (mm) | 0.65 (0.02) | 0.92 (0.07)* | 0.99 (0.07)* |
| FS (%) | 39.6 (2.2) | 35.3 (2.6) | 26.2 (1.9)*§ |
| EF (%) | 70.4 (2.8) | 65.2 (3.7) | 51.4 (3.1)*§ |
| Relative WT (mm mm−1) | 0.35 (0.01) | 0.54 (0.03)* | 0.47 (0.03)* |
| LV mass (mg) | 70.8 (2.4) | 102.2 (11.1)* | 120.0 (8.4)* |
bpm, beats per minute; EF, ejection fraction; FS, fractional shortening; HR, heart rate; IVS, interventricular septum; LV, left ventricular; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; N, number of mice; PW, posterior wall; relative WT, relative wall thickness; TAC, transverse aortic constriction.
Data are presented as mean (SEM).
P < 0.05 vs. sham.
P < 0.05 vs. 1-week TAC.
Fig 1.
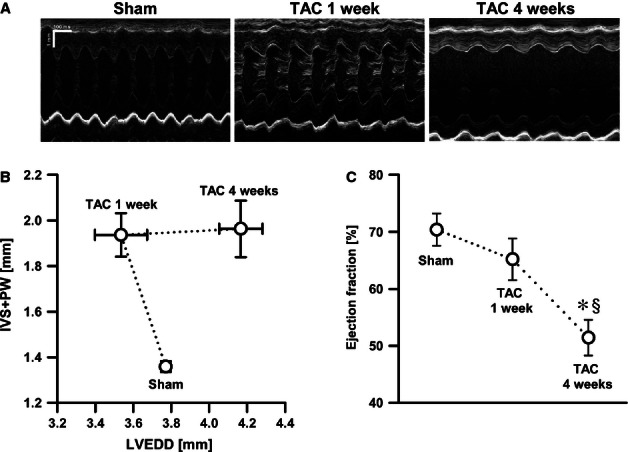
Pressure overload-induced hypertrophy. (A) Echocardiographic M-mode images. (B) The sum of interventricular septum (IVS) and posterior wall (PW) thickness vs. left ventricular end-diastolic diameter (LVEDD) as a measure of hypertrophy vs. dilatation, respectively. Mice (N = 8–11) displayed compensated hypertrophy [transverse aortic constriction (TAC) 1 week] followed by the transition to HF (TAC 4 weeks). (C) Systolic function measured as ejection fraction showed a progressive reduction in TAC mice. *P < 0.05 vs. sham; §P < 0.05 vs. TAC 1 week.
One- and 4-week TAC induced a similar increase in cardiac hypertrophy as determined by an increased heart weight (normalized to left tibia length) compared with sham-operated mice (P < 0.05). However, the relative lung weight was significantly increased 4 weeks after TAC compared with 1 week after TAC, indicating pulmonary oedema (Table 2). In conjunction with the echocardiography, these results demonstrated that sustained pressure overload induced by TAC resulted in early onset of concentric hypertrophy followed by progressive cardiac dilatation and ultimately HF.
Table 2.
Gravimetric analysis of mice after 1 and 4 weeks sham/TAC
| Sham | 1-week TAC | 4-weeks TAC | |
|---|---|---|---|
| Final − initial body weight (g) | 0.85 (0.47) | 0.19 (1.17) | −0.73 (0.51)* |
| Tibia length (mm) | 17.0 (0.45) | 16.9 (0.4) | 17.2 (0.6) |
| Wet HW (mg) | 108.2 (19.3) | 241.3 (20.8)* | 300.6 (35.8)*§ |
| HW/BW (mg g−1) | 5.17 (0.80) | 11.56 (1.63)* | 13.32 (1.89)* |
| HW/TL (mg mm−1) | 6.35 (1.11) | 14.28 (1.40)* | 17.50 (2.23)*§ |
| Dry LW (mg) | 51.8 (4.9) | 50.8 (5.0) | 94.9 (35.7)*§ |
| LW/BW (mg g−1) | 2.49 (0.29) | 2.42 (0.19) | 4.29 (1.50)*§ |
| LW/TL (mg mm−1) | 3.05 (0.30) | 3.01 (0.31) | 5.48 (1.96)*§ |
HW, heart weight; HW/BW, heart weight-to-body weight ratio; HW/TL, heart weight-to-tibia length ratio; LW, lung weight; LW/BW, lung weight-to-body weight ratio; LW/TL, lung weight-to-tibia length ratio; TAC, transverse aortic constriction.
Data are presented as mean (SD).
P < 0.05 vs. sham.
P < 0.05 vs. 1-week TAC.
Cardiomyocytes
In line with the increased left ventricular dimensions, the volume of cardiomyocytes was significantly increased at 4 weeks after TAC compared with the other groups. The interstitial volume tended to be higher at 1 and 4 weeks after TAC, but was not statistically different compared with sham. Accordingly, the ultrastructure of the cardiomyocytes was changed with significant elevation of myofibril and mitochondria volume at 4 weeks after TAC. The volume of myofibrils was significantly increased already in 1-week TAC hearts with respect to the sham group. The ratio between myofibril and mitochondria volume increased substantially at 1 week after TAC, and similar values were obtained at 4 weeks after TAC. On the other hand, no significant changes were observed in sarcoplasmic or nuclear volume (Figs 2–4 Table 3).
Fig 2.
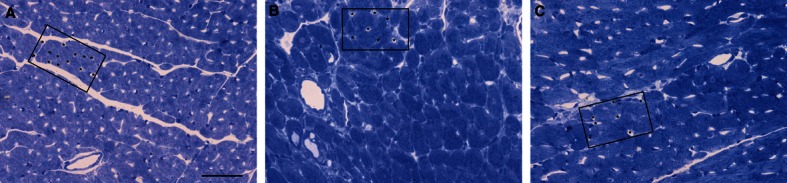
Myocardial histology. LM images of transversely sectioned left ventricular myocardium from sham (A) and TAC groups (B and C: 1 week and 4 weeks after TAC, respectively). The increase in cardiomyocyte diameter and the decrease in capillary profile number per area are clearly visible at 1 and 4 weeks after TAC. The decrease in capillary density is marked by asterisks labelling capillaries within an equal area. Scale bar: 50 μm.
Fig 4.

Stereological data. (A) Total volume of cardiomyocytes. (B) Total volume of myofibrils. (C) Ratio between myofibril and mitochondrial volume. (D) Mean number of axon profiles per nerve fibre profile. (E) Total length of axons. (F) Total length of capillaries. (G) Reciprocal of length density of capillaries. (H) Thickness of capillary endothelium. Each point represents data from one animal, the horizontal bars indicate mean values for each group. Horizontal lines at the top of the diagrams indicate significant differences between groups (P < 0.05). TAC, transverse aortic constriction.
Table 3.
Stereological analysis of cardiomyocytes after 1 and 4 weeks sham/TAC
| Sham | 1-week TAC | 4-weeks TAC | |
|---|---|---|---|
| VV(myo/lv) (%) | 82.56 (6.25) | 76.72 (9.30) | 83.00 (6.23) |
| V(myo,lv) (mm³) | 72.54 (14.43) | 95.48 (15.41) | 133.15 (25.69)*§ |
| VV(int/lv) (%) | 17.44 (6.25) | 23.28 (9.30) | 17.00 (6.23) |
| V(int,lv) (mm³) | 14.85 (4.01) | 31.05 (17.92) | 26.42 (7.99) |
| VV(mf/myo) (%) | 51.85 (3.15) | 58.29 (1.83)* | 59.52 (2.67)* |
| V(mf,lv) (mm³) | 37.30 (5.77) | 55.77 (9.91)* | 79.35 (16.17)*§ |
| VV(mi/myo) (%) | 31.10 (1.02) | 27.80 (2.64)* | 28.44 (2.39)* |
| V(mi,lv) (mm³) | 22.55 (4.57) | 26.46 (4.57) | 37.85 (8.00)*§ |
| VV(sp/myo) (%) | 15.1 (3.58) | 11.89 (1.53)* | 10.48 (1.22)* |
| V(sp,lv) (mm³) | 11.29 (4.36) | 11.32 (1.99) | 13.92 (2.85) |
| VV(nuc/myo) (%) | 2.03 (0.73) | 2.03 (0.56) | 1.55 (0.62) |
| V(nuc,lv) (mm³) | 1.46 (0.60) | 1.92 (0.59) | 2.01 (0.83) |
| V(mf)/V(mi) | 1.67 (0.12) | 2.12 (0.25)* | 2.11 (0.25)* |
int, interstitium; lv, left ventricle; mf, myofibrils; mi, mitochondria; myo, cardiomyocytes; nuc, nucleus; sp, sarcoplasm; TAC, transverse aortic constriction; V, total volume of a compartment within the left ventricle; VV(compartment/reference volume), volume fraction of a compartment related to a specific reference volume.
Data are presented as mean (SD).
P < 0.05 vs. sham.
P < 0.05 vs. 1-week TAC.
Fig 3.

Ultrastructure of cardiomyocytes. Transmission EM images of longitudinally sectioned cardiomyocytes from sham (A) and TAC groups (B and C: 1 week and 4 weeks after TAC, respectively). Note that the distance between capillaries and the sarcomere width are increased due to TAC. cap, capillary; mf, myofibrils; mi, mitochondria; nuc, nucleus. Scale bar: 5 μm.
Cardiac nerve fibres
The total length of nerve fibres as well as the total length of axons was significantly decreased in both TAC groups compared with sham hearts, but did not differ between TAC groups (Fig. 4). Interestingly, the reduction in total nerve fibre length was accompanied by a decrease of the mean number of axons within one nerve fibre (Fig. 5; Table 4). The distribution of nerve fibres was altered in both TAC groups with a shift towards areas lacking nerve fibres, resulting in increased heterogeneity of the innervation (Fig. 6). All nerve fibres stained by PGP 9.5 were positive for tyrosine hydroxylase, the key enzyme of noradrenalin synthesis (Fig. 7).
Fig 5.
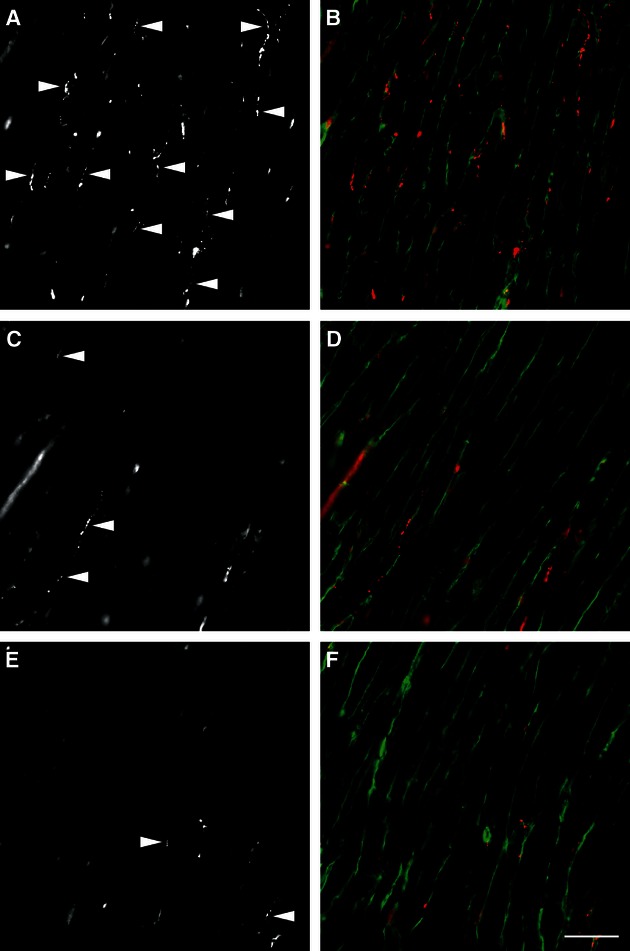
Distribution of nerve fibres stained by immunohistochemistry. The nerve fibre density was decreased in both TAC groups (C, D: 1 week after TAC; E, F: 4 weeks after TAC) compared with sham (A and B). (A, C and E) PGP 9.5 staining only; (B, D and F) merged channels with red = PGP 9.5, green = wheatgerm agglutinin. Arrowheads in (A, C and E) label some PGP 9.5-positive neurons. Scale bar: 40 μm. Images were taken using a Zeiss Axiophot microscope.
Table 4.
Stereological analysis of nerve fibres in mice after 1 and 4 weeks sham/TAC
| Sham | 1-week TAC | 4-weeks TAC | |
|---|---|---|---|
| L(nf, lv) (m) | 45.52 (17.68) | 21.20 (12.55)* | 15.34 (4.63)* |
| L(ax) (m) | 81.0 (29.2) | 27.0 (15.8)* | 17.3 (4.6)* |
| QQ(ax/nf) | 1.78 (0.16) | 1.28 (0.03)* | 1.15 (0.14)* |
ax, axon; L, length; lv, left ventricle; nf, nerve fibre; QQ(ax/nf), number of axon profiles per nerve fibre profile; TAC, transverse aortic constriction.
Data are presented as mean (SD).
P < 0.05 vs. sham.
Fig 6.
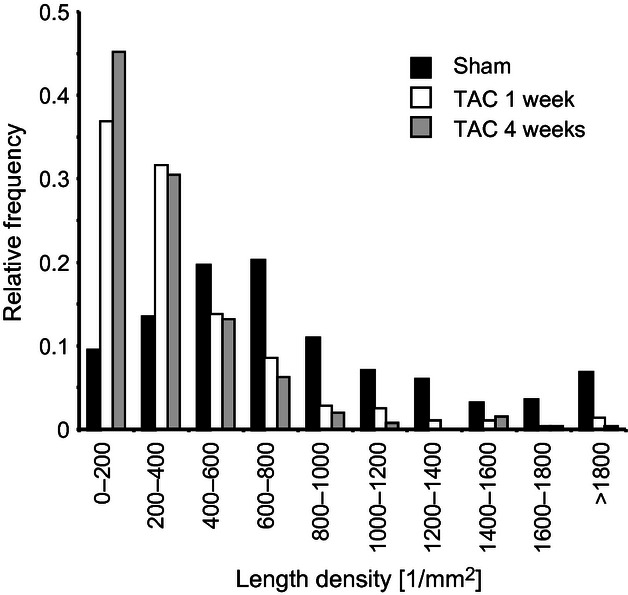
Distribution of nerve fibre length density. Histogram demonstrating the relative distribution of length density of nerve fibres. Due to the decreased innervation density, the heterogeneity of myocardial areas is increased in both transverse aortic constriction (TAC) groups.
Fig 7.

PGP 9.5-positive neurons are also positive for tyrosine hydroxylase. In sham (A–C), 1 week after TAC (D–F) and 4 weeks after TAC (G–I), all observed PGP 9.5-positive nerve fibres were also stained by tyrosine hydroxylase (arrowheads). (A, D and G) Tyrosine hydroxylase staining; (B, E and H) PGP 9.5 staining; (C, F and I) the merged channel including tyrosine hydroxylase (green), PGP 9.5 (red) and nuclei (blue). Scale bar: 50 μm. Images were taken using a Zeiss confocal laser-scanning microscope (LSM 510 Meta).
Capillaries
The total length of capillaries was similar in all groups. The reciprocal of the length density, a parameter that indicates the supply area of capillaries, showed a tendency to elevated levels in both TAC groups compared with sham mice. The volume and the mean cross-sectional area of the capillaries and the capillary lumen were not different among the groups; however, the volume and the arithmetic mean thickness of the capillary endothelium was significantly larger in the 4-weeks TAC group compared with sham (Table 5).
Table 5.
Stereological analysis of capillaries in mice after 1 and 4 weeks sham/TAC
| Sham | 1-week TAC | 4-weeks TAC | |
|---|---|---|---|
| L(cap,lv) (m) | 387 (75) | 459 (136) | 520 (191) |
| 1/LV(cap/lv) (µm²) | 231 (48) | 288 (67) | 342 (113) |
| V(caplum,lv) (mm³) | 3.17 (1.73) | 2.65 (1.26) | 3.42 (1.29) |
| V(endo,lv) | 2.22 (0.39) | 3.56 (0.95)* | 4.48 (0.80)* |
| A(cap) (µm) | 14.2 (5.7) | 13.4 (0.8) | 17.4 (7.8) |
| A(caplum) (µm²) | 8.33 (4.48) | 5.50 (1.45) | 7.96 (5.72) |
| τ(endo) (nm) | 395 (124) | 509 (163) | 556 (139)* |
A, mean cross-sectional area; cap, capillary; caplum, capillary lumen; endo, capillary endothelium; L, length; lv, left ventricle; LV, length density; V, volume; τ, arithmetic mean thickness.
Data are presented as mean (SD).
P < 0.05 vs. sham.
Discussion
The major finding of the present study was that murine left ventricles subjected to pressure overload had already reduced myocardial innervation at the stage of compensated hypertrophy. Furthermore, both total axon length and the mean number of axons per nerve fibre were progressively reduced after aortic banding, indicating further aggravation of the denervation process during the transition to HF. These findings revealed that hypoinnervation and regional heterogeneity precede left ventricular HF.
Previous reports suggested either hypoinnervation (Himura et al. 1993; Ungerer et al. 1998; Kaye et al. 2000; Ogita et al. 2001) or hyperinnervation (Cao et al. 1999; Kimura et al. 2007) as part of the structural remodelling process of the nervous system in experimental and human HF. Several reasons may contribute to this disparity, including methodological approaches that were employed to determine the innervation density and different HF aetiologies. In our study, we took advantage of design-based stereology and made use of a recently established method by which the total length of nerve fibres, the total length of axons and the mean number of axons per nerve fibre are estimated within a defined reference volume. The advantage of these methods is that they are not influenced by changes in the reference volume, viz. the volume of the left ventricle, or other parameters (Mühlfeld et al. 2010a,b). Furthermore, HF aetiology was controlled by the use of an experimental pressure overload model, that allowed the separation of morphological alterations in myocardial innervation at the stage of compensated hypertrophy vs. manifest HF (Balakumar et al. 2007; Jacobshagen et al. 2008). Accordingly, left ventricular murine myocardium subjected to mechanical overload induced by TAC effectively compensated the hypertrophic response at 1-week post-TAC. Concentric hypertrophy subsequently deteriorated into eccentric hypertrophy associated with progressive left ventricular dilatation and reduced ejection fraction, ultimately resulting in body weight reduction and HF.
Regional or distributional changes of the nerve fibres are involved in the generation of ventricular arrhythmias and sudden cardiac death as they may induce foci with varying adrenergic stimulus (Cao et al. 2000). We found that the regional variation of nerve fibre density in the setting of pressure overload was shifted towards lower relative frequencies of areas with specific nerve fibre length densities, indicating a substantial increase in areas with a very low degree of innervation. All nerve fibres stained by the pan-neuronal marker PGP 9.5 were also positive for tyrosine hydroxylase, the key enzyme of noradrenalin synthesis, indicating that there was not a major degree of transdifferentiation of the neurons to a different transmitter expression as observed by others (Kanazawa et al. 2010).
Altogether these data confirm that the regional heterogeneity of the sympathetic innervation increases in hypertrophy and HF induced by pressure overload (Cao et al. 2000). Considering the preserved left ventricular ejection fraction in the compensated hypertrophy together with increased occurrence of arrhythmias (Artham et al. 2009), it is possible that both the decreased total innervation and the increased spatial heterogeneity contribute to unbalanced neural activation and increased arrhythmogenic potential during the progression of hypertrophy and HF.
The stereological analysis confirmed an increase in cardiomyocyte and myofibrillar volume as a hypertrophic response underlying increased relative heart weight due to mechanical overload. From the present data it cannot be concluded whether the increased total volume of cardiomyocytes is caused by increased cardiomyocyte size, number or a combination of both mechanisms. Although it has been widely accepted for many years that cardiomyocytes are terminally differentiated cells, recent work has questioned this dogma (Kajstura et al. 2004, 2012). Additionally, apoptosis was postulated to be a major contributor to the transition from hypertrophy to HF (Bing, 1994). As there are currently no conclusive design-based stereological studies on pressure-induced cardiac hypertrophy with respect to cardiomyocyte number, this important topic warrants further attention.
Although the total length of capillaries showed a trend to elevated levels, there was also a trend to an elevated average capillary supply area (reciprocal of capillary length density). This indicates that the increase in capillarization associated with hypertrophy did not follow the increase in cardiomyocyte volume, suggesting inadequate capillary supply area in the cardiac tissue. Increased thickness of the capillary endothelium at the early stage of hypertrophy is typical for hypertension, but is also observed in diabetic hearts, and is interpreted as a degenerative process (Okruhlicova et al. 2005), or an adaptation to angiogenesis or increased blood flow (Masuda et al. 2003). In addition to the increase in myocyte size, the increase in endothelial thickness may attenuate the oxygen diffusion properties and further the progression from hypertrophy to HF.
Concluding remarks
In conclusion, this study provides quantitative morphological evidence for deleterious reduction of the myocardial innervation in early stages of LVH induced by pressure overload. We suggest that decreased nerve fibre density and increased regional heterogeneity may contribute to the enhanced arrhythmogenic potential in hypertrophy induced by pressure overload.
Acknowledgments
The authors thank Eva-Maria Gutschi, the staff of the Institute for Biomedical Research Animal Facility and Center of Biomedical Research (ZMF) for excellent technical assistance. In addition, the authors are thankful to Tamara Papadakis, Gerhard Kripp, Gerd Magdowski (Gießen) and Susanne Kuhlmann (Hannover) for expert technical assistance with the preparation of the microscopic sections. The authors declare that they have no conflict of interest.
Author contributions
CM, JS, HH, AS, BP and SS contributed to concept/design, data analysis and interpretation, drafting of the manuscript, critical revision and approval of the manuscript. CM and JS performed microscopic acquisition of data, AS carried out the echocardiography, and SS performed the animal experiments.
References
- Anversa P, Beghi C, Levicky V, et al. Morphometry of right ventricular hypertrophy induced by strenuous exercise in rat. Am J Physiol. 1982;243:H856–H861. doi: 10.1152/ajpheart.1982.243.6.H856. [DOI] [PubMed] [Google Scholar]
- Anversa P, Ricci R, Olivetti G. Quantitative structural analysis of the myocardium during physiologic growth and induced cardiac hypertrophy: a review. J Am Coll Cardiol. 1986;7:1140–1149. doi: 10.1016/s0735-1097(86)80236-4. [DOI] [PubMed] [Google Scholar]
- Artham SM, Lavie CJ, Milani RV, et al. Clinical impact of left ventricular hypertrophy and implications for regression. Prog Cardiovasc Dis. 2009;52:153–167. doi: 10.1016/j.pcad.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Balakumar P, Singh AP, Singh M. Rodent models of heart failure. J Pharmacol Toxicol Meth. 2007;56:1–10. doi: 10.1016/j.vascn.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Bing OH. Hypothesis: apoptosis may be a mechanism for the transition to heart failure with chronic pressure overload. J Mol Cell Cardiol. 1994;26:943–948. doi: 10.1006/jmcc.1994.1115. [DOI] [PubMed] [Google Scholar]
- Cao JM, Qu Z, Kim YH, et al. Spatiotemporal heterogeneity in the induction of ventricular fibrillation by rapid pacing: importance of cardiac restitution properties. Circ Res. 1999;84:1318–1331. doi: 10.1161/01.res.84.11.1318. [DOI] [PubMed] [Google Scholar]
- Cao JM, Fishbein MC, Han JB, et al. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101:1960–1969. doi: 10.1161/01.cir.101.16.1960. [DOI] [PubMed] [Google Scholar]
- Chien KR. Stress pathways and heart failure. Cell. 1999;98:555–558. doi: 10.1016/s0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- Eisele JC, Schaefer IM, Nyengaard JR, et al. Effect of voluntary exercise on number and volume of cardiomyocytes and their mitochondria in the mouse left ventricle. Basic Res Cardiol. 2008;103:12–21. doi: 10.1007/s00395-007-0684-x. [DOI] [PubMed] [Google Scholar]
- Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- Gruber C, Kohlstedt K, Loot AE, et al. Stereological characterization of left ventricular cardiomyocytes, capillaries, and innervation in the nondiabetic, obese mouse. Cardiovasc Pathol. 2012a;21:346–354. doi: 10.1016/j.carpath.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Gruber C, Nink N, Nikam S, et al. Myocardial remodelling in left ventricular atrophy induced by caloric restriction. J Anat. 2012b;220:179–185. doi: 10.1111/j.1469-7580.2011.01453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbenkian S, Wharton J, Polak JM. The visualisation of cardiovascular innervation in the guinea pig using an antiserum to protein gene product 9.5 (PGP 9.5) J Auton Nerv Syst. 1987;18:235–247. doi: 10.1016/0165-1838(87)90122-6. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Himura Y, Felten SY, Kashiki M, et al. Cardiac noradrenergic nerve terminal abnormalities in dogs with experimental congestive heart failure. Circulation. 1993;88:1299–1309. doi: 10.1161/01.cir.88.3.1299. [DOI] [PubMed] [Google Scholar]
- Hu P, Zhang D, Swenson L, et al. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signalling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–H1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- Jacobshagen C, Grüber M, Teucher N, et al. Celecoxib modulates hypertrophic signalling and prevents load-induced cardiac dysfunction. Eur J Heart Fail. 2008;10:334–342. doi: 10.1016/j.ejheart.2008.02.013. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Leri A, Castaldo C, et al. Myocyte growth in the failing heart. Surg Clin North Am. 2004;84:161–177. doi: 10.1016/S0039-6109(03)00215-9. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Rota M, Cappetta D, et al. Cardiomyogenesis in the aging and failing human heart. Circulation. 2012;126:1869–1881. doi: 10.1161/CIRCULATIONAHA.112.118380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kanazawa H, Ieda M, Kimura K, et al. Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J Clin Invest. 2010;120:408–421. doi: 10.1172/JCI39778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye DM, Vaddadi G, Gruskin SL, et al. Reduced myocardial nerve growth factor expression in human and experimental heart failure. Circ Res. 2000;86:e80–e84. doi: 10.1161/01.res.86.7.e80. [DOI] [PubMed] [Google Scholar]
- Kiatchoosakun S, Restivo J, Kirkpatrick D, et al. Assessment of left ventricular mass in mice: comparison between two-dimensional and m-mode echocardiography. Echocardiography. 2002;19:199–205. doi: 10.1046/j.1540-8175.2002.00199.x. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ieda M, Kanazawa H, et al. Cardiac sympathetic rejuvenation. A link between nerve function and cardiac hypertrophy. Circ Res. 2007;100:1755–1764. doi: 10.1161/01.RES.0000269828.62250.ab. [DOI] [PubMed] [Google Scholar]
- Leyton RA, Sonnenblick EH. The ultrastructure of the failing heart. Am J Med Sci. 1969;258:304–327. [PubMed] [Google Scholar]
- Manning WJ, Wei JY, Katz SE, et al. In vivo assessment of LV mass in mice using high-frequency cardiac ultrasound: necropsy validation. Am J Physiol. 1994;266:H1672–H1675. doi: 10.1152/ajpheart.1994.266.4.H1672. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Ferrans VJ, Roberts WC. Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am J Pathol. 1975;79:387–434. [PMC free article] [PubMed] [Google Scholar]
- Masuda H, Kawamura K, Nanjo H, et al. Ultrastructure of endothelial cells under flow alteration. Microsc Res Techn. 2003;60:2–12. doi: 10.1002/jemt.10237. [DOI] [PubMed] [Google Scholar]
- Mattfeldt T, Mall G, Gharehbaghi H, et al. Estimation of surface area and length with the orientator. J Microsc. 1990;159:301–317. doi: 10.1111/j.1365-2818.1990.tb03036.x. [DOI] [PubMed] [Google Scholar]
- Mendez J, Keys A. Density and composition of mammalian muscle. Metabolism. 1961;9:184–188. [Google Scholar]
- Mühlfeld C, Nyengaard JR, Mayhew TM. A review of state-of-the-art stereology for better quantitative 3D morphology in cardiac research. Cardiovasc Pathol. 2010a;19:65–82. doi: 10.1016/j.carpath.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Mühlfeld C, Papadakis T, Krasteva G, et al. An unbiased stereological method for efficiently quantifying the innervation of the heart and other organs based on total length estimations. J Appl Physiol. 2010b;108:1402–1409. doi: 10.1152/japplphysiol.01013.2009. [DOI] [PubMed] [Google Scholar]
- Nyengaard JR, Gundersen HJ. The isector: a simple and direct method for generating isotropic, uniform random sections from small specimens. J Microsc. 1992;165:427–431. [Google Scholar]
- Ogita H, Shimonogata T, Fukunami M, et al. Prognostic significance of cardiac (123)I metaiodobenzylguanidine imaging for mortality and morbidity in patients with chronic heart failure: a prospective study. Heart. 2001;86:656–660. doi: 10.1136/heart.86.6.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okruhlicova L, Tribulova N, Weismann P, et al. Ultrastructure and histochemistry of rat myocardial capillary endothelial cells in response to diabetes and hypertension. Cell Res. 2005;15:532–538. doi: 10.1038/sj.cr.7290322. [DOI] [PubMed] [Google Scholar]
- Pearlman ES, Weber KT, Janicki JS. Quantitative histology of the hypertrophied human heart. Fed Proc. 1981;40:2042–2047. [PubMed] [Google Scholar]
- Rakusan K, Tomanek RJ. Distribution of mitochondria in normal and hypertrophied myocytes from the rat heart. J Mol Cell Cardiol. 1986;18:299–305. doi: 10.1016/s0022-2828(86)80412-6. [DOI] [PubMed] [Google Scholar]
- Rakusan K, Flanagan MF, Geva T, et al. Morphometry of human coronary capillaries during normal growth and the effect of age in left ventricular pressure-overload hypertrophy. Circulation. 1992;86:38–46. doi: 10.1161/01.cir.86.1.38. [DOI] [PubMed] [Google Scholar]
- Toischer K, Rokita AG, Unsöld B, et al. Differential cardiac remodeling in preload vs. afterload. Circulation. 2010;122:993–1003. doi: 10.1161/CIRCULATIONAHA.110.943431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomanek RJ, Searls JC, Lachenbruch PA. Quantitative changes in the capillary bed during developing, peak, and stabilized cardiac hypertrophy in the spontaneously hypertensive rat. Circ Res. 1982;51:295–304. doi: 10.1161/01.res.51.3.295. [DOI] [PubMed] [Google Scholar]
- Ungerer M, Hartmann F, Karoglan M, et al. Regional in vivo and in vitro characterization of autonomic innervation in cardiomyopathic human heart. Circulation. 1998;97:174–180. doi: 10.1161/01.cir.97.2.174. [DOI] [PubMed] [Google Scholar]
- Wei S, Guo A, Chen B, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER. Stereological Methods. Practical Methods for Biological Morphometry. New York: Academic Press; 1979. Vol. 1. [Google Scholar]