

Abstract
Enzymatic activities in vivo occur in a crowded environment composed of many macromolecules. This environment influences DNA replication by increasing the concentration of the constituents, desolvation, decreasing the degrees of freedom for diffusion and hopping of proteins onto DNA, and enhancing binding equilibria and catalysis. However, the effect of macromolecular crowding on protein structure is poorly understood. Here we examine macromolecular crowding using the replication system of bacteriophage T7 and we show that it affects several aspects of DNA replication; the activity of DNA helicase increases and the sensitivity of DNA polymerase to salt is reduced. We also demonstrate, using SAXS analysis, that the complex between DNA helicase and DNA polymerase/trx is far more compact in a crowded environment. The highest enzymatic activity corresponds to the most compact structure. Better knowledge of the effect of crowding on structure and activity will enhance mechanistic insight beyond information obtained from NMR and X-ray structures.
Keywords: Macromolecular crowding, T7 replisome, DNA polymerase, Helicase, SAXS
Enzymatic processes observed in dilute buffer are often assumed to represent those in the cellular environment. However, most often this is not the case since the cell is filled with macromolecules in high density, leading to macromolecular crowding1. The in vivo concentration of molecules in the cytoplasm is between 50–400 mg/ml, ~70% being proteins2. Such crowding affects enzymatic activities both in vitro and in vivo. It is not always feasible to study the macromolecules under cellular conditions. To mimic “cellular conditions” inert crowding agents such as polyethylene glycol, dextran, and Ficoll are often used. Proteins can also be used as crowding agents with bovine serum albumin representing a common additive. There are–many examples of the effect of co–solutes on the properties of macromolecules and protein–nucleic acid complexes3. These co–solutes are thought to mimic the crowding effect in the cell that arises from DNA, RNA, and protein complexes. Macromolecular crowding and the thermodynamic effects have been recognized since the 1960's by the studies of Ogston4. Notwithstanding the wealth of information on the effect of macromolecular crowding, the effect of macromolecular crowding on structure-function relationships is poorly understood. Macromolecular crowding stabilizes the native state of a protein by destabilizing the unfolded state, thus compensating for the energetically unfavorable “folded” conformation5,6. An experimental and computational analysis has examined the effect of macromolecular crowding on structural changes between the folded and unfolded states of apoflavodoxin. These studies demonstrated that the effects of macromolecular crowding on protein structure are observable7. To assess the interactions between DNA molecules Parsegian and co-workers applied osmotic pressure in ordered arrays of DNA molecules and used X–ray diffraction to measure the structural changes8. The osmotic pressure method they developed utilizes the equivalent of the mechanical work (osmotic pressure) needed to bring macromolecules closer in spite of their repulsive interactions and the chemical work (removing of the solute) needed to concentrate the macromolecular subphase. In a different instance using far–UV circular dichroism spectroscopy Stagg et al9 showed the structural effects of elevated amounts of Ficoll 70 on apoflavodoxin. The theoretical radius of gyration (Rg) of apoflavodoxin indicates compactness in the crowded environment.
There are limited reports on the effect of molecular crowding on DNA replication. Previous studies revealed effects on several aspects of DNA metabolism among them: oriC replication10, blunt–end DNA ligation by DNA ligase11, activities of phage T4 DNA kinase12, the association of accessory proteins13, and the assembly of a phage T4 DNA polymerase holoenzyme14. Macromolecular crowding has also been used as a tool to detect interactions within the T4 replisome15.
In the present study we examine the effect of macromolecular crowding on the proteins that mediate leading-strand DNA synthesis. Phage T7 has evolved an economical mechanism for the replication of its DNA16. In contrast to eukaryotes or even E. coli, where tens of proteins are required, in the T7 system only four proteins account for the major reactions that occur at the replication fork: (i) the gene 5 DNA polymerase (gp5), and (ii) its processivity factor, the host encoded thioredoxin (trx), (iii) the gene 4 helicase–primase (gp4), and (iv) the gene 2.5 ssDNA binding protein (gp2.5). The limited number of proteins enables: (1) reconstitution of a replisome, (2) determination of the structures of functional complexes, (3) examination of the composition of active replisomes, and (4) studying the effect of macromolecular crowding. The structure of interacting species within the replisome is not known although extensive effort has been made in this direction17. Therefore alternative methods that provide low resolution structure such as small angle X-ray scattering (SAXS) are useful.
RESULTS
Macromolecular crowding reduces diffusion
A computer simulation of the random walk of lysozyme within the volume of an E. coli cell in the presence or the absence of 18,000 ribosomes is shown in Figure 1a. Restriction of spatial movement caused by decreased diffusion is observed in the crowded environment. One important consequence of this effect is the stabilization of weakly bound complexes. In addition to the effect on diffusion, macromolecules occupy between 5–40% of the cell volume. This volume is not accessible to other molecules and the resulting effect is designated as the excluded volume effect18 whose magnitude depends on the size of the molecule in question. Another major effect of macromolecular crowding is solvent entropy. In a mixture of macromolecules and smaller molecular components the loss of entropy of the large molecules is compensated by the gain in entropy of the smaller molecules. Thus, desolvation around macromolecules enhances the entropy that compensates for the loss in the conformational entropy that accompanies folding or complex formation (for review see19).
Figure 1. Effects of macromolecular crowding.

The most widely used crowding agent is polyethylene glycol (PEG). PEG is a straight–chain polymer containing simple repeating subunits; PEG 1 kDa, used in the present study, has 21 repeating units and is best modeled as a spherical particle20. PEG and other crowding agents can increase the rate of enzymatic reactions21, alter reaction products1, protect macromolecules from thermal denaturation22, accelerate protein folding23, and facilitate nucleic acids renaturation24. PEG also induces precipitation of proteins at high concentration (30%) and is used in protein purification. We calculated the volume that PEG molecules of different sizes occupy at different concentrations (Fig. 1b). For example, at a concentration of 4% PEG 1 kDa, 8% of the solution is occupied by PEG.
Macromolecular crowding alters the binding properties and rate constants of a number of enzymes including DNA polymerases. We have examined the effect of increasing the concentration of PEG on the structure of the replication proteins and on the reactions they catalyze. To determine if the macromolecular crowding effect of PEG is comparable to that found in vivo on the diffusion of a protein, we used NMR to measure the diffusion of lysozyme (15 kDa) (Fig. 1c). The diffusion coefficient in the presence of PEG is reduced 13-fold, a ratio similar to that observed in cells25.
Macromolecular crowding enhances the activity of the gp5/trx complex
The processivity factor, E. coli trx, binds with high affinity (5 nM) to T7 gp5 in a 1:1 stoichiometry26 to increase the binding of gp5 to a primer–template 20–80–fold26. The increased affinity enhances the processivity of gp5 from less than 50 nucleotides per binding event to approximately 80026 resulting in a dramatic increase in the macroscopic rate of DNA synthesis. We have examined the effect of PEG on the polymerase activity of gp5 when mixed with trx in a ratio of 1:1 (Fig. 2b, left). PEG increases the activity of gp5 as much as trx in the same molar ratio relative to gp5 (Fig. 2b, right).
Figure 2. Gp5/trx polymerase activity and macromolecular crowding.
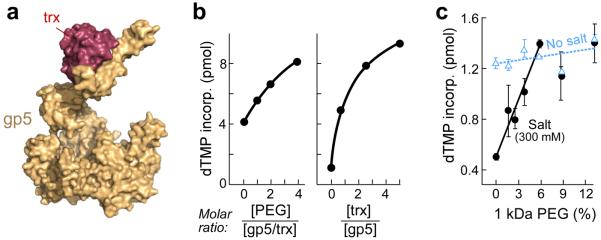
(a) T7 DNA polymerase (gp5, yellow) bound to its processivity factor E. coli thioredoxin (trx, red) polymerizes nucleotides continuously on the leading strand. The crystal structure of gp5/trx bound to primer template and an incoming nucleotide (PDB id code: 1t8e34) in a view from the side. The figure was created using PyMOL (http://www.pymol.org). (b) Polymerase activity of gp5 with increasing amounts of trx (right). The activity was measured in a standard reaction containing 40 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 10 mM DTT, 50 mM potassium glutamate, 0.25 mM dATP, dCTP, dGTP, and [α–32P] dTTP, 20 nM primed M13 DNA, 20 nM gp5, and the indicated amount of trx. After incubation at 37 °C for 10 min the amount of [α–32P] dTMP incorporated into DNA was measured. Increased polymerase activity of gp5 and trx premixed in a ration of 1:1 is observed as the content of PEG is increased (left). (c) Effect of macromolecular crowding by PEG on polymerase activity of gp5/trx in the presence or the absence of salt. Polymerase activity was measured under similar buffer conditions as in (a). Gp5 (5 nM) was mixed with trx (25 nM) in the presence of 300 mM NaCl (black) or in absence of NaCl (cyan). The error bars were derived from three independent experiments.
We have shown previously that the activity of gp5/trx is reduced as the NaCl concentration is increased from 50 mM to 300 mM27. At 125 mM NaCl, gp5/trx retains 50% activity. The high salt presumably masks the charged residues at the DNA binding surface. We proposed that the difference in the behavior of gp5 and gp5/trx at high salt concentration arises from a different distribution of charged amino acids on the DNA binding interface. The binding of trx to gp5 leads to a conformational change leading to a different electrostatic potential27.
We examined the effect of PEG on the activity of gp5/trx in the absence or presence of 300 mM NaCl, a concentration that diminished the activity and the binding of gp5/trx to DNA in the earlier studies. Macromolecular crowding does not affect the processivity of DNA polymerase15 and indeed increasing PEG concentrations result in only a slight (<10%) increase in polymerase activity in the absence of NaCl (Fig. 2c, cyan). However, in the presence of 300 mM NaCl there is a 3-fold increase upon the addition of PEG 1 kDa up to 4% where gp5/trx retains full activity (Fig. 2c, black).
Macromolecular crowding enhances helicase but not primase activity of gp4
Gp4 is a multifunctional enzyme bearing helicase and primase activities in the same polypeptide. We have used individual domains of gp4 to focus on specific activities (Fig. 3a, bottom). Gp4A (residues 1–566, 63-kDa) is the full-length protein. Gp4B (residues 64–566, 56-kDa) arises from an internal initiation codon and is present in phage-infected cells in amounts equivalent to the full length protein. Gp4B lacks the N-terminal zinc-binding subdomain, an essential component for primer synthesis.
Figure 3. The effect of macromolecular crowding on gp4 activity.

(a) Front (top panel) and side View (middle panel)of a gp4 model. Gp4 has both primase and helicase activities in the same polypeptide chain. Subunits of the heptameric crystal structure of gp4B (PDB entry 1q5749) were aligned with the hexameric helicase fragment (PDB ID code: 1e0k30) and primase fragment (PDB entry 1nui44) of gp4. Schematic representation of gp4 constructs (bottom). The boundaries for the helicase and primase domains of gp4 are depicted45. The three constructs containing the C-terminal helicase domain of gp4 are denoted as gp4A, B, and D. Residue numbers are as indicated. The figure was created using PyMOL (http://www.pymol.org). (b) Effect of PEG on oligonucleotide synthesis by primase fragment. The standard reaction contained the oligonucleotide 5'-GGGTCA10-3' containing the primase recognition sequence, 200 μM [α-32P]-CTP and ATP, and increasing amounts of 1kDa PEG (0, 1.25, 2.5, 5, 10%) in a buffer containing 40 mM Tris-HCl (pH 7.5), 10 mM MnCl2, 10 mM DTT, and 50 mM potassium glutamate. The quenched samples were loaded onto 25% polyacrylamide sequencing gel containing 3 M urea and visualized using autoradiography. (c) Effect of PEG on the DNA unwinding activity of gp4B. The DNA fork depicted (right) was prepared by partially annealing a 5'–32P labeled 45-mer oligonucleotide to a 65-mer oligonucleotide. DNA unwinding activity was performed in a standard reaction containing 40 mM Tris–HCl (pH 7.5), 50 mM potassium glutamate, increasing amounts of PEG 1 kDa (0, 0.25, 0.5, 1, 2, 4 and 8%), and 400 nM of gp4B. The gel shows the separation of unwound ssDNA (bottom) from the dsDNA substrate in a 10% non-denaturing gel. (d) Effect of highly crowding conditions (8% PEG) on the DNA-dependent dTTPase activity of gp4B. The dTTPase activity was measured in the presence of 5 mM dTTP and 8% PEG and with various concentrations of gp4B (0–200 nM). Difference in the dTTPase activity in the presence and absence of high concentration of PEG is denoted as residual.
The effect of macromolecular crowding on primase and helicase activities was examined (Fig. 3b–d). On a DNA template containing the primase recognition site 5'-GGGTC-3' the primase fragment catalyzes the synthesis of the di-, tri-, and tetraribonucleotides pppAC, pppACC, and pppACCC28. We examined the synthesis of oligoribonucleotides in the presence of PEG. The reaction conditions involve incubating the primase fragment (gp4, residues 1–271, 30-kDa) with an oligonucleotide containing a primase recognition sequence, [α-32P] CTP, and ATP, and increasing amounts of PEG (0–10%). The radioactively labeled oligoribonucleotide are separated on a denaturing polyacrylamide gel and the radioactivity measured on an autoradiogram. Macromolecular crowding does not affect tetramer synthesis but slightly enhances trinucleotide formation (Fig. 3b). In the unwinding assay a radiolabeled DNA strand partially annealed to a complementary strand creates a fork structure (see cartoon in Fig. 3c). Unwinding of the DNA releases the radiolabeled strand that has a mobility in 10% TBE gels greater than that of the fork structure. In the experiment (Fig. 3c) the DNA was incubated with 400 nM gp4B, dTTP, and increasing amounts of PEG 1 kDa (0 – 8%). Increasing amounts of PEG results in enhancement of unwinding activity as observed by the radiolabeled DNA strand that migrates more rapidly than the fork substrate. Concentrations of PEG higher than 4% result in protein precipitation (Fig. 3c, lane 8).
The DNA-dependent dTTP hydrolysis activity of gp4B in 8% PEG was examined (Fig. 3d). At this high concentration of PEG the dTTP hydrolysis activity of gp4B is enhanced up to 70 nM of the enzyme. The dotted line represents the theoretical activity taking the excluded volume effect of PEG into account. The activity in the presence of PEG is higher than that predicted, suggesting that excluded volume is not the only factor contributing to the increased activity. Increasing amounts of PEG enhances not only the DNA dependent dTTPase activity but also the DNA–independent dTTPase (Supplementary Fig. S1b), presumably due to an enhancement of oligomerization of gp4B.
Macromolecular crowding enhances the concerted activity of gp4 and gp5/trx
Leading- and lagging-strand synthesis involves interactions between gp5/trx and the helicase and primase. On the leading-strand the helicase activity unwinds the DNA ahead of gp5/trx to which it is tightly bound. The effect of PEG on leading-strand synthesis was measured using circular M13 DNA containing a replication fork (Fig.4a, top). Gp5/trx and gp4B (helicase but no primase activity) carry out strand-displacement synthesis (Fig. 4a). The addition of increasing amounts of PEG increases the rate of DNA synthesis with a 3-fold increase occurring at 4% PEG.
Figure 4. The effect of macromolecular crowding on gp5/trx and gp4 interactions.

The three proteins move the replication fork at a rate of approximately 150 nt/sec at 25 degrees. The bifunctional gene 4 helicase-primase (gp4) assembles on the lagging-strand as a hexamer where it forms a complex with gp5/trx and unwinds the DNA duplex. (a) Effect of PEG on strand-displacement DNA synthesis mediated by gp5/trx and gene 4 helicase (gp4B). Using M13 dsDNA with a 5' ssDNA tail (top), the efficiency of strand-displacement DNA synthesis in the presence of PEG was determined. The standard reaction contained the dsM13 template (10 nM), 0.3 mM dATP, dGTP, dCTP and [α–32P] dTTP (0.1 μCi), 10 nM gp5/trx, 200 nM monomeric concentrations of gp4B, and increasing amounts of PEG 1 kDa (0–8%). After incubation for 30 min at 37 °C and the amount of DNA synthesis was determined by the amount of [α–32P] dTTP incorporated into DNA. (b) Primase-dependent DNA synthesis. The reaction was similar to (a) except that gp4A replaced gp4B and 10 nM M13 ssDNA replaced the dsM13 DNA. The reaction buffer also contained ATP and CTP (100 μM each). The amounts of primase-dependent DNA synthesis was determined by measuring the incorporation of [32P] dTMP into DNA. The error bars were derived from three independent experiments.
Lagging-strand synthesis requires gp4A, the full length gp4, for the synthesis of olioribonucleotides to initiate the synthesis of Okazaki fragments28. To examine the synthesis of primers and their transfer to gp5/trx we have used M13 ssDNA for the primase to synthesize oligoribonucleotides and for gp5/trx to extend the primers (Fig.4b, top). In this assay, the full-length gp4 (gp4A) was used. To initiate DNA synthesis the primase must first synthesize tetraribonucleotides on the DNA and then transfer them to gp5/trx. ATP and CTP are provided in addition to the four dNTPs since the primers synthesized are pppACCC, pppACAC, and pppACCA. As shown in Figure 4b primase-dependent DNA synthesis increases up to 4-fold upon the addition of PEG 1 kDa. However, oligoribonucleotide synthesis is not affected (Fig. 3b). The decrease of both strand-displacement DNA synthesis and primase-dependent DNA synthesis at PEG concentration above 4 % is due to precipitation of the proteins (Supplementary Fig. S2).
Macromolecular crowding makes gp5/trx-gp4 complex more compact
During leading-strand synthesis gp5/trx and gp4 form a stable complex that increases processivity16. Two interactions of gp5/trx and gp4 are responsible for this increase in processivity. A tight interaction of the two proteins results in a processivity of approximately 5000 nt. The processivity is limited to 5000 nucleotides by the occasional dissociation of gp5/trx. A second interaction of the C-terminal tail of gp4 with a basic patch on gp5 captures gp5/trx in the event it dissociates and allows it to return to the primer, increasing the processivity to greater than 17,000 nucleotides. We have used SAXS to examine the structure of this later complex and to determine the effect of macromolecular crowding on the complex. Gp4D (residues 241–566, 36-kDa) is the minimal fragment of the helicase that forms hexameric rings (Fig. 3a, grey). Gp4D contains the helicase domain and the linker connecting the helicase and primase domains. Although gp4D has less helicase activity than that of gp4A or gp4B it forms stable hexamers in the presence of ssDNA and dTTP and it binds gp5/trx at the replication fork to form a stable complex29. All of the features of gp4D and its available hexameric crystal structure30 facilitate solution structural experiments using SAXS. To assure the stability of the hexamer of gp4D we have used a 15 nucleotide oligonucleotide to which gp4D binds tightly in the presence of the non-hydrolyzable analogue β,γ methylene dTTP31.
It is crucial to ascertain that the complex between gp5/trx and gp4D can be identified prior to the addition of PEG. Therefore, we carried out SAXS analysis on samples containing a constant amount of gp4D but increasing amounts of gp5/trx. This procedure enables the detection of weakly interacting high molecular weight protein complexes. We recently applied this method to translation initiation complexes32. This method also provides information on the stoichiometry of proteins within the complex. As shown in the scheme presented in Figure 5a mixing of two interacting proteins will result in an increase at the Rg values. The amount of one protein needed to obtain saturation of the radius of gyration (Rg) value provides information on the tendency of the species to form a complex. Thus, strong complexes are characterized by a sharp rise of the Rg value until saturation with only small amount of protein that is titrated over the other, and vice versa. If the proteins do not form a complex Rg value represent an intermediate value of the two.
Figure 5. SAXS reconstitution assay of the gp5/trx/gp4D complex.

(a) Schematic view of the experimental design. Samples containing proteins A and B are premixed in various ratios and placed in the sample cell. Rg values were extracted from the SAXS data. Increased Rg values indicate that a higher order specie is formed. Intermediate Rg value from the individual proteins in the mixture is indicative of non-interacting species. (b–c) Small angle X–Ray scattering (SAXS) curves of hexameric gp4D (5 μM) premixed with increasing amounts of gp5/trx (0, 2, 4, 8, 12, 24 μM). Raw SAXS data (b) and the corresponding Guinier plots (c) for every sample. Colors indicate a gradual increase in the gp5/trx concentration (red to yellow). (c) The Rg values for the complex formed derived from these data and determined using Guinier plots. The insets in both (b) and (c) represent the theoretical SAXS curves of linearly combined spectra (from the available crystal structures) of the putative complex and the free proteins in solution. The data represent a complex between gp4D and gp5/trx in a molar ratio of 1:2, respectively. The theoretical SAXS curves of gp4D bound to gp5trx in a molar ratio of 1:1, 1:2, 1:3, and 1:4, respectively, are presented in Supplementary Figure S4. (d) Scattering intensities (I0) shown as a function of concentration of gp5/trx. Data presents scattering at zero angle of gp4D (5 μM) and increasing amounts of gp5/trx (0–24 μM), corresponding to the amount of electron scatterers at any sample. (e) SAXS results plotting the radius of gyration (Rg) against gp5/trx and gp4D ratios. Experimental SAXS data were collected for gp4D (5 μM) and increasing amounts of gp5/trx (0–24 μM) in a buffer containing 20 mM Tris–HCl (pH 7.5), 50 mM potassium glutamate, and 2 mM DTT. Gp4D was premixed with 6.6 μM 15mer ssDNA and 0.5 mM β, γ methylene dTTP to form hexameric molecules. Radius of gyration serves as an indicator for the formation of higher-order protein complexes. The dashed line represents a theoretical curve of Rg values that would be obtained if the components did not interact.
The Rg values, corresponding to the size of the measured particles, were derived from the Guinier plots (Fig. 5c) that represent the linear small angle part of the X–ray scattering data (Fig. 5b). A typical binding curve is observed upon plotting of Rg against the ratio between gp5/trx and gp4D providing information on complex formation (Fig. 5e). The Rg values are saturated above the ratio of 2:1 for gp5/trx to gp4D hexameric ring, suggesting at least 2 gp5/trx per hexamer (Fig. 5e). I0 is proportional to the molecular mass (number of atomic scatterers). An increase in I0 due to the increase in the molecular mass is observed (Fig. 5d). There is no evidence of aggregation from Guinier analysis (Fig. 5c). We have linearly combined artificial SAXS data to merge the contribution of a theoretical SAXS signal of the complex and the individual proteins (Supplementary Fig. S4). The artificial SAXS data was generated using CRYSOL33 from the crystal structure of gp5/trx (PDB ID code: 1t8e34) and gp4D (PDB ID code: 1e0k30) and a model of the complex. These artificial SAXS spectra serve as a reference for the reconstitution using SAXS to determine how many gp5/trx are bound to one hexameric gp4D. The settings of a theoretical complex by which 2 polymerases are bound to one helicase hexameric ring fit the data.
The effect of PEG on the structure of gp4D and gp5/trx in solution was examined by SAXS. No change in the Rg of gp4D or gp5/trx is observed upon titration PEG to the sample (Supplementary Fig. S3) indicating that there is no conformational transition of the individual proteins by PEG.
Rg of gp5/trx (without primer-template DNA or nucleotides) mixed with gp4D at the ratio of 1:1 is equal to 63 Å (Fig. 5e)). This Rg value fits well with the dimension obtained from a structural model of the complex constructed by the crystal structures of gp5/trx and gp4D29. Figure 6 shows the Rg values of the complex formed with a mixture of gp4D hexamer and gp5/trx at a ratio of 1:1 and increasing amounts of PEG (0–10%). Up to 4% PEG there is a gradual decrease in the Rg indicating compactness of the volume of the gp5/trx-gp4 complex (Fig. 6). The results demonstrate conformational changes in the complex as a result of macromolecular crowding.
Figure 6. The effect of PEG 1kDa on the Rg of gp4D bound to gp5/trx.

The sample contained 4.5 μM gp4D (hexamer) premixed with 6.6 μM 15-mer ssDNA, 0.5 mM β, γ methylene dTTP and 5 μM gp5/trx. The reaction buffer contained 20 mM Tris-HCl (pH 7.5), 50 mM potassium glutamate, 2mM DTT, and increasing amounts of PEG 1 k (0–10%). The Rg values for the samples were derived from the SAXS data and determined using Guinier plots. SAXS Guinier plots at high PEG concentrations are presented in the inset.
DISCUSSION
We have examined the effect of macromolecular crowding on several of the reactions that occur at the replication fork. Addition of PEG cancels the inhibitory effect of salt on the activity of DNA polymerase (gp5/trx). This effect probably results from an increase in the surface of interaction with DNA, an interaction crucial for increasing processivity27.
Traditionally reconstitution of the gp5/trx complex has used a molar excess of trx although the reported Kd for trx binding to gp5 is 5 nM35. It is now evident that the requirement for an excess of trx is due to macromolecular crowding at higher protein concentration. The use of an inert crowding agent such as PEG or bovine serum albumin can eliminate the need for excessive concentrations of trx.
The active site in the helicase where hydrolysis of dTTP occurs is formed at the interface of two subunits (Rec A-like domains). Consequently, the helicase hexamer has a total of six active site clefts where nucleotides bind. PEG increases the efficiency of hydrolysis of dTTP and unwinding activity of the helicase. Does the increase in dTTPase activity result from an apparent increase of gp4 concentration? To answer this question we used 8% PEG to provide a high crowing environment and examined the DNA independent and the DNA-dependent dTTPase activity of gp4B. The apparent concentration of gp4 increased from 70 nM to 76 nM as the PEG concentration increased from 4% to 8% resulting in a decrease in volume of 8% (see Fig. 1b). In this new apparent concentration of gp4 the DNA-dependent dTTPase activity increased 2-fold. Therefore, the increase in the DNA-dependent dTTPase activity is not governed only by the increase in the apparent concentration of gp4B but also by an increase in hexamer formation.
Macromolecular crowding affects the two activities of gp4 differently although both activities reside in the same polypeptide chain. PEG does not increase tetraribonucleotide synthesis by primase fragment while increasing helicase activity. Interestingly, primer synthesis of primase fragment is not affected by PEG whereas utilization of ATP/CTP is increased (trimer formation) presumably due to an increase in the affinity of the primase to ATP/CTP. The weak binding of the primase fragment lacking the helicase domain to a DNA template has been observed previously using surface plasmon resonance36. The molecular basis for sequence recognition has not yet been elucidated. However, the negligible effect of PEG may indicate transient association and dissociation of the primase to its DNA template, a process affected by diffusion. In the context of the full length gp4, the priming activity is no longer bimolecular, and therefore, the influence of crowding is expected to be negligible. The effect of PEG on strand-displacement and primase-dependent DNA synthesis along with SAXS analysis of the structures suggest that addition of PEG promotes the association of gp4 to gp5 to form a stable replisome.
Protein folding and protein–protein interactions are driven by the same forces. These processes are governed by hydrophobic interactions (polymerization mode), salt-dependent polar (electrostatic mode) and hydrogen–bond interactions. To adapt to environmental stress cells have evolved an array of effective countermeasures. Osmotic stress is counterbalanced by “osmolytes”, small, charged or polar solutes that balance the stress. One direct effect of osmotic stress is that protein side chains are affected by water depletion37. Charged residues must search for other interactions when water is absent leading to interactions with other charged residues. We have found, using NMR, that the relaxation of protein residues is affected by macromolecular crowding presumably due to water depletion (Akabayov et al., manuscript submitted).
The loss of conformational entropy in a protein during folding or complex formation is an outcome of the release of water molecules to minimize exposure of hydrophobic patches to solvent38. Therefore water has a central role in protein assembly and thermodynamics is the driving forces involving the motion of water molecules. Release of water molecule by PEG increases the entropy that compensates for the decrease in the entropy upon complex formation. The depletion of water from both DNA polymerase and DNA helicase would presumably increase the contact interaction to DNA and alter the conformation of the protein to enhance its movement on DNA.
Another challenge is to characterize the structure of weakly binding protein counterparts. Traditionally, attention has been directed toward the structure of stable complexes of macromolecules. Most biophysical characterizations are dependent on the acquisition of a stable species, a requirement not easily met with weakly interacting macromolecules. Interest in the structure of weakly interacting components in complexes has risen significantly over the years, particularly with regard to the high molecular weight transient complexes involving DNA replication. Weak interactions between T7 DNA polymerase and gp4 are not short lived39. We assume that complex formation between DNA polymerase and gp4 remain stable (although governed by weak interactions) to enable copying the entire genome.
We have shown how SAXS can be used to probe high molecular weight complexes such as a gp5/trx-gp4. The number of DNA polymerases in a functional replisome is not known. SAXS can provide an estimate of the stoichiometry of gp5/trx on a hexamer of gp4D within a reconstituted complex. We have used SAXS to determine the number of gp5/trx molecules bound to a hexamer of gp4 when neither protein is in a replication mode. Such a complex represents that formed the replication fork and involves the C-terminal tail of gp4 and a basic patch on gp5/trx. It is this complex that provides a backup gp5/trx in the event that gp5/trx dissociates into solution, an exchange that assures processivity. Our data supported by linear combination analysis of theoretical SAXS spectra show that there are two DNA polymerases per hexamer of gp4.
The leading- and lagging-strand gp5/trx are bound via different interactions and would thus add two more gp5/trx to the replisome. Using this SAXS based assay we have determined the best conditions for subsequent experiment with PEG. We have used a similar approach to characterize the interactions between the C–terminal domain of eIF5 and eIF132. This approach is not necessarily limited to SAXS. We have identified interactions of Mn–dNTP to the active site of T7 DNA polymerase using X-ray absorption near edge spectroscopy utilizing the same principle40. In the later experiment we were able to distinguish between signal originating from Mn2+ that is free in solution and Mn2+ bound to the active site of gp5. These methods together with biochemical and other biophysical methods provide an opportunity to investigate the structure of high molecular weight complexes that cannot be crystallized due to weak binding interactions or have size limitations for NMR.
Although there is considerable information on the effect of macromolecular crowding on bimolecular reaction there is little information on its effect on the structure of the molecules involved. FlgM, an unstructured protein. folds to adopt a structure in a macromolecular crowded environment41. Likewise, the two unrelated proteins: VlsE and α/β flavodoxin increase their structural content by folding of the unstructured regions7.
We have shown that the complex between gp5/trx and gp4D adopts a compact conformation in an environment with macromolecular crowding. We suggest that crystal structure or NMR solution structures may not represent precisely the in vivo structure if no consideration of the macromolecular crowding effect is taken into account.
METHODS
Protein expression and purification
Chemicals were from Sigma. ATP and CTP were from Roche Molecular Biochemicals. dNTPs were from USB Corp. Pre-made gels (10–20% linear gradients) were from BioRad (Hercules, CA). T7 gp5, gp4, E. coli trx were overproduced and purified as described26,46. M13 ssDNA was prepared as described previously47. [γ–32P] dATP (800 Ci/mmol), [α–32P] CTP, and dTTP (800 Ci/mmol) were from Perkin Elmer.
DNA unwinding assay
The unwinding activity of gp4 was measured using a mini replication fork consisting of 5' 32P–labeled 45mer oligonucleotide (5'–ATGACTCTATGCACATTGACATGCTTCAGATTCGTATTGTACACT–3') partially annealed to a 65mer oligonucleiotide (5' T20AGTCGTAATCCGACCTCGAGGCATTGTCAATGTGCATAGAGTCAT–3'). Reactions contain 400 nM monomeric gp4B, 100 nM DNA, 40 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 0.1 mM DTT, 50 mM potassium glutamate, 1 mM dTTP, and the indicated concentrations of PEG 1 kDa. The reaction was incubated for 5 min at 37 °C and terminated by adding 5× stop buffer to a final concentration of 0.4% SDS, 40 mM EDTA, 8% glycerol and 0.1% bromophenol blue. Samples were loaded onto 10% non-denaturing polyacrylamide gel. The amount of radiolabeled single-stranded 45-mer formed was measured using a Fuji BAS Bioimaging analyzer.
dTTPase activity
The dTTPase activity of gp4 was determined in a reaction containing 0–200 nM gp4B, 1 nM M13 ssDNA, 5 mM dTTP, 0.1 mCi of [α–32P] dTTP, 40 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 50 mM potassium glutamate, 0.1 mM DTT, and 8% PEG 1 kDa. After 10 min at 37 °C the reaction was halted using 40 mM EDTA and 0.4 μL was spotted onto polyethyleneimine TLC plate (EMD). Reaction products were separated using 0.5 M formate and 0.5 M LiCl. The plate was dried, subjected to autoradiography, and the products measured.
Oligoribonucleotide Synthesis
Synthesis of oligoribonucleotides by primase was measured as described48 in reactions containing 5 μM T7 primase fragment. Reactions (10uL) contained 5 μM DNA templates (5'-GGGTCA10), 200 μM ATP, 200 μM [α-32P]-CTP, primase fragment, 40 mM Tris-HCl pH 7.5, 10 mM MnCl2, 10 mM DTT, 50 mM potassium glutamate, and 1 kDa PEG (0, 1.25, 2.5, 5, 10%). After incubation at room temperature for 10 minutes, the reaction was terminated by adding an equal volume of sequencing buffer containing 98 % formamide, 0.1 % bromophenolblue, and 20 mM EDTA. The samples were loaded onto 25% polyacrylamide sequencing gel containing 3 M urea and visualized using autoradiography.
DNA polymerase assay
Polymerase activity was measured in a reaction containing 20 nM M13 ssDNA annealed to a 24mer primer, 40 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 10 mM DTT, 50 mM potassium glutamate, 0.25 mM dTTP, dGTP, dCTP, and [α–32P] dATP (5 cpm/pmol). Gp5 (or gp5/trx) and PEG 1 kDa were added at the indicated amount. The reaction was incubated at 37 °C for 10 min and terminated by the addition of EDTA to 40 mM. Aliquots were spotted on DE–81 filters (Whatman), washed extensively with 0.3 M ammonium formate (pH 8.0), and the radioactivity retained measured.
Strand-displacement synthesis
DNA templates were prepared by annealing a primer containing 5' non-complementary ssDNA region (underlined) (5'–(T36)AATTC GTAAT CATGG TCATAGCTGT TTCCT–3') to M13 DNA. The reaction contained 10 nM template DNA, 0.3 mM dNTPs, 0.1 μCi [α–32P] dTTP, 20 nM T7 gp5/trx, 200 nM gp4B, and the indicated concentrations of PEG 1 kDa. The reaction was incubated for 30 min at 37 °C and the amount of DNA synthesis determined as in the polymerase assay.
Primase-dependent DNA synthesis
RNA primers made by gp4A were extended by gp5/trx. The reaction contained 10 nM M13 ssDNA, 0.3 mM dNTPs, 0.1 μCi [α–32P] dCTP, 20 nM gp5/trx, 200 nM monomeric gp4A, and PEG 1 kDa (0–8%). The reaction was incubated for 30 min at 37 °C. and amounts of DNA synthesis were determined as described in the polymerase assay.
Small angle X–ray scattering (SAXS)
Measurements were made at the National Synchrotron Light Source (Upton, NY) on beamline X–9. Sample loading, data collection and processing were performed as described49.
The magnitude of the scattering vector (q) is defined as:
-
(1)
where 2θ is the scattering angle
The SAXS data showed a linear behavior (see Fig. 5c and Fig. 6- except samples containing 6 and 10% PEG) in the low q, Guinier region, indicating that the proteins did not aggregate. Radii of gyration (Rg) were derived from data in the qRg < 1.3 region using the Guinier approximation embedded in PRIMUS_ENREF_49_ENREF_47_ENREF_4550:
-
(2)
To ensure binding of gp5/trx to gp4D we used samples prepared as followed. Binding of gp5/trx (A) to gp4D (B) is a single step reaction of the type A + B ⇆ AB. The dissociation constant is given by
Where [A] is the concentration of gp5/trx and [B] is the concentration of gp4D hexamers and [AB] is the concentration of the complex in equilibrium.
Expressing the equilibrium concentrations of the separate components in terms of initial concentrations (indicated by a subscript Zero) and the concentration of the complex
We obtain
If we wish to have x fraction of A0 in the complex AB:
Then the following amount of B is required to have AB complex:
-
(3)
B0, the concentration of gp4D was kept constant at 5 μM for all samples. KD, the dissociation constant is 90 nM39. Therefore, to obtain a 1:1 complex 5.3 μM gp5/trx was mixed with 5μM gp4D. To examine whether binding of multiple gp5/trx occur per one hexamer of gp4D, gp5/trx with various concentrations (0, 2, 4, 8, 12, 24 μM) was added. The components were mixed prior to the SAXS measurement, centrifugated for 1 min to eliminate scattering from precipitated protein and measured. The samples of gp5/trx and gp4D were measured in 20 mM Tris–HCl (pH 7.5), 50 mM potassium glutamate, 2 mM DTT.
To simulate the scattering profile and Rg for a mixture of gp5/trx and gp4D, we calculated the scattering intensity using the following formula:
-
(4)
with q= scattering vector, I0 is the extrapolated intensity at zero scattering angle, Rg the radius of gyration of the respective particle. The intensity at zero scattering angle was calculated according to
where N is the number of particles per unit volume and V the volume of the particle with:
The apparent Rg of the mixture was then calculated from slope of the curve of ln(I) vs. q2 according to the Guinier approximation:
NMR Spectroscopy
We carried out DOSY-NMR experiments using a stimulated spin echo sequence on lysozyme in the presence or the absence of PEG to evaluate the effect of PEG on diffusion coefficients. The decrease in magnetization with increasing gradient strength can be analyzed using the following formula:
where D is the diffusion coefficient with , kB is the Boltzmann constant, T the temperature, η the viscosity, F the dimensionless Perrin factor, rS the hydrodynamic radius of the molecule q = γδg, Δ is the separation of the gradient echo, δ the gradient duration, γ the gyromagnetic ratio of the nucleus and g the strength of the gradient. The duration of the gradient in this experiment was set to 2.5 msec, the separation of the gradient echo to 150 msec. The gradient strength was varied from 15 to 80 G/cm. Thirty two points were recorded in the indirect dimension. DMSO was used as an internal reference.
Simulation of Brownian Motion
We used Matlab to perform a simple simulation of Brownian motion in a space of the size of an E. coli cell with and without ribosomes as crowding agents.
We calculated 18,000 random positions of non-overlapping spheres with a radius of 11 nm in E. Coli cell space (0.8 × 2 μm). We used the diffusion coefficient with kB=Boltzmann constant, T=Temperature (293 K), η=viscosity inside an E. coli cell with η=3.5*10−3 kg/m/sec43 and d = radius of the particle, here d=30 Å, to calculate the average displacement squared with k = 2 Dnτ,where D is the diffusion coefficient, n the number of dimensions (3) and τ the time interval (here 0.0001 sec). The displacement vector for each time interval was determined by multiplying a random number by the displacement factor using Matlab's randn function for normal distribution of random numbers. If the new position of the molecule overlapped with a ribosome position of if the position was outside the confines of the given space, a new position was calculated in the same manner. For simplicity, the positions of the ribosome molecules were kept fixed.
Excluded volume calculation
To calculate the excluded volume, we assumed spherical shape of the PEG molecules. The specific volume was calculated using the following equation
-
(5)
with rh=hydrodynamic radius, NA=Avogadro constant, MW=molecular weight. To calculate the volume occupied by PEG at different concentrations, the specific volume is multiplied by the density of PEG and the concentration. The values for the hydrodynamic radius of PEG with different sizes was taken from 43.
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by National Institutes of Health Grants GM54397 (to C.C.R.) and by Agilent Foundation (to G.W.). We thank L. Yang and M. Allier (Beamline X-9 NSLS, BNL) at Brookhaven National Laboratory. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. We thank Richard Gillilan (Cornell High Energy Synchrotron Source) for helpful discussion.
Footnotes
AUTHOR CONTRIBUTIONS BA, SA, GW and CCR designed research, wrote paper, and performed the studies. SL contributed materials.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Supplementary information includes 4 figures is available at Nature Communications website.
REFERENCES
- 1.Zimmerman SB, Minton AP. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- 2.Zimmerman SB, Trach SO. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-v. [DOI] [PubMed] [Google Scholar]
- 3.Minton AP. How can biochemical reactions within cells differ from those in test tubes? J. Cell Sci. 2006;119:2863–9. doi: 10.1242/jcs.03063. [DOI] [PubMed] [Google Scholar]
- 4.Ogston AG, Phelps CF. Exclusion of inulin from solutions of hyaluronic acid. Nature. 1960;187:1024. doi: 10.1038/1871024a0. [DOI] [PubMed] [Google Scholar]
- 5.Minton AP. Effect of a concentrated “inert” macromolecular cosolute on the stability of a globular protein with respect to denaturation by heat and by chaotropes: a statistical-thermodynamic model. Biophys. J. 2000;78:101–9. doi: 10.1016/S0006-3495(00)76576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minton AP, Wilf J. Effect of macromolecular crowding upon the structure and function of an enzyme: glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 1981;20:4821–6. doi: 10.1021/bi00520a003. [DOI] [PubMed] [Google Scholar]
- 7.Perham M, Stagg L, Wittung-Stafshede P. Macromolecular crowding increases structural content of folded proteins. FEBS Lett. 2007;581:5065–9. doi: 10.1016/j.febslet.2007.09.049. [DOI] [PubMed] [Google Scholar]
- 8.Strey HH, Podgornik R, Rau DC, Parsegian VA. DNA--DNA interactions. Curr. Opin. Struct. Biol. 1998;8:309–13. doi: 10.1016/s0959-440x(98)80063-8. [DOI] [PubMed] [Google Scholar]
- 9.Stagg L, Zhang SQ, Cheung MS, Wittung-Stafshede P. Molecular crowding enhances native structure and stability of alpha/beta protein flavodoxin. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18976–81. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuller RS, Kaguni JM, Kornberg A. Enzymatic replication of the origin of the Escherichia coli chromosome. Proc. Natl. Acad. Sci. U. S. A. 1981;78:7370–4. doi: 10.1073/pnas.78.12.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pheiffer BH, Zimmerman SB. Polymer-stimulated ligation: enhanced blunt- or cohesive-end ligation of DNA or deoxyribooligonucleotides by T4 DNA ligase in polymer solutions. Nucleic Acids Res. 1983;11:7853–71. doi: 10.1093/nar/11.22.7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison B, Zimmerman SB. T4 polynucleotide kinase: macromolecular crowding increases the efficiency of reaction at DNA termini. Anal. Biochem. 1986;158:307–15. doi: 10.1016/0003-2697(86)90555-5. [DOI] [PubMed] [Google Scholar]
- 13.Jarvis TC, Ring DM, Daube SS, von Hippel PH. “Macromolecular crowding”: thermodynamic consequences for protein-protein interactions within the T4 DNA replication complex. J. Biol. Chem. 1990;265:15160–7. [PubMed] [Google Scholar]
- 14.Reddy MK, Weitzel SE, von Hippel PH. Assembly of a functional replication complex without ATP hydrolysis: a direct interaction of bacteriophage T4 gp45 with T4 DNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 1993;90:3211–5. doi: 10.1073/pnas.90.8.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy MK, Weitzel SE, Daube SS, Jarvis TC, von Hippel PH. Using macromolecular crowding agents to identify weak interactions within DNA replication complexes. Methods Enzymol. 1995;262:466–76. doi: 10.1016/0076-6879(95)62038-9. [DOI] [PubMed] [Google Scholar]
- 16.Hamdan SM, Richardson CC. Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 2009;78:205–43. doi: 10.1146/annurev.biochem.78.072407.103248. [DOI] [PubMed] [Google Scholar]
- 17.Bailey S, Eliason WK, Steitz TA. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science. 2007;318:459–63. doi: 10.1126/science.1147353. [DOI] [PubMed] [Google Scholar]
- 18.Minton AP. Excluded volume as a determinant of macromolecular structure and reactivity. Biopolymers. 1981;20:2093–120. [Google Scholar]
- 19.Levy Y, Onuchic JN. Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 20.Samiotakis A, Wittung-Stafshede P, Cheung MS. Folding, stability and shape of proteins in crowded environments: experimental and computational approaches. Int. J. Mol. Sci. 2009;10:572–88. doi: 10.3390/ijms10020572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minton AP. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 2001;276:10577–80. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Liang KC, Czader A, Waxham MN, Cheung MS. The effect of macromolecular crowding, ionic strength and calcium binding on calmodulin dynamics. PLoS Comput. Biol. 2011;7:e1002114. doi: 10.1371/journal.pcbi.1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van den Berg B, Wain R, Dobson CM, Ellis RJ. Macromolecular crowding perturbs protein refolding kinetics: implications for folding inside the cell. EMBO J. 2000;19:3870–5. doi: 10.1093/emboj/19.15.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sikorav JL, Church GM. Complementary recognition in condensed DNA: accelerated DNA renaturation. J. Mol. Biol. 1991;222:1085–108. doi: 10.1016/0022-2836(91)90595-w. [DOI] [PubMed] [Google Scholar]
- 25.Konopka MC, Shkel IA, Cayley S, Record MT, Weisshaar JC. Crowding and confinement effects on protein diffusion in vivo. J. Bacteriol. 2006;188:6115–23. doi: 10.1128/JB.01982-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huber HE, Tabor S, Richardson CC. Escherichia coli thioredoxin stabilizes complexes of bacteriophage T7 DNA polymerase and primed templates. J. Biol. Chem. 1987;262:16224–32. [PubMed] [Google Scholar]
- 27.Akabayov B, et al. Conformational dynamics of bacteriophage T7 DNA polymerase and its processivity factor, Escherichia coli thioredoxin. Proc. Natl. Acad. Sci. U. S. A. 2010;107:15033–8. doi: 10.1073/pnas.1010141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frick DN, Richardson CC. DNA primases. Annu. Rev. Biochem. 2001;70:39–80. doi: 10.1146/annurev.biochem.70.1.39. [DOI] [PubMed] [Google Scholar]
- 29.Kulczyk AW, et al. An Interaction between DNA Polymerase and Helicase is Essential for the High Processivity of the Bacteriophage T7 Replisome. J. Biol. Chem. 2012 doi: 10.1074/jbc.M112.410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singleton MR, Sawaya MR, Ellenberger T, Wigley DB. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell. 2000;101:589–600. doi: 10.1016/s0092-8674(00)80871-5. [DOI] [PubMed] [Google Scholar]
- 31.Guo S, Tabor S, Richardson CC. The linker region between the helicase and primase domains of the bacteriophage T7 gene 4 protein is critical for hexamer formation. J. Biol. Chem. 1999;274:30303–9. doi: 10.1074/jbc.274.42.30303. [DOI] [PubMed] [Google Scholar]
- 32.Luna RE, et al. The C-terminal domain of eukaryotic initiation factor 5 promotes start codon recognition by its dynamic interplay with eIF1 and eIF2beta. Cell Rep. 2012;1:689–702. doi: 10.1016/j.celrep.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svergun DI, Barberato C, Koch MHJ. CRYSOL– a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules fromAtomic Coordinates. J. Appl. Cryst. 1995;28:768–73. [Google Scholar]
- 34.Brieba LG, et al. Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J. 2004;23:3452–61. doi: 10.1038/sj.emboj.7600354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber HE, Russel M, Model P, Richardson CC. Interaction of mutant thioredoxins of Escherichia coli with the gene 5 protein of phage T7. The redox capacity of thioredoxin is not required for stimulation of DNA polymerase activity. J. Biol. Chem. 1986;261:15006–12. [PubMed] [Google Scholar]
- 36.Lee SJ, Zhu B, Hamdan SM, Richardson CC. Mechanism of sequence-specific template binding by the DNA primase of bacteriophage T7. Nucleic Acids Res. 2010;38:4372–83. doi: 10.1093/nar/gkq205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertini I, et al. Solid-state NMR of proteins sedimented by ultracentrifugation. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10396–9. doi: 10.1073/pnas.1103854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harano Y, Kinoshita M. Large gain in translational entropy of water is a major driving force in protein folding. Chem. Phys. Lett. 2004;399:342–8. [Google Scholar]
- 39.Hamdan SM, et al. A unique loop in T7 DNA polymerase mediates the binding of helicaseprimase, DNA binding protein, and processivity factor. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5096–101. doi: 10.1073/pnas.0501637102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akabayov B, Richardson CC. Binding of Mn-deoxyribonucleoside triphosphates to the active site of the DNA polymerase of bacteriophage T7. Powder Diffraction. 2011;2:159–163. doi: 10.1154/1.3583156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dedmon MM, Patel CN, Young GB, Pielak GJ. FlgM gains structure in living cells. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12681–4. doi: 10.1073/pnas.202331299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bereiter-Hahn J, Anderson OR, Reif WE. Cytomechanics: The Mechanical Basis of Cell Form and Structure. Springer-Verlag; Berlin: 1987. pp. 3–30. [Google Scholar]
- 43.Kuga S. Pore size distribution analysis of gel substances by size exclusion chromatography. Journal of Chromatography. 1981;206:449–461. [Google Scholar]
- 44.Kato M, Ito T, Wagner G, Richardson CC, Ellenberger T. Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol. Cell. 2003;11:1349–60. doi: 10.1016/s1097-2765(03)00195-3. [DOI] [PubMed] [Google Scholar]
- 45.Ilyina TV, Gorbalenya AE, Koonin EV. Organization and evolution of bacterial and bacteriophage primase-helicase systems. J. Mol. Evol. 1992;34:351–7. doi: 10.1007/BF00160243. [DOI] [PubMed] [Google Scholar]
- 46.Notarnicola SM, Mulcahy HL, Lee J, Richardson CC. The acidic carboxyl terminus of the bacteriophage T7 gene 4 helicase/primase interacts with T7 DNA polymerase. J. Biol. Chem. 1997;272:18425–33. doi: 10.1074/jbc.272.29.18425. [DOI] [PubMed] [Google Scholar]
- 47.Nakai H, Richardson CC. Interactions of the DNA polymerase and gene 4 protein of bacteriophage T7. Protein-protein and protein-DNA interactions involved in RNA-primed DNA synthesis. J. Biol. Chem. 1986;261:15208–16. [PubMed] [Google Scholar]
- 48.Mendelman LV, Richardson CC. Requirements for primer synthesis by bacteriophage T7 63-kDa gene 4 protein. Roles of template sequence and T7 56-kDa gene 4 protein. J. Biol. Chem. 1991;266:23240–50. [PubMed] [Google Scholar]
- 49.Toth EA, Li Y, Sawaya MR, Cheng Y, Ellenberger T. The crystal structure of the bifunctional primase-helicase of bacteriophage T7. Mol. Cell. 2003;12:1113–1123. doi: 10.1016/s1097-2765(03)00442-8. [DOI] [PubMed] [Google Scholar]
- 50.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–128. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.