

Abstract
We have developed a novel assay to detect the cytosolic localization of protein domains by inserting a short consensus sequence for phosphorylation by protein kinase A. In transfected COS-1 cells, this sequence was labeled efficiently with [32P]phosphate only when exposed to the cytosol and not when translocated into the lumen of the endoplasmic reticulum. The phosphorylation state of this sequence can therefore be used to determine the topology of membrane proteins. This assay is sufficiently sensitive to detect even the transient cytosolic exposure of the N-terminal domain of a membrane protein with a reverse signal-anchor sequence. The extent of phosphorylation per newly synthesized polypeptide was shown to reflect the time of exposure to the cytosol, which depends on translation, targeting and translocation of the N-terminus. By altering the length of the N-terminal domain or manipulating the translation rate, it was determined that protein targeting is rapid and requires only a few seconds. The rate of N-terminal translocation was estimated to be ∼1.6 times the rate of translation.
Keywords: endoplasmic reticulum/phosphorylation/protein targeting/protein topology/translation
Introduction
In mammalian cells, protein targeting to the endoplasmic reticulum (ER) is mostly co-translational (Walter and Johnson, 1994). A hydrophobic signal sequence is recognized in the context of the nascent chain–ribosome complex by signal recognition particle (SRP), which in turn interacts with the SRP receptor on the ER membrane. During this process, both SRP and SRP receptor hydrolyze GTP (Rapiejko and Gilmore, 1997; Song et al., 2000) and release the signal sequence, which then interacts directly with the Sec61α subunit of the translocon (Mothes et al., 1994, 1998) and initiates the translocation of the flanking polypeptide.
Targeting and translocation depend on several events occurring in a defined order and time frame. In vitro translation/insertion experiments using pre-prolactin have shown that SRP binding can only occur when the nascent polypeptide is shorter than ∼140 residues (Siegel and Walter, 1988). In vitro, SRP binding reduces the translation rate (Wolin and Walter, 1989; Ogg and Walter, 1995; Mason et al., 2000), a function that may serve to prevent the growing polypeptide from losing translocation competence due to premature folding or to chain termination and disassembly of the translation complex. Evidence for a physiological role for SRP-induced translation slow-down has been reported recently for yeast (Mason et al., 2000). The kinetics of SRP binding to the nascent chain–ribosome complex and of subsequent interaction with the SRP receptor are thus important for efficient ER targeting and translocation. However, very little is known about the rate of these processes in relation to the rate of the competing process of translation under in vivo conditions.
Two basic types of signal sequence can be distinguished that induce translocation of either the C- or N-terminal end across the ER membrane (Spiess, 1995). Examples of the first type are the cleaved signals of secretory proteins and the signal-anchors of type II membrane proteins with a cytosolic N-terminus and an exoplasmic C-terminus (Ncyt/Cexo), such as the transferrin receptor. Examples of the second type are the reverse signal-anchor sequences of Nexo/Ccyt proteins such as the microsomal cytochromes P-450. Both types of signals are recognized by SRP and utilize the Sec61 translocation machinery (High et al., 1991). Major determinants for the orientation of the signal are the charged residues flanking its hydrophobic core (the more positive end tends to stay cytosolic; von Heijne, 1984; Hartmann et al., 1989; Beltzer et al., 1991), and the hydrophobicity and length of the core (longer and more hydrophobic sequences favor N-terminal translocation; Sakaguchi et al., 1992; Wahlberg and Spiess, 1997; Eusebio et al., 1998; Rösch et al., 2000). An additional determinant is the folding behavior of the N-terminal hydrophilic sequence, which is completed and potentially folded before the signal emerges from the ribosome and translocation is initiated (Denzer et al., 1995). In multispanning proteins, the orientation of the initial signal may also depend on the topogenic information in subsequent transmembrane segments (Gafvelin et al., 1997; Goder et al., 1999; Nilsson et al., 2000).
Since the insertion of polypeptides into the membrane is the result of multiple determinants, it is often not trivial to predict the topology of proteins—particularly of multispanning ones—from their primary sequence. A number of techniques have been developed to determine the localization of individual hydrophilic segments experimentally (Wessels et al., 1991). Sensitivity to proteases and accessibility to sequence-specific antibodies can be used to assay exposure to the cell exterior or the cytosol. However, cleavage patterns of multispanning proteins may be difficult to interpret and efficient antibodies against short loops hard to generate. The insertion of specific cleavage sites, e.g. for factor Xa (e.g. Wilkinson et al., 1996), or of known antigenic epitopes (e.g. Kast et al., 1996) at various positions throughout the protein has been successfully used to map protein topology. Alternatively, diagnostic cysteines for biotinylation with an impermeant sulfhydryl reagent have been inserted for this purpose (Loo and Clarke, 1995). In addition, N-glycosylation at natural or engineered glycosylation sites provides conclusive evidence for the lumenal localization of a sequence.
In this study, we have developed a novel assay to detect the cytosolic disposition of a polypeptide segment. A short consensus sequence for phosphorylation by cAMP-dependent protein kinase (PKA), inserted into the polypeptide, is labeled efficiently with [32P]phosphate only when exposed to the cytosol. This assay thus yields information complementary to that of an inserted glycosylation site, which positively identifies lumenal exposure. Furthermore, the high sensitivity of the assay allows detection of even transient exposure of protein domains to the cytosol. Within a defined range, the extent of phosphorylation per polypeptide reflects the time of cytosolic exposure. Using this approach, we were able to determine the in vivo kinetics of SRP-dependent targeting to the ER and of translocation of an N-terminal domain across the ER membrane.
Results
A phosphorylation tag as a sensor of permanent and transient cytosolic localization
Phosphorylation of amino acid side chains is a major cytosolic modification of proteins and therefore of potential value as a tool to differentiate between cytosolic and lumenal localization of a protein domain. For this, we took advantage of an established consensus sequence for phosphorylation by PKA, LeuArgArgAlaSerLeuGly (Hjelmquist et al., 1974; Kemp et al., 1977), which has been shown to be phosphorylated efficiently at its serine side chain as a free peptide as well as in various sequence contexts within longer polypeptides and proteins. To test its value as a sensor of cytosolic localization, this heptapeptide sequence was inserted at different positions into the sequence of the H1 subunit of the asialoglycoprotein receptor. H1 is a type II membrane protein composed of a 40 residue N-terminal portion exposed to the cytosol, an uncleaved signal-anchor sequence spanning the membrane and a C-terminal, exoplasmic portion of ∼230 amino acids (Figure 1A; Spiess and Lodish, 1986). Glycosylation at two sites within the C-terminal portion of the protein clearly indicates the lumenal disposition of the C-terminus. Constructs were transfected into COS-1 cells and analyzed for expression by labeling the cells for 40 min with [35S]methionine, and for phosphorylation by labeling for 40 min with [32P]phosphate in the presence of 20 µM forskolin, an indirect activator of PKA. The cells were then extracted with 0.1% saponin to remove any soluble products that may have failed to be integrated into the membrane (Beltzer et al., 1991; Denzer et al., 1995) and to reduce background by extracting a large fraction of endogenous soluble phosphoproteins. The residual cellular material was then immunoprecipitated, separated by SDS–gel electrophoresis and subjected to autoradiography or quantitation by PhosphorImager analysis.

Fig. 1. Schematic representation of the model membrane proteins analyzed. H1 (A), H1-PC (B) and H1-PN (C) are Ncyt/Cexo proteins with a type II signal-anchor sequence (open rectangle) and a glycosylated (Y) C-terminal domain. H1rev (D), H1rev-PC (E) and H1rev-PN (F) contain a reverse signal-anchor sequence (hatched rectangle) and insert to ∼95% as unglycosylated Nexo/Ccyt proteins. The circle symbolizes the consensus sequence for phosphorylation by PKA shown below (with the target serine indicated by an arrow).
Upon labeling with [35S]methionine, H1 was immunoprecipitated as a 40 kDa glycoprotein that could be deglycosylated with endo-β-d-N-acetyl glucosaminidase H (endo H) to a polypeptide of 34 kDa (Figure 2A, lanes 1 and 2). No radioactivity was incorporated upon labeling with [32P]phosphate (Figure 2A, lanes 3 and 4). H1 has previously been shown to contain two weak sites for phosphorylation by protein kinase C in the presence of phorbol esters (Geffen et al., 1991). However, under the conditions used here, this activity was negligible. When the phosphorylation sequence was inserted into the C-terminal portion of H1 (H1-PC; Figure 1B), no 32P-labeling could be detected (Figure 2B, lanes 3 and 4). This was consistent with the notion that the phosphorylation sequence was transferred co-translationally from the ribosome directly through the translocon into the ER lumen, without exposure to the cytosol. When the phosphorylation tag was inserted at the very N-terminus of H1 in construct H1-PN (Figure 1C), the tagged protein was labeled efficiently with [32P]phosphate (Figure 2C). Both endo H-sensitive and (partially or completely) endo H-resistant forms of the protein were labeled, indicating that the protein was phosphorylated both in the ER and in later compartments of the secretory pathway.

Fig. 2. Expression and phosphorylation pattern of the model proteins (A–F) with opposite orientations. Transfected COS-1 cells were labeled with [35S]methionine or [32P]phosphate for 40 min and subjected to immunoprecipitation. Samples were incubated with (+) or without (–) endo H and analyzed by SDS–gel electrophoresis and fluorography. The asterisks indicate the position of a small percentage of proteins with a reverse signal-anchor that nevertheless inserted as type II (Ncyt/Cexo) membrane proteins. The arrowhead points to the labeled Nexo/Ccyt proteins. All 35S-samples and 32P-samples were exposed identically.
As controls, we constructed a series of proteins corresponding to H1, H1-PC and H1-PN but with the opposite orientation in the membrane: a lumenal N-terminus and a cytoplasmic C-terminus. This was accomplished by replacing the signal-anchor sequence by a reverse signal-anchor in H1rev, H1rev-PC and H1rev-PN (Figure 1D–F). It has been shown previously that replacing the charged residues flanking the hydrophobic domain of the signal (Arg34, Arg40, Glu67, Glu68) with amino acids of opposite charge (Asp34, Asp34, Lys67, Lys68) and exchanging the hydrophobic sequence LLLLSLGLSLLLLVVVCVI for a Leu25 sequence resulted in 95% N-terminal translocation (H1rev; this construct was called H1-4Leu25 in Wahlberg and Spiess, 1997). Accordingly, upon expression in COS-1 cells, H1rev was almost completely unglycosylated except for a minute fraction of polypeptides that were still inserted as type II proteins (Figure 2D, lanes 5 and 6, asterisks). The products were not labeled with [32P]phosphate, indicating the absence of any cryptic phosphorylation sites within the C-terminal portion of the protein (lanes 7 and 8). In contrast, the C-terminal phosphorylation tag of H1rev-PC was phosphorylated efficiently (Figure 2E, lanes 7 and 8), demonstrating that the tag at this position is functional if exposed to the cytosol (compare with Figure 2B). As expected, H1rev-PN with the N-terminal phosphorylation tag was 32P-labeled in the small fraction of glycosylated type II polypeptides present in the cells (Figure 2F, lanes 7 and 8). However, it was also labeled in its unglycosylated form, which finally extends its N-terminus into the ER lumen (arrowhead in lane 7). This suggests that the N-terminal tag sequence was phosphorylated during the short, transient exposure to the cytosol between its emergence from the ribosome and its translocation across the membrane.
To test whether this phosphorylation was indeed restricted to proteins that are synthesized during the labeling period, phosphate incorporation into H1rev-PN was analyzed in the presence or absence of the translation inhibitor cycloheximide at a concentration of 100 µg/ml for 2 h before as well as during the labeling period. [35S]methionine labeling showed that incubation with cycloheximide completely blocked translation (Figure 3, lanes 2 and 6). For H1rev-PN, the total amount of protein of either orientation, as estimated by western analysis, was reduced approximately by half as a result of degradation during the cycloheximide incubation (lanes 9 and 10). Pre-existing glycosylated Ncyt/Cexo polypeptides of H1-PN (lane 4) and H1rev-PN (lane 8) were still 32P-phosphorylated efficiently. The unglycosylated form of H1rev-PN, however, was not labeled with [32P]phosphate (lane 8, asterisk). H1rev-PN with an Nexo/Ccyt orientation was therefore labeled exclusively during the transient exposure of the N-terminal tag sequence to the cytosol before its translocation into the ER lumen.

Fig. 3. Co-translational phosphorylation of H1rev-PN. COS-1 cells transfected with H1-PN or H1rev-PN were incubated with (+) or without (–) 100 µg/ml cycloheximide for 2 h before and 40 min during labeling with [35S]methionine or [32P]phosphate. After saponin extraction, the membrane proteins were immunoprecipitated and analyzed by SDS–gel electrophoresis and fluorography. The positions of phosphorylated H1rev-PN with Ncyt/Cexo and Nexo/Ccyt orientation are indicated by an asterisk and an arrowhead, respectively. To visualize the total H1rev-PN in the transfected cells, western analysis was performed on a parallel sample (W). A non-specific band detected upon western analysis is indicated by a circle.
Phosphorylation as a timer of cytosolic exposure
The time period during which the N-terminal phosphorylation tag in H1rev-PN is exposed to the cytosol is determined by four processes (Figure 4): (A) translation of the polypeptide from the emergence of the tag sequence from the ribosome until the signal sequence is exposed sufficiently to be recognized by SRP (∼10 hydrophobic residues); (B) SRP binding to the nascent chain–ribosome complex; (C) targeting of the ternary complex to the ER membrane; and (D) translocation of the N-terminal portion across the membrane. If phosphorylation is slow relative to these processes, the extent of phosphorylation will reflect the time of cytosolic exposure of the tag. As a result, lengthening the spacer peptide between the phosphorylation tag and the reverse signal-anchor, which increases the time for processes (A) and (B), will increase specific phosphorylation, whereas shortening the spacer will reduce it.

Fig. 4. The phosphorylation tag acts as a timer of cytosolic exposure. The time of cytosolic exposure of the N-terminal phosphorylation sequence is determined by the time of translation of the N-terminal domain from the appearance of the tag until the signal has emerged sufficiently for interaction with SRP (A), the kinetics of SRP binding (B) and of targeting of the complex to the ER (C), and by the time required to translocate the N-terminal domain across the membrane (D). The phosphorylation tag is shown as a circle and its average modification is indicated by its filled portion.
To test this concept, we constructed variants of H1rev-PN with shorter or longer spacer peptides. The N-terminal sequence between the phosphorylation tag and the reverse signal-anchor was shortened from 40 residues in the original H1rev-PN to 20 residues in H1rev20-PN or extended to 85 residues in H1rev85-PN. Transfected COS-1 cells were then labeled for 40 min in parallel either with [35S]methionine or with [32P]phosphate (Figure 5A). Upon immunoprecipitation and gel electrophoresis, the radioactivity in the unglycosylated Nexo/Ccyt form was quantified by phosphorimaging and the specific phosphorylation (32P/35S) determined. As shown in Figure 5A, specific phosphorylation was indeed increased with the extended spacer sequence in H1rev85-PN and reduced with the shortened spacer in H1rev20-PN.
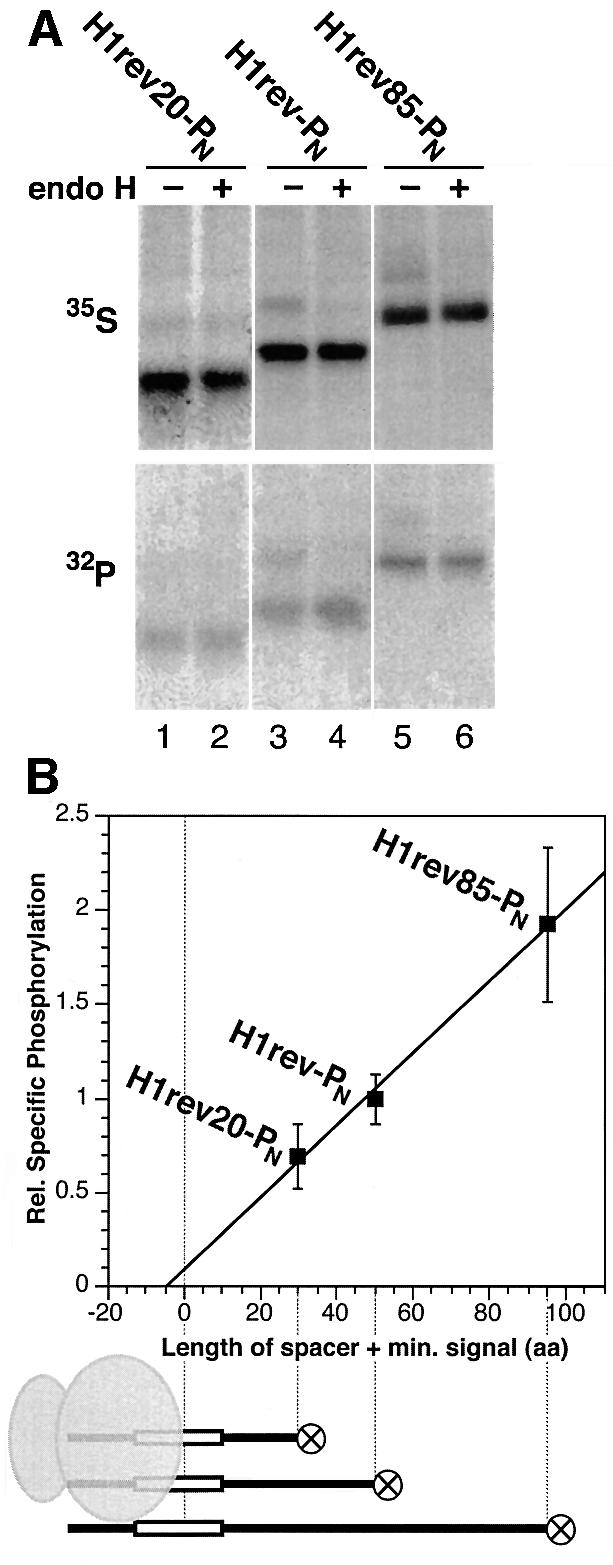
Fig. 5. Specific phosphorylation as a function of the spacer length between the phosphorylation tag and the signal sequence. (A) H1rev20-PN, H1rev-PN and H1rev85-PN, which have spacers of 20, 40 and 85 residues, respectively, were expressed in COS-1 cells, labeled with [35S]methionine or [32P]phosphate, extracted with saponin, immunoprecipitated and, after incubation with (+) or without (–) endo H, analyzed by gel electrophoresis and autoradiography. Incorporation of 32P in the unglycosylated Nexo/Ccyt form of the proteins increased relative to the incorporation of 35S with increasing spacer length. (B) The specific phosphorylation ([32P]/[35S]) was quantified for three series of co-labeling experiments with a total of 10 samples for each construct, normalized to the specific phosphorylation of H1rev-PN, and plotted against the length of the spacer plus the minimal size of the signal to be recognized by SRP (∼10 residues, as illustrated below the graph).
To eliminate variability between parallel samples and to standardize quantitation, the experiment was subsequently performed with a modified protocol in which cells were labeled simultaneously with [32P]phosphate and [35S]methionine. The contribution of the two isotopes in the resulting double-labeled bands was then determined by successive exposure of the gel to a PhosphorImager screen with or without a transparency film in between, which was calibrated to block >99% of 35S, but only 25% of 32P. The same results were also obtained by exposing the gels twice for identical times within a period of 2 weeks and calculating the contributions based on the isotopes’ half-lives. Quantitation of three series of experiments with a total of 10 individual determinations per construct is shown in Figure 5B. Phosphorylation indeed depends on the length of the N-terminal domain of the model proteins, confirming that the phosphorylation tag acts as a timer for its cytosolic exposure.
The time required to synthesize the N-terminal domain and the time to translocate it across the membrane [processes (A) and (D) in Figure 4] depend on the length of the N-terminal domain. For process (A), the spacer between the phosphorylation tag and the reverse signal-anchor plus the minimal length of a signal to be recognized by SRP (∼10 apolar residues) is relevant. This amounts to 30, 50 and 95 residues for the three constructs analyzed and also corresponds approximately to the size of the N-terminal domain to be translocated in process (D). In contrast, the time for SRP binding and targeting [processes (B) and (C)] can be expected to be constant. Extrapolation of the specific phosphorylation to a length of zero (Figure 5B) should therefore yield an estimate of the phosphorylation taking place during the time of SRP binding and targeting. The phosphorylation occurring during these events is small in comparison with that during translation and translocation of the N-terminal domain in the three constructs. SRP binding and targeting are thus relatively fast and take approximately the same time as is needed to translate and translocate five amino acids.
Estimating the time of SRP binding and targeting, and of N-terminal translocation
Of the four steps that determine the time that the phosphorylation sequence spends in the cytosol, the first one, translation of the N-terminal domain, can be manipulated easily by incubation with low concentrations of the elongation inhibitor cycloheximide. Reducing the translation rate increases the time required to synthesize the polypeptide from the N-terminal phosphorylation tag until the hydrophobic signal has emerged sufficiently to be recognized by SRP [process (A) in Figure 4]. This rate reduction should not affect the subsequent processes of SRP binding, targeting and translocation of the N-terminal domain. The time required by these latter processes is therefore a constant contribution that should become apparent upon extrapolation of the specific phosphorylation of a single Nexo/Ccyt protein to infinite translation rate (zero translation time).
COS-1 cells expressing H1rev20-PN or H1rev85-PN were incubated with different cycloheximide concentrations. The resulting translation rates were reflected directly in the amount of [35S]methionine incorporated into the protein of interest during the labeling period of 40 min (Figure 6A) and covered a range of a factor of ∼8. The specific phosphorylation at different translation rates was determined by labeling the cells simultaneously with [32P]phosphate and [35S]methionine, and measuring the contribution of 32P and 35S in the immunoprecipitated protein as described above. The results were plotted against the time of translation expressed in multiples of the translation time in the absence of cycloheximide (Figure 6B and C). A linear increase in specific phosphorylation with increasing translation time was obtained for both constructs, consistent with the concept of a phosphorylation timer. For H1rev20-PN (Figure 6B), extrapolation to zero translation time yielded slightly more than half the specific phosphorylation observed under normal conditions. Therefore, in the absence of cycloheximide, ∼45% of the [32P]phosphate incorporation occurred during translation of the N-terminal domain and ∼55% during SRP binding, targeting and translocation. This indicates that the latter three processes together take only slightly longer than the translation of 30 residues. At a translation rate of ∼5 amino acids/s, as has been determined previously for mammalian cell lines (reviewed by Hershey, 1991), this amounts to 6 s for translation and ∼7 s for SRP binding, targeting and translocation (Table I; Figure 6B, lower scale on x-axis).

Fig. 6. Specific phosphorylation as a function of translation rate. (A) Elongation was reduced by incubation of transfected COS-1 cells with different concentrations of cycloheximide during 40 min of [35S]methionine labeling. Saponin extraction, immunoprecipitation and fluorography were then performed. (B and C) COS-1 cells expressing H1rev20-PN or H1rev85-PN were labeled with [35S]methionine and [32P]phosphate, saponin extracted, immunoprecipitated and analyzed by gel electrophoresis. The specific phosphorylation (average with standard deviation from three determinations each) normalized to that determined in the absence of inhibitor is plotted against the relative translation time of the N-terminal domain from the emergence of the phosphorylation tag to the appearance of a minimal signal (translation of 30 and 95 residues, respectively). The lower scale provides absolute times of translation for a translation rate of 5 amino acids/s.
Table I. Estimated values for the kinetics of targeting and translocation.
| Length of N-terminal domain L | 30 aa | 95 aa | |
| Determined from Figure 6: | |||
| time for SRP binding, targeting and N-terminal translocation relative to the translation time: | 1.20 ± 0.33 | 0.81 ± 0.27 | |
| translocation rate relative to the translation rate: | 1.59 ± 0.27 | ||
| time for SRP binding and targeting relative to the time of translation per residue: | 17.2 ± 7.2 aa | ||
| At a translation rate of 5 aa/s: | |||
| translation time: | 6.0 s | 19.0 s | |
| time for SRP binding, targeting and N-terminal translocation: tbc + td | 7.2 ± 2.0 s | 15.4 ± 5.1 s | |
| time for SRP binding and targeting: tbc | 3.4 ± 1.4 s | ||
| translocation time: td | 3.8 ± 0.6 s | 11.9 ± 1.9 s | |
| translocation rate: vd | 8.0 ± 1.4 aa/s | ||
The kinetics derived from Figure 6 are relative to those of translation. The rate of translation in eukaryotic cells was estimated to be ∼5 amino acids/s (aa/s) (Hershey, 1991). ![]() , tbc and td are the times of translation (without inhibitor), of SRP binding and targeting, and of N-terminal translocation, respectively [processes A–D in Figure 4]. The relevant peptide length L is from the point of complete emergence of the phosphorylation tag from the ribosome to when there is sufficient exposure of the signal sequence for SRP recognition. This is also approximately the length of polypeptide translocated across the membrane.
, tbc and td are the times of translation (without inhibitor), of SRP binding and targeting, and of N-terminal translocation, respectively [processes A–D in Figure 4]. The relevant peptide length L is from the point of complete emergence of the phosphorylation tag from the ribosome to when there is sufficient exposure of the signal sequence for SRP recognition. This is also approximately the length of polypeptide translocated across the membrane. ![]() and vd are the rates of translation without inhibitor and of N-terminal translocation, respectively. See Materials and methods for details.
and vd are the rates of translation without inhibitor and of N-terminal translocation, respectively. See Materials and methods for details.
For construct H1rev85-PN (Figure 6C), ∼55% of phosphorylation occurred during the translation time of the N-terminal domain of ∼95 residues, and ∼45% was attributable to the subsequent events. This corresponds to ∼19 s for translation and ∼15 s for SRP binding, targeting and translocation. Based on the assumptions that the translocation time is proportional to the length of the translocated domain and that the kinetics of SRP binding and targeting are the same for the two constructs, a translocation rate of ∼8 amino acids/s can be calculated from these values (summarized in Table I). The average time of SRP binding and targeting then turns out to be quite short, only 3–4 s.
Discussion
We have used a consensus sequence for phosphorylation by PKA to probe in vivo protein exposure to the cytosol. This reporter sequence can be used in two ways, either as a sensor of permanent cytoplasmic exposure or as a timer for the duration of its transient presence in the cytoplasm. As a sensor, it can be applied to determine the topology of membrane proteins. In our model system, the tag was introduced at the N-terminus or into the C-terminal domain of a single-spanning membrane protein, which, depending on the signal-anchor sequence, inserted either as an Ncyt/Cexo or an Nexo/Ccyt protein. In either sequence context, the tag was strongly phosphorylated only when it was presented to the cytosolic side of the membrane. Insertion of a phosphorylation tag is thus comparable with but complementary to the insertion of potential glycosylation sites, which monitor luminal localization of a sequence. The combined use of glycosylation and phosphorylation sequences may therefore yield positive evidence for either disposition of a sequence of interest.
Glycosylation sites require a minimal distance from the membrane of ∼12 residues in order to be functional (Nilsson and von Heijne, 1993). We have not tested yet how close to the membrane (e.g. within cytoplasmic loops between transmembrane segments) the phosphorylation tag is still functional. With only seven residues, the phosphorylation tag is similar in size to or even smaller than most epitope tags, such as the Myc or hemagglutinin (HA) epitope sequences. The fact that it contains charged residues, i.e. two arginines which are essential for phosphorylation, calls for some caution, since the tag sequence might affect the topology of a polypeptide if inserted close to topogenic transmembrane segments. This disadvantage may be alleviated by compensating with negative residues flanking the tag sequence. In summary, insertion of a phosphorylation sequence may be a useful additional method to analyze protein topology.
In addition to providing information on the static disposition of the tagged sequence, the phosphorylation sequence can serve as a tool to measure the kinetics of intracellular targeting and translocation in living cells. Aspects of in vivo kinetics of protein insertion and translocation have been analyzed previously in bacteria. In bacterial systems, pulse–chase experiments with a resolution in the order of ∼10 s are possible, and translocated sequences are directly accessible from the outside of the cells. It has been shown that membrane integration of bacteriorhodopsin is co-translational in Halobacterium salinarium and that the extracellular segments of the protein are translocated sequentially from the N- to the C-terminus (Dale and Krebs, 1999; Dale et al., 2000). Our assay allows time- and localization-dependent modification of a substrate in the interior of compartmentalized mammalian cells. The phosphorylation tag has made it possible to measure events in the time scale of a few seconds by analyzing essentially stable products at the end of a much longer experimental period.
Phosphorylation is sufficiently rapid to incorporate easily detectable amounts of phosphate into the tag sequence of proteins that present their N-terminal domain only transiently to the cytosol during biosynthesis. According to our model, the time of cytosolic presentation (and thus of phosphorylation) depends on the distance between the tag and the internal signal-anchor sequence, the rate of translation, the kinetics of SRP binding and targeting to the ER membrane, and finally on the rate of N-terminal translocation. In agreement with this concept, the specific phosphorylation increased with both increasing length of the N-terminal domain and decreasing elongation rate. This also showed that in our system, the rate of phosphorylation is slow relative to translation, targeting and translocation. As a result, the amount of phosphate incorporated reflects the time consumed by these processes. Specific phosphorylation increased linearly with reduced elongation rate over a range of almost a factor of eight. This experiment covered a time period of ∼2.5 min (using H1rev85-PN; Figure 6C) during which the timer was linear.
Analyzing the specific phosphorylation as a function of translation rate (Figure 6B and C) revealed that under normal conditions, approximately half the phosphorylation of H1rev20-PN and H1rev85-PN occurred after the signal sequence had emerged from the ribosome. This indicates that the phosphorylation sequence was translocated across the membrane and thus disappeared from the cytosol when the ribosome had progressed at most another 30 or 95 residues, respectively. Completion of N-terminal translocation thus clearly occurred co-translationally.
Since phosphorylation is dependent on the rate of translation and the length of the N-terminal domain, it is possible to deduce estimates for the rates of N-terminal translocation and for the average time for SRP binding and targeting (Table I; Materials and methods). The rate of translocation was estimated to be ∼1.6 times the rate of translation. The time for SRP binding and targeting is quite short, in the order of only a few seconds, whereas completion of translation takes 10–20 times longer. This result, based on manipulation of the elongation rate (Figure 6), is in excellent agreement with the result obtained by changing the length of the N-terminal domain (Figure 5), thus confirming the underlying model.
At first glance, the short time of SRP binding and targeting seems to exclude an important role for an SRP-induced translation slow-down, since translation would proceed <20 residues at the normal rate during this period. However, it represents the average time for all ribosomes. Initial targeting of a polysome may take significantly longer than targeting of the subsequent ribosomes in a polysome that is already tethered to the ER membrane. In addition, COS cells have an extensive ER that spreads throughout the cell. Other cell types with low secretion activity (e.g. keratinocytes) have a much smaller ER, and initial targeting of a polysome may take considerably longer. SRP-induced reduction of the translation rate may be more important in these cell types and possibly for other signal sequences.
Evidence for a physiological role for elongation slow-down by SRP has been reported only very recently for yeast (Mason et al., 2000). A mutant version of the 14 kDa subunit of SRP defective for elongation arrest in vitro led to temperature-sensitive growth. Small amounts of untargeted Pho8p could be detected at the restrictive temperature, but not of DPAPB, another yeast protein strongly dependent on SRP for targeting. However, using the ubiquitin-assisted translocation assay (Johnsson and Varshavsky, 1994), a reduced targeting rate could also be observed for the DPAPB signal sequence.
The short time of only a few seconds for SRP binding and targeting in COS cells is also corroborated by a previous experiment in which we tested the efficiency of ER targeting of truncated polypeptides with an N-terminal cleavable signal (Goder et al., 1999). The shortest constructs contained only 55 residues following the signal sequence. Since at least 35 of these are hidden within the ribosome, the stop codon was reached and the ribosome disassembled when only 20 residues behind the signal had emerged. More than 90% of the protein with the signal sequence of the vasopressin precursor was nevertheless targeted to the ER and translocated. Since translation of 20 residues takes ∼4 s, most nascent chains must have recruited SRP within this period. If SRP did not affect elongation, this time would also include targeting to the ER membrane. An elongation slow-down by SRP binding would gain additional time for targeting of the ternary complex to the ER. Similar constructs with the signal of influenza HA were targeted less efficiently, with ∼60, 75 and 90% for proteins with 55, 75 and 95 residues, respectively, following the signal sequence (Goder et al., 1999). SRP binding (or SRP binding and targeting) was therefore somewhat slower with a half-time of ∼4 s (the translation time of ∼20 residues). This experiment illustrates that the SRP binding kinetics vary for different signal sequences.
A relevant feature of a signal for rapid recruitment of SRP may be its hydrophobicity. Using the wheat germ in vitro translation system and canine SRP, it was shown that SRP binding, as judged by elongation arrest, increases with increasing length of an oligoleucine signal sequence (Hatsuzawa et al., 1997). The reverse signal-anchor used in our constructs is very hydrophobic as it consists of a stretch of 25 leucines. Whether the time for SRP binding and targeting is significantly increased with a less hydro phobic signal in vivo remains to be tested.
The phosphorylation timer may be of general use to analyze a number of different processes under in vivo conditions, among them the targeting to various compartments within the cell. In addition, it could also be useful to detect the import of proteins into the cytosol from outside the cell or their export from organelles.
Materials and methods
DNA constructs
H1-PN, H1rev-PN, H1-PC and H1rev-PC. To insert the consensus heptapeptide sequence for phosphorylation by PKA, LRRASLG, at the N-terminus of the coding sequence of the cDNAs of wild-type H1 (Spiess et al., 1985) and of the mutant H1rev (identical to H1-4Leu25; Wahlberg and Spiess, 1997), the partially complementary oligonucleotides AGCTTACCATGTTAAGAAGAGCTAGCTTAGGAAC and CTTGGTTCCTAAGCTAGCTCTTCTTAACATGGTA were used. Upon annealing, they produced sticky ends to replace the HindIII–StyI fragments of the cDNAs easily. For insertion of the heptapeptide into the C-terminal domain, the partially complementary oligonucleotides GTGCTTAGAAGAGCTAGCTTAGGGCC and CTAAGCTAGCTCTTCTAAG were annealed and exchanged for the DraIII–ApaI fragment of the constructs, which encodes residues 190–220 of the original coding sequences.
H1rev20-PN and H1rev85-PN. These constructs were generated by PCR with Vent polymerase (New England Biolabs). The N-terminal domain of H1-PN between the phosphorylation tag and the hydrophobic signal was shortened to 20 residues using H1rev-PN as the template and a primer that placed the StyI site 20 residues towards the C-terminus. To extend the spacer to 85 residues, we used the construct DmA4 (Denzer et al., 1995) as the template and two primers to amplify residues 144–188 of mouse dihydrofolate reductase for insertion between the NheI site within the phosphorylation sequence and the StyI site at the start of the H1 sequence. The final constructs for in vivo expression were subcloned into the vector pECE (Ellis et al., 1986) and verified by sequencing.
In vivo expression and labeling
Cell culture reagents were from Life Technologies, Inc. COS-1 cells were grown in modified Eagle’s minimal essential medium supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C with 7.5% CO2. Transient transfection was performed with lipofectin (Life Technologies, Inc.) according to the manufacturer’s instructions in 6-well clusters. The cells were processed 2 days after transfection. For in vivo labeling with [35S]methionine, transfected cells were starved for 40 min in methionine-free medium, labeled for 40 min at 37°C with 100 µCi/ml [35S]methionine in starvation medium, and washed with cold phosphate-buffered saline (PBS). The cells were then incubated for 20 min with 0.1% saponin in PBS, which permeabilizes the membranes to release soluble proteins. The cells were then lysed, and immunoprecipitated using a rabbit antiserum directed against a synthetic peptide corresponding to residues 277–287 near the C-terminus of H1 (anti-H1C). The immune complexes were isolated with protein A–Sepharose (Pharmacia, Sweden) and analyzed by SDS–PAGE and fluorography. For deglycosylation, the immune complexes were released from protein A–Sepharose by boiling in 50 mM Na citrate pH 6, 1% SDS, and incubated with 1 mU endo H for 5 h at 37°C. Quantitation was performed using a PhosphorImager (Molecular Dynamics Inc.).
For in vivo labeling with [32P]phosphate, cells were starved in phosphate-free medium and labeled for 40 min with 100 µCi/ml [32P]phosphate in the presence of 20 µM forskolin. For co-labeling, cells were starved for 40 min in medium without methionine and phosphate, and then labeled for 40 min with 100 µCi/ml [35S]methionine and [32P]phosphate. Cells were extracted with 0.1% saponin for 30 min at 4°C in the presence of phosphatase inhibitors (500 µM nitrophenyl phosphate, 50 µM sodium orthovanadate, 1 mM sodium fluoride, 1 mM EDTA) and processed further as described above. For quantitation, the gels were exposed to a PhosphorImager plate twice for identical times with and without a transparency film in between, which was calibrated to block >99% of 35S, but only 25% of 32P.
To determine the effect of cycloheximide on the translation of H1rev20-PN and H1rev85-PN (Figure 6A), transfected cells were labeled with [35S]methionine for 40 min in the presence of 0–10 µg/ml cycloheximide, extracted with 0.1% saponin in PBS for 20 min, and the remaining material was immunoprecipitated, analyzed by gel electrophoresis and quantified using a PhosphorImager. To normalize the values for cell density in the different wells, they were corrected based on the protein content in the saponin extracts as measured using the bicinchoninic acid method (Pierce). A titration curve from duplicate measurements was plotted and used for the subsequent analysis of specific phosphorylation versus translation time (Figure 6B).
For western analysis, SDS gels were blotted to nitrocellulose membranes. H1 derivatives were detected using the same primary antibody as for immunoprecipitation, a goat anti-rabbit IgG secondary antibody coupled to horseradish peroxidase, and the ECL detection kit (Amersham-Pharmacia).
Data analysis
Specific phosphorylation p is defined by the rate of phosphorylation vp, the time of translation ta = L/va (L, length to be translated; va, translation rate), the time of SRP binding and targeting tbc, the time for translocation of the N-terminal sequence td = L/vd (vd, translocation rate) and a proportionality factor k, which includes the specific activities of the radiolabeled precursors and the counting efficiencies:
p = kvp(ta + tbc + td) = kvp(L/va + tbc + L/vd)
At variable translation rate, the equation can be normalized to the translation time ![]() or the translation rate
or the translation rate ![]() in the absence of inhibitor using
in the absence of inhibitor using 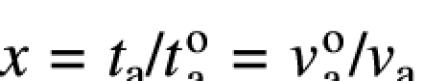 :
:
![]() ]
]
The parameter (tbc + td)/![]() was determined by linear regression from the data sets of H1rev20-PN and H1rev85-PN (Figure 6A and B) for p = 0:
was determined by linear regression from the data sets of H1rev20-PN and H1rev85-PN (Figure 6A and B) for p = 0:
![]()
It is important to note that neither the proportionality factor, which may vary between different experiments, nor the actual rate of phosphorylation, which may vary between different constructs, affects 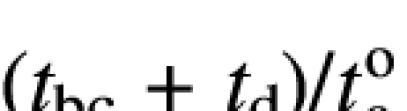 . This term, estimated for separate constructs with different L, can therefore be used to determine vao/vd and
. This term, estimated for separate constructs with different L, can therefore be used to determine vao/vd and ![]() by linear regression of the equation:
by linear regression of the equation:
![]()
Acknowledgments
Acknowledgements
We thank Dr Dieter Walz for advice on data analysis, and Drs Vivienne Laird and Natasha Kralli for critically reading the manuscript. This work was supported by Grant 31-43483.95 from the Swiss National Science Foundation.
References
- Beltzer J.P., Fiedler,K., Fuhrer,C., Geffen,I., Handschin,C., Wessels,H.P. and Spiess,M. (1991) Charged residues are major determinants of the transmembrane orientation of a signal-anchor sequence. J. Biol. Chem., 266, 973–978. [PubMed] [Google Scholar]
- Dale H. and Krebs,M.P. (1999) Membrane insertion kinetics of a protein domain in vivo. The bacterioopsin N terminus inserts co-translationally. J. Biol. Chem., 274, 22693–22698. [DOI] [PubMed] [Google Scholar]
- Dale H., Angevine,C.M. and Krebs,M.P. (2000) Ordered membrane insertion of an archaeal opsin in vivo. Proc. Natl Acad. Sci. USA, 97, 7847–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzer A.J., Nabholz,C.E. and Spiess,M. (1995) Transmembrane orientation of signal-anchor proteins is affected by the folding state but not the size of the N-terminal domain. EMBO J., 14, 6311–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis L., Clauser,E., Morgan,D.O., Edery,M., Roth,R.A. and Rutter,W.J. (1986) Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose. Cell, 45, 721–732. [DOI] [PubMed] [Google Scholar]
- Eusebio A., Friedberg,T. and Spiess,M. (1998) The role of the hydrophobic domain in orienting natural signal sequences within the ER membrane. Exp. Cell Res., 241, 181–185. [DOI] [PubMed] [Google Scholar]
- Gafvelin G., Sakaguchi,M., Andersson,H. and von Heijne,G. (1997) Topological rules for membrane protein assembly in eukaryotic cells. J. Biol. Chem., 272, 6119–6127. [DOI] [PubMed] [Google Scholar]
- Geffen I., Fuhrer,C. and Spiess,M. (1991) Endocytosis by the asialoglycoprotein receptor is independent of cytoplasmic serine residues. Proc. Natl Acad. Sci. USA, 88, 8425–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V., Bieri,C. and Spiess,M. (1999) Glycosylation can influence topogenesis of membrane proteins and reveals dynamic reorientation of nascent polypeptides within the translocon. J. Cell Biol., 147, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann E., Rapoport,T.A. and Lodish,H.F. (1989) Predicting the orientation of eukaryotic membrane spanning proteins. Proc. Natl Acad. Sci. USA, 86, 5786–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsuzawa K., Tagaya,M. and Mizushima,S. (1997) The hydrophobic region of signal peptides is a determinant for SRP recognition and protein translocation across the ER membrane. J. Biochem., 121, 270–277. [DOI] [PubMed] [Google Scholar]
- Hershey J.W.B. (1991) Translational control in mammalian cells. Annu. Rev. Biochem., 60, 717–755. [DOI] [PubMed] [Google Scholar]
- High S., Flint,N. and Dobberstein,B. (1991) Requirements for the membrane insertion of signal-anchor type proteins. J. Cell Biol., 113, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmquist G., Andersson,J., Edlund,B. and Engstroom,L. (1974) Amino acid sequence of a (32P) phosphopeptide from pig liver pyruvate kinase phosphorylated by cyclic 3′,5′-AMP-stimulated protein kinase and γ-(32P)ATP. Biochem. Biophys. Res. Commun., 61, 559–563. [DOI] [PubMed] [Google Scholar]
- Johnsson N. and Varshavsky,A. (1994) Ubiquitin-assisted dissection of protein transport across membranes. EMBO J., 13, 2686–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kast C., Canfield,V., Levenson,R. and Gros,P. (1996) Transmembrane organization of mouse P-glycoprotein determined by epitope insertion and immunofluorescence. J. Biol. Chem., 271, 9240–9248. [DOI] [PubMed] [Google Scholar]
- Kemp B.E., Graves,D.J., Benjamini,E. and Krebs,E.G. (1977) Role of multiple basic residues in determining the substrate specificity of cyclic AMP-dependent protein kinase. J. Biol. Chem., 252, 4888–4894. [PubMed] [Google Scholar]
- Loo T.W. and Clarke,D.M. (1995) Membrane topology of a cysteine-less mutant of human P-glycoprotein. J. Biol. Chem., 270, 843–848. [DOI] [PubMed] [Google Scholar]
- Mason N., Ciufo,L.F. and Brown,J.D. (2000) Elongation arrest is a physiologically important function of signal recognition particle. EMBO J., 19, 4164–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothes W., Prehn,S. and Rapoport,T.A. (1994) Systematic probing of the environment of a translocating secretory protein during translocation through the ER membrane. EMBO J., 13, 3973–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothes W., Jungnickel,B., Brunner,J. and Rapoport,T.A. (1998) Signal sequence recognition in cotranslational translocation by protein components of the endoplasmic reticulum membrane. J. Cell Biol., 142, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson I.M. and von Heijne,G. (1993) Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem., 268, 5798–5801. [PubMed] [Google Scholar]
- Nilsson I., Witt,S., Kiefer,H., Mingarro,I. and von Heijne,G. (2000) Distant downstream sequence determinants can control N-tail translocation during protein insertion into the endoplasmic reticulum membrane. J. Biol. Chem., 275, 6207–6213. [DOI] [PubMed] [Google Scholar]
- Ogg S.C. and Walter,P. (1995) SRP samples nascent chains for the presence of signal sequences by interacting with ribosomes at a discrete step during translation elongation. Cell, 81, 1075–1084. [DOI] [PubMed] [Google Scholar]
- Rapiejko P.J. and Gilmore,R. (1997) Empty site forms of the SRP54 and SRα GTPases mediate targeting of ribosome–nascent chain compexes to the endoplasmic reticulum. Cell, 89, 703–713. [DOI] [PubMed] [Google Scholar]
- Rösch K., Naeher,D., Laird,V., Goder,V. and Spiess,M. (2000) The topogenic contribution of uncharged amino acids on signal sequence orientation in the endoplasmic reticulum. J. Biol. Chem., 275, 14916–14922. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M., Tomiyoshi,R., Kuroiwa,T., Mihara,K. and Omura,T. (1992) Functions of signal and signal-anchor sequences are determined by the balance between the hydrophobic segment and the N-terminal charge. Proc. Natl Acad. Sci. USA, 89, 16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel V. and Walter,P. (1988) The affinity of signal recognition particle for presecretory proteins is dependent on nascent chain length. EMBO J., 7, 1769–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Raden,D., Mandon,E.C. and Gilmore,R. (2000) Role of Sec61α in the regulated transfer of the ribosome–nascent chain complex from the signal recognition particle to the translocation channel. Cell, 100, 333–343. [DOI] [PubMed] [Google Scholar]
- Spiess M. (1995) Head or tails—what determines the orientation of proteins in the membrane. FEBS Lett., 369, 76–79. [DOI] [PubMed] [Google Scholar]
- Spiess M. and Lodish,H.F. (1986) An internal signal sequence: the asialoglycoprotein receptor membrane anchor. Cell, 44, 177–185. [DOI] [PubMed] [Google Scholar]
- Spiess M., Schwartz,A.L. and Lodish,H.F. (1985) Sequence of human asialoglycoprotein receptor cDNA. An internal signal sequence for membrane insertion. J. Biol. Chem., 260, 1979–1982. [PubMed] [Google Scholar]
- von Heijne G. (1984) Analysis of the distribution of charged residues in the N-terminal region of signal sequences: implications for protein export in prokaryotic and eukaryotic cells. EMBO J., 3, 2315–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg J.M. and Spiess,M. (1997) Multiple determinants direct the orientation of signal-anchor proteins: the topogenic role of the hydrophobic signal domain. J. Cell Biol., 137, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P. and Johnson,A.E. (1994) Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu. Rev. Cell Biol., 10, 87–119. [DOI] [PubMed] [Google Scholar]
- Wessels H.P., Beltzer,J.P. and Spiess,M. (1991) Analysis of protein topology in the endoplasmic reticulum. Methods Cell Biol., 34, 287–302. [DOI] [PubMed] [Google Scholar]
- Wilkinson B.M., Critchley,A.J. and Stirling,C.J. (1996) Determination of the transmembrane topology of yeast Sec61p, an essential component of the endoplasmic reticulum translocation complex. J. Biol. Chem., 271, 25590–25597. [DOI] [PubMed] [Google Scholar]
- Wolin S.L. and Walter,P. (1989) Signal recognition particle mediates a transient elongation arrest of preprolactin in reticulocyte lysate. J. Cell Biol., 109, 2617–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]