

Abstract
Mutations of CCM3/PDCD10 cause 10-15% of hereditary cerebral cavernous malformations. The phenotypic characterization of CCM3-mutated patients has been hampered by the limited number of patients harboring a mutation in this gene. This is the first report on molecular and clinical features of a large cohort of CCM3 patients. Molecular screening for point mutations and deletions was used to identify 54 CCM3-mutated index patients. Age at referral and clinical onset, type of inaugural events and presence of extra-axial lesions were investigated in these 54 index patients and 22 of their mutated relatives. Mean age at clinical onset was 23.0 ± 16 years. Clinical onset occurred before 10 years in 26% of the patients, and cerebral hemorrhage was the initial presentation in 72% of these patients. Multiple extra-axial, dural-based lesions were detected in 7 unrelated patients. These lesions proved to be meningiomas in 3 patients who underwent neurosurgery and pathological examination. This ‘multiple meningiomas’ phenotype is not associated with a specific CCM3 mutation. Hence, CCM3 mutations are associated with a high risk of early-onset cerebral hemorrhage and with the presence of multiple meningiomas.
Key Words : Cavernous angioma, CCM, CCM1, CCM2, KRIT1, Meningioma, PDCD10
Cavernous angiomas, also known as cerebral cavernous malformations (CCM; OMIM 116860), affect roughly 0.5% of the population and are characterized by abnormal enlarged capillary cavities without intervening brain parenchyma [Russell and Rubinstein, 1989]. CCM lesions are mostly parenchymal lesions affecting the brain and to a lesser extent the spinal cord. In the brain, CCM lesions are usually found in the subcortical white matter. However, CCM may arise in some patients from the dura mater. These dural-based lesions are mostly unique and most often located in the middle cranial fossa, but they may more rarely be encountered outside the middle cranial fossa, as in the convexity [Biondi et al., 2002].
Clinical CCM symptoms include recurrent headaches, focal neurological deficits, hemorrhagic stroke, and seizures [Zabramski et al., 1994; Zabramski et al., 1999; Labauge et al., 2007], but CCM can also be asymptomatic. CCM are sporadic or familial, and over 90% of the patients have a mutation in one of the 3 known genes CCM1/KRIT1 (OMIM 604214) [Laberge-le Couteulx et al., 1999; Sahoo et al., 1999], CCM2/MGC4607 (OMIM 607929) [Liquori et al., 2003; Denier et al., 2004] or CCM3/PDCD10 (OMIM 609118) [Bergametti et al., 2005]. Linkage data on 20 large North American families suggested that 40% of the CCM families were linked to CCM3 loci, but after identification of the gene, the proportion of CCM3-mutated families appeared to be much lower.
Less than 40 CCM3 families have been reported so far [Guclu et al., 2005; Verlaan et al., 2005; Denier et al., 2006; Gault et al., 2006; Liquori et al., 2006, 2008; Lee et al., 2008; Choe et al., 2010]. This limited number of CCM3-mutated patients hampered the analysis of genotype-phenotype correlations. In a previous study on 333 CCM mutation carriers including 28 CCM3 patients belonging to 17 families, we suggested that the proportion of CCM3 patients with a childhood onset was higher compared to CCM1 and CCM2 patients [Denier et al., 2006]. We also showed in 4 unrelated CCM3 patients the occurrence of multiple dural-based lesions; however, the pathological nature of these lesions was unknown since none of these patients had undergone neurosurgery [Labauge et al., 2009].
Herein, we conducted a molecular and clinical study on a larger CCM3 patient cohort, focusing on the age at clinical onset, the type of inaugural event and the association with extra-axial dural-based lesions.
Patients and Methods
Genomic DNA sequencing and quantitative multiplex PCR of short fragments of the 3 known CCM genes allowed us to identify a consecutive series of 300 unrelated CCM probands harboring a loss of function mutation in 1 of the 3 CCM genes. Among these patients who were either enrolled in research programs or referred for molecular diagnosis, 189 were mutated in CCM1, 57 in CCM2 and 54 in CCM3. Our study group was composed of these 54 probands and 22 of their mutated relatives. Partial information on some of these patients have been previously reported [Denier et al., 2006; Ahdab et al., 2008; Labauge et al., 2009; Choe et al., 2010].
CCM diagnosis was established on the basis of cerebral MRI and pathological data. Clinical information was collected through direct interview and reviewing of the medical and MRI charts. Clinical assessment included information on age at referral, age at clinical onset and characteristics of inaugural manifestations. MRI investigations were used to search for extra-axial lesions. Pathological data from the 3 patients who underwent surgery for extra-axial lesions were reviewed by experienced neuropathologists (F.C., S.M.-P., C.B).
Results
CCM3 Mutations
A CCM3 mutation was identified in 54 probands (table 1). Thirteen had a deletion encompassing one or several coding exons, including 8 whole gene deletions. Forty-one probands had a point mutation leading to a premature stop codon (table 1). Thirty-one different point mutations were identified including 6 recurrent ones (3 found twice, 2 found 3 times, and 1 found 4 times). Point mutations leading to an abnormal splicing accounted for 42% (13/31); nonsense mutation and small insertions or deletions leading to a frameshift and premature stop codon accounted each for 29% (9/31). Two deletions involved only noncoding exons. One was a deletion of exons 1-3 that happened de novo in a 5-year-old boy (C270) with typical multiple cerebral cavernomas (fig. 1). The second one was a deletion of exons 1 and 2 that cosegregated with the affected phenotype within family C085. They were both considered as probably deleterious. The de novo appearance of mutations was established in 4 probands (C052, C146, C107, and C270) and was deduced from familial haplotype analysis for the father of proband C044.
Table 1.
CCM3 mutations
| Exons | Mutation (NM_007217) | Effect on splicing (cDNA analysis) | Theoretic effect on the protein | Family number | Reported in other families |
|---|---|---|---|---|---|
| 01 – 02 | deletion of exons 1 – 2 | absence of protein synthesis | C085 | ||
| 01 – 03 | deletion of exons 1 – 3 | absence of protein synthesis | C270 | ||
| 01 – 10 | whole gene deletion | absence of protein synthesis | C024 C045a C052a C146a C224 C399c C474 C556 | Liquori et al. [2008] | |
| 03 – 04 | deletion of exons 3 – 4 | absence of protein synthesis | C518 | ||
| 04 | c.81delT | p.P28LfsX6 | C436 | ||
| 05 | c.97-2A>G | exon 5 deletion | p.L33_K50del | C601 | |
| 05 | c.103C>T | p.R35X | C055a C377 C446 | Lee et al. [2008] | |
| 05 | c.113_114delGT | p.S39CfsX13 | C211a | ||
| 05 | c.129_138 del | p.R45SfsX17 | C560 | ||
| 05 | deletion of exon 5 | p.L33_K50del | C043a | ||
| 06 | c.151-1G>C | exon 6 deletion | p.A51SfsX3 | C523 | |
| 06 | c.175dupA | p.T59NfsX17 | C594 | ||
| 06 | c.196delA | Guclu et al. [2005] | |||
| 06 | c.212insA | p.K71Xfs | C304 | ||
| 06 | c.223delA | Guclu et al. [2005] | |||
| 06 | c.268 + 1dupG | several transcripts | C474 | ||
| 07 | c.269-1G>A | exon 7 deletion | p.E90GfsX3 | C493 | |
| 07 | c.275delT | p.M92Xfs | C418 | ||
| 07 | c.283C>T | p.R95X | C254a C589 | Guclu et al. [2005]; Verlaan et al. [2005] | |
| 07 | c.322C>T | p.R108X | C571 C467 C313 | ||
| 07 | c.372_73delAG | p.R124Xfs | C357 | ||
| 07 | c.385C>T | p.Q129X | C031a | ||
| 07 | c.394_395delinsGATT | exon 7 deletion | p.E90GfsX3 | C615 | |
| 08–10 | deletion of exons 8 – 10 | C433 | |||
| 08 | c.396-31_396-13delinsA | exon 8 deletion | p.D133HfsX10 | C596 | |
| 08 | c.396-3C>G | exon 8 deletion | p.D133HfsX10 | C238a | |
| 08 | c.396-2A>G | Verlaan et al. [2005] | |||
| 08 | c.401_402dupTA | p.A135X | C070a | ||
| 08 | c.418G>T | p.D133HfsX10 | C502 | ||
| 08 | c.456T>G | p.Y152X | C128a | ||
| 08 | c.474 + 1G>A | Liquori et al. [2006] | |||
| 08 | c.474 + 1G>C | C327 | |||
| 08 | c.474 + 2T>C | exon 8 deletion | p.D133HfsX10 | C223a | |
| 08 | c.474 + 5G>A | exon 8 deletion | p.D133HfsX10 | C158b C189 | Liquori et al. [2006] |
| 09 | c.475-1G>A | exon 9 deletion | p.A159GfsX42 | 007a, b | |
| 09 | c.496G>T | p.E166X | C319 C572 | ||
| 09 | c.514A>T | p.K172X | C122 | ||
| 09 | c.534delA | Guclu et al. [2005] | |||
| 09 | c.538delA | p.T180fs188X | C161b, d | ||
| 09 | c.557 + 1G>C | ? | C141a | ||
| 09 | c.557 + 3_557 + 6del | exon 9 deletion | C044a | ||
| 10 | c.586C>T | p.R196X | C030a,b C077a C346 C520 | ||
| 10 | c.608T>G | p.L203X | Gault et al. [2006]; Liquori et al. [2006] | ||
Patients reported in Denier et al. [2006].
Patients reported in Labauge et al. [2009].
Patient reported in Choe et al. [2010].d Patient reported in Ahdab et al. [2008].
Fig. 1.
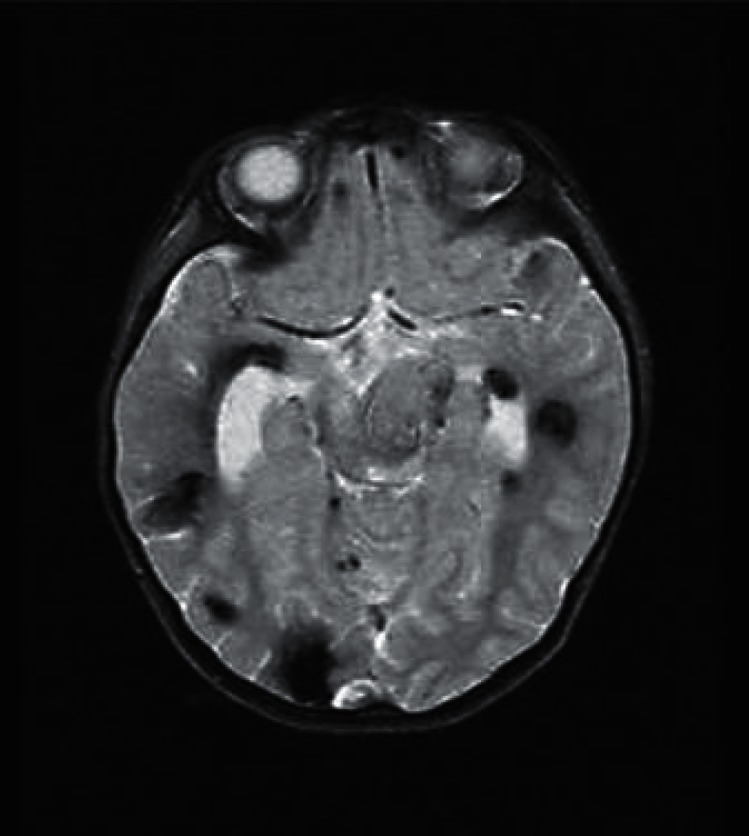
Cerebral MRI of proband C270. Multiple CCM lesions in a 5-year-old boy harboring a de novo mutation removing CCM3 exons 1-3.
CCM3 Mutations Are Associated with Early-Onset Cerebral Hemorrhage
Thirty of the 54 probands were of the female sex (55.5%), and 24 were of the male sex (44.5%). Among the 54 probands, 5 were asymptomatic and were referred after the fortuitous discovery of CCM on an MRI performed for another reason. Twenty-five probands had at least one symptomatic relative (familial cases), and 27 probands reported having no symptomatic relative (isolated cases). Information was not available for 2 probands. Analysis of both parents of 6 clinically sporadic cases was performed and showed that the mutation appeared de novo for 4 patients and was inherited from an asymptomatic parent for the other 2. Forty-eight probands had multiple lesions on MRI, and 3 had a single lesion. Two of them were aged 1.2 and 13 years at onset respectively; the third proband was a 10-year-old boy for whom cavernoma was discovered in a context of developmental delay and known multiple cavernoma in an aunt.
Age at referral ranged between 1.2 and 71.8 years (mean age at referral: 28.9 ± 16.8 years). Age at clinical onset was available for 43 probands. Mean age at clinical onset was 23.0 ± 16.0 years. The first manifestation was a cerebral hemorrhage for 21 patients (49%), but the nature of the first clinical manifestation was different depending on the age at clinical onset (table 2, fig. 2). Cerebral hemorrhage was the main inaugural manifestation in patients under 20 years of age, and it was the first event in 72% of the children younger than 10. The proportion of patients whose first clinical manifestation was a cerebral hemorrhage decreased with age. Among the 13 patients with a clinical onset later than 30 years, only 3 patients started their clinical manifestations with a cerebral hemorrhage.
Table 2.
Age at onset and inaugural manifestation
| Age at onset years | Probands | Inaugural manifestation | ||
|---|---|---|---|---|
| cerebral hemorrhage | epilepsy | other | ||
| 0 – 10 | 11 | 8 | 3 | 0 |
| 11 – 20 | 9 | 5 | 2 | 2 |
| 21 – 30 | 10 | 5 | 4 | 1 |
| 31 – 40 | 10 | 3 | 3 | 4 |
| 41 – 50 | 1 | 0 | 0 | 1 |
| >51 | 2 | 0 | 2 | 0 |
Fig. 2.

Proportion of patients with an inaugural cerebral hemorrhage depending on age at clinical onset.
A CCM3 mutation was detected in 22 relatives. Mean age at referral was 34.8 ± 19.3 years. Ten of them were symptomatic for cavernomas, 11 were asymptomatic, and the clinical status was unknown for the last one. Sixteen had multiple cavernomas, and one had a single cavernoma lesion and MRI. Data were not available for 4 patients.
CCM3 Mutations Are Associated with Extra-Axial Lesions and Multiple Meningiomas
Multiple intracranial, dural-based lesions were detected in 7 unrelated patients from families C30, C107, C158, C161, C346, C399, and C615 (fig. 3a, b). Three of these patients (from families C346, C399 and C616) underwent neurosurgery. Pathological analysis of the lesions identified in all 3 patients typical grade I transitional meningiomas (fig. 3b).
Fig. 3.

Families with multiple dural-based lesions. a Pedigrees of the 7 families with multiple dural-based lesions. Squares and circles indicate males and females, respectively. The probands are indicated by arrows. b Cerebral MRI and pathological data of 2 patients with pathologically proven meningiomas. A-D Mother of proband C399. Axial T2-weighted gradient-echo sequences (A) show multiple parenchymal cavernous angiomas. Sagittal (B) and axial (C) gadolinium-enhanced dural lesions on T1-weighted images. D Paraffin sections colored with hematein eosin. Meningothelial meningioma with typical whorl formations (arrowheads). Inset A high-power magnification showing central clearing of nuclei (arrow). Scale bar = 100 µm. E-H Father of proband C615. Axial T2-weighted gradient-echo sequences (E) show multiple parenchymal cavernous angiomas. Axial (F) and coronal (G) gadolinium-enhanced T1-weighted sequences (E) show left frontal and sphenoidal dural lesions. H Typical pattern of a transitional menigioma: prominent whorls and several psammoma bodies (hematoxylin-eosin-saffron staining, magnification: ×100).
Within family C346, the mother of the proband, born in 1950, had multiple parenchymal CCM lesions but was asymptomatic with regard to her CCM lesions. However, she underwent surgery for a supra-sellar meningioma at age 43. Pathological analysis showed a typical grade I transitional meningioma. Her daughter, born in 1974, had a peripheral facial palsy leading to the identification of an extra-axial cavernous angioma of the internal auditory canal and multiple parenchymal cerebral CCM lesions.
Within family C399, the mother of the proband, born in 1953, presented in 2007 a loss of vision of her right eye and a right trochlear nerve palsy. Cerebral MRI showed multiple parenchymal CCM lesions, multiple dural-based lesions seen as hypointense lesions on gadolinium-enhanced T1 sequences with a dural tail sign and a meningioma of the right optical canal nerve that was surgically removed (fig. 3b, A-C). Pathological analysis showed a typical meningothelial meningioma (fig. 3b, C). Her daughter, born in 1970, was symptomatic and her MRI showed a spinal and multiple parenchymal CCM lesions. No extra-axial dural-based lesion was detected.
Within family C615, the father of the proband, born in 1953, underwent neurosurgery for parenchymal CCM in 1992 and 2001 (fig. 3b, E-G). In 1993, a meningioma of the right larger sphenoidal wing was detected which was operated upon. In 2005, 3 novel meningiomas (left frontal, left small sphenoidal wing and left pterional bone) were surgically removed. Pathological analysis of all 4 lesions showed grade I transitional meningiomas (fig. 3b, H). His daughter, born in 1988, developed paresthesias of the right leg, arm and trunk, and was diagnosed as having multiple cerebral parenchymal CCM lesions in 2010. Her MRI does not show extra-axial lesions.
The 4 remaining patients were the probands of families C30, C107, C158, and C161 (3 women and 1 man; mean age: 48 years, range: 42-64 years). MRI of these patients were highly stereotyped and showed: (1) multiple parenchymal cavernous angiomas, and (2) multiple dural-based lesions with similar MR patterns, i.e. hypointense on T1 and hyperintense on T2 sequences, markedly enhanced by gadolinium injection, and with a diffuse dural enhancement. The number of dural lesions increased with age in 3 of them (C30, C161 and C158). Since none of these 4 patients underwent surgery, it was therefore not possible to establish the pathological nature of these lesions which may either be extra-axial, dural-based cavernous angiomas mimicking meningiomas or true meningiomas.
The association of multiple extra-dural lesions and multiple meningiomas was not detected so far in any of the probands mutated in CCM1 (189 patients) or CCM2 (57 patients).
Discussion
We report herein the largest series of CCM3-mutated patients. The analysis of this consecutive series of 54 probands and their relatives allowed us (1) to confirm the association of CCM3 mutations with early-onset cerebral hemorrhage, (2) to show for the first time that these mutations may be associated in some patients with the development of multiple meningiomas, and (3) to show that this association is specifically encountered in CCM3-mutated patients and not detected in CCM1 or CCM2 mutants.
We present 7 patients with both parenchymal CCM lesions and multiple dural-based lesions, and we show that this very unusual phenotype is strongly associated with mutations in CCM3. These lesions, multiple in all cases except one (mother of proband C346), have the typical MRI features of meningiomas. They are isointense on T1, slightly hyperintense on T2 sequences, and strongly and homogenously enhanced by gadolinium injection with a dural tail sign in most of them. However, these features have been previously reported in patients showing extra-axial histologically proven cavernous angiomas [Perry et al., 1993; Kanaan et al., 2001; Biondi et al., 2002; Guermazi et al., 2005]. In most cases, these patients were preoperatively considered as having a meningioma. These previously reported patients had only one single dural-based lesion and no parenchymal CCM lesion except one patient showing both multiple parenchymal CCM and dural-based histopathologically proven cavernous angiomas [Lewis et al., 1994]. Herein, we have been able to show for the first time that in some patients, these dural-based lesions are typical meningiomas and not extra-axial cavernous angiomas. In one of our patients (father of proband C615), 4 distinct meningiomas were surgically removed. In 2 other patients, surgery of one of their multiple lesions was performed, and pathological analysis disclosed a typical meningioma. Altogether, these data strongly suggest that CCM3 mutations are associated with both multiple meningiomas and multiple extra-axial cavernous angiomas. The penetrance of this ‘meningioma’ phenotype is incomplete.
It is also of note that the mutations observed in patients with multiple meningiomas do not differ from those observed in patients showing solely parenchymal cerebral cavernous angiomas. Indeed, the same mutation, R196X, was identified in one family with multiple meningiomas (C346), but also in families C077 and C520 in which no dural-based lesion was detected. The complete CCM3 deletion is associated with multiple meningiomas in family C399, whereas in 7 other families, showing a complete deletion of CCM3, no dural-based lesion was detected in any affected member. The absence of dural-based lesions in patients carrying similar mutations suggests that these mutations have either an incomplete penetrance or, most likely, involve modifying genes.
Meningiomas are the most frequent primary central nervous system tumors in adult patients, and numerous studies have been conducted to identify the genes and mechanisms involved in their genesis and progression [Mawrin and Perry, 2010]. Loss of function mutations in the NF2 gene have been involved in the majority of these tumors. A variety of additional cytogenetic anomalies, mutations of several other tumor suppressor genes and genes involved in the Wnt signaling pathway have been involved in both NF2-mutated and NF2-nonmutated meningiomas [Mawrin and Perry, 2010; Clark et al., 2013]. However, to our knowledge, neither the CCM3 locus nor the CCM3 gene have previously been involved in sporadic or familial meningioma. CCM3 encodes an adaptor protein which is part of the cytosolic CCM complex, but CCM3 is also part of several multiprotein complexes including serine/threonine kinases STK24, STK35 and MST4, and the STRIPAK complex [Ma et al., 2007; Voss et al., 2007; Goudreault et al., 2009; Faurobert and Albiges-Rizo, 2010]. Many components of this complex are involved in cell cycle regulation and apoptosis with CCM3 having a proapoptotic role in vitro [Chen et al., 2009; Fidalgo et al., 2010]. In addition, Ccm3 ablation in the mouse has recently been shown to be responsible for increased cell proliferation and cell survival/resistance to apoptosis through an activation of the Akt signalling pathway [Louvi et al., 2011]. At present, the mechanisms leading from CCM3 mutations to meningioma are unknown. Future work including genomic analysis of the meningioma lesions of our patients should help to decipher the mechanisms and signaling pathways which may be involved. It may also help to unravel specific functions of CCM3 since neither CCM1 nor CCM2 mutations lead to this phenotype.
Acknowledgements
This work was supported by the Programme Hospitalier de Recherche Clinique (AOR3031). We are indebted to B. Fontaine, J.P. Neau (MD), A. Lannuzel, and M. Clanet for their help in the clinical characterization of some of these patients and to M. Arnould for her help in the molecular analysis.
References
- Ahdab R, Riant F, Brugieres P, Roujeau JC, Hodel J, Hosseini H. Familial cerebral cavernomatous malformations associated with palmar capillary telangiectasias. Neurology. 2008;71:861–862. doi: 10.1212/01.wnl.0000325474.61048.ae. [DOI] [PubMed] [Google Scholar]
- Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi A, Clemenceau S, Dormont D, Deladoeuille M, Ricciardi GK, et al. Intracranial extra-axial cavernous (HEM) angiomas: tumors or vascular malformations? J Neuroradiol. 2002;29:91–104. [PubMed] [Google Scholar]
- Chen L, Tanriover G, Yano H, Friedlander R, Louvi A, Gunel M. Apoptotic functions of PDCD10/CCM3, the gene mutated in cerebral cavernous malformation 3. Stroke. 2009;40:1474–1481. doi: 10.1161/STROKEAHA.108.527135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe CU, Riant F, Gerloff C, Tournier-Lasserve E, Orth M. Multiple cerebral cavernous malformations and a novel CCM3 germline deletion in a German family. J Neurol. 2010;257:2097–2098. doi: 10.1007/s00415-010-5648-7. [DOI] [PubMed] [Google Scholar]
- Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO Science 2013, E-pub ahead of print. [DOI] [PMC free article] [PubMed]
- Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74:326–337. doi: 10.1086/381718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550–556. doi: 10.1002/ana.20947. [DOI] [PubMed] [Google Scholar]
- Faurobert E, Albiges-Rizo C. Recent insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. FEBS J. 2010;277:1084–1096. doi: 10.1111/j.1742-4658.2009.07537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidalgo M, Fraile M, Pires A, Force T, Pombo C, Zalvide J. CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J Cell Sci. 2010;123:1274–1284. doi: 10.1242/jcs.061341. [DOI] [PubMed] [Google Scholar]
- Gault J, Sain S, Hu LJ, Awad IA. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery. 2006;59:1278–1284. doi: 10.1227/01.NEU.0000249188.38409.03. discussion 1284-1285. [DOI] [PubMed] [Google Scholar]
- Goudreault M, D'Ambrosio LM, Kean MJ, Mullin MJ, Larsen BG, et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics. 2009;8:157–171. doi: 10.1074/mcp.M800266-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guclu B, Ozturk AK, Pricola KL, Bilguvar K, Shin D, et al. Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery. 2005;57:1008–1013. doi: 10.1227/01.neu.0000180811.56157.e1. [DOI] [PubMed] [Google Scholar]
- Guermazi A, Lafitte F, Miaux Y, Adem C, Bonneville JF, Chiras J. The dural tail sign – beyond meningioma. Clin Radiol. 2005;60:171–188. doi: 10.1016/j.crad.2004.01.019. [DOI] [PubMed] [Google Scholar]
- Kanaan I, Jallu A, Alwatban J, Patay Z, Hessler R. Extra-axial cavernous hemangioma: two case reports. Skull Base. 2001;11:287–295. doi: 10.1055/s-2001-18635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labauge P, Denier C, Bergametti F, Tournier-Lasserve E. Genetics of cavernous angiomas. Lancet Neurol. 2007;6:237–244. doi: 10.1016/S1474-4422(07)70053-4. [DOI] [PubMed] [Google Scholar]
- Labauge P, Fontaine B, Neau JP, Bergametti F, Riant F, et al. Multiple dural lesions mimicking meningiomas in patients with CCM3/PDCD10 mutations. Neurology. 2009;72:2044–2046. doi: 10.1212/WNL.0b013e3181a92b13. [DOI] [PubMed] [Google Scholar]
- Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–193. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- Lee ST, Choi KW, Yeo HT, Kim JW, Ki CS, Cho YD. Identification of an Arg35X mutation in the PDCD10 gene in a patient with cerebral and multiple spinal cavernous malformations. J Neurol Sci. 2008;267:177–181. doi: 10.1016/j.jns.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Lewis AI, Tew JM, Jr, Payner TD, Yeh HS. Dural cavernous angiomas outside the middle cranial fossa: a report of two cases. Neurosurgery. 1994;35:498–504. doi: 10.1227/00006123-199409000-00019. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–1464. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Berg MJ, Squitieri F, Ottenbacher M, Sorlie M, et al. Low frequency of PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus. Hum Mutat. 2006;27:118. doi: 10.1002/humu.9389. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Penco S, Gault J, Leedom TP, Tassi L, et al. Different spectra of genomic deletions within the CCM genes between Italian and American CCM patient cohorts. Neurogenetics. 2008;9:25–31. doi: 10.1007/s10048-007-0109-x. [DOI] [PubMed] [Google Scholar]
- Louvi A, Chen L, Two AM, Zhang H, Min W, Günel M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci USA. 2011;108:3737–3742. doi: 10.1073/pnas.1012617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Zhao H, Shan J, Long F, Chen Y, et al. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007;18:1965–1978. doi: 10.1091/mbc.E06-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawrin C, Perry A. Pathological classification and molecular genetics of meningiomas. J Neurooncol. 2010;99:379–391. doi: 10.1007/s11060-010-0342-2. [DOI] [PubMed] [Google Scholar]
- Perry JR, Tucker WS, Chui M, Bilbao JM. Dural cavernous hemangioma: an under-recognized lesion mimicking meningioma. Can J Neurol Sci. 1993;20:230–233. [PubMed] [Google Scholar]
- Russell DS, Rubinstein L. Pathology of tumours of the nervous system. ed 5. Baltimore: Williams and Wilkins; 1989. [Google Scholar]
- Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8:2325–2333. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- Verlaan DJ, Roussel J, Laurent SB, Elger CE, Siegel AM, Rouleau GA. CCM3 mutations are uncommon in cerebral cavernous malformations. Neurology. 2005;65:1982–1983. doi: 10.1212/01.wnl.0000188903.75144.49. [DOI] [PubMed] [Google Scholar]
- Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, et al. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8:249–256. doi: 10.1007/s10048-007-0098-9. [DOI] [PubMed] [Google Scholar]
- Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422–432. doi: 10.3171/jns.1994.80.3.0422. [DOI] [PubMed] [Google Scholar]
- Zabramski JM, Henn JS, Coons S. Pathology of cerebral vascular malformations. Neurosurg Clin N Am. 1999;10:395–410. [PubMed] [Google Scholar]