

Abstract
Purpose of Review
This review offers a concise summary of the most recent experimental advances in vascular development using the mouse as a model organism.
Recent Findings
Recent mouse studies have revealed a spread of phenotypic diversity between endothelia of distinct developmental origins and organs. For example, expression of unique transcription factors distinguishes hemogenic from non-hemogenic endothelium within the same vessel. Vasculature of the brain is particularly susceptible to endothelial malformations due to combinatorial germline and somatic mutations; surprisingly these mutations can afflict the endothelium by either cell autonomous or paracrine effects. Mutant mice have been used to understand how multiple signaling pathways integrate and refine cellular responses. In particular, we learned how VEGFR3 regulates Notch signaling and EphrinB2 coordinates VEGFR2 responses. The regulation of Prox1 by miR181 highlighted the contribution of microRNAs in the induction of lymphatic endothelium. Information gained on heterotypic interactions has further clarified the influence of blood vessels on the morphogenesis of parenchyma and contributed to our understanding of organ-specific endothelial differentiation. Finally, mouse models have uncovered endothelial cell polarity as a keystone for successful vascular lumenization.
Summary
Our understanding of the process of vascular development has gained significant refinement in the last two years and has clarified the origin of several disorders rooted in development.
Keywords: Alagille syndrome, cerebral cavernous malformations, hemogenic endothelium, macrophage, pericyte, vascular development, vascular lumen
Introduction
Efforts in experimental research during the last two years have continued to seek molecular information to better explain the morphogenesis of the vasculature, its plasticity and ability to provide unique functions to specific organs. We have identified five areas where the use of the mouse model has particularly excelled our understanding of vascular development. In this review, we summarize the highlights in these areas and place the latest advancements in the context of existent knowledge.
Hemogenic endothelium: the origin of definitive hematopoietic cells
Endothelial and hematopoietic lineages are spatially and chronologically linked at the onset of development [1]. This close relationship has long prompted the hypothesis that hematopoietic and endothelial cells emerge from a common progenitor: the hemangioblast [2]. However, blood emergence has been observed in close contact or “budding” from endothelium in a number of vascular beds including the dorsal aorta, the aorta-gonad-mesonephros region (AGM), the vitelline artery and the placenta [3]. These observations elicited the historical speculation that a subset of endothelial cells hold hematopoetic potential, (hemogenic endothelium). During the last four years, a preponderance of experimental evidence supports the notion that definitive hematopoietic stem cells (HSC) do in fact originate from a specialized endothelium with temporally restricted hemogenic capacity [3,4,5,6,7]. In vitro work further verifies that hematopoietic cells originate from an endothelial intermediate. Live culture imaging of mouse flk-1+ hemangioblasts derived from embryonic stem cells demonstrate an endothelial stage prior to HSC emergence. Cultured hemangioblasts first generate phenotypically defined endothelial cells which then begin to express hematopoietic transcription factors, such as Runx1. Subsequent culture of these Runx1+ endothelial cells results in the emergence of rounded cells with hematopoietic markers [6]. Importantly, in human tissues, hematopoietic cells in the AGM were found to co-express Vascular Endothelial (VE) cadherin and CD45 showing concurrent endothelial and hematopoietic traits [5].
Curiously, the hemogenic capacity of endothelial cells is limited to specific vascular beds, and temporally restricted to a narrow developmental window. This begs the question: How is this endothelium specified to such a remarkable function? While the full answer to this question remains incomplete, lineage tracing experiments revealed that all hemogenic endothelium of the embryo originates from the lateral plate mesoderm (Hox6B positive); while non-hemogenic endothelium does not express Hox6B [*8]. Furthermore, hemogenic endothelium lineage restriction has been shown to rely on the expression of Runx1 and absence of HoxA3 (Figure 1) [**9]. HoxA3 is expressed in embryonic vasculature in a mutually exclusive pattern to Runx1. Induction of HoxA3 expression in embryonic stem cells and ex vivo cultures repressed hematopoiesis, but not endothelial fate, indicating that expression of HoxA3 limits areas of hemogenic endothelium [**9]. Interestingly, Runx1 is able to “rescue” hematopoiesis and de-regulate endothelial markers, which corresponds to the transient Runx1 expression on budding hematopoietic cells found at hemogenic sites. 3
Figure 1. Hemogenic potential of the dorsal aorta.
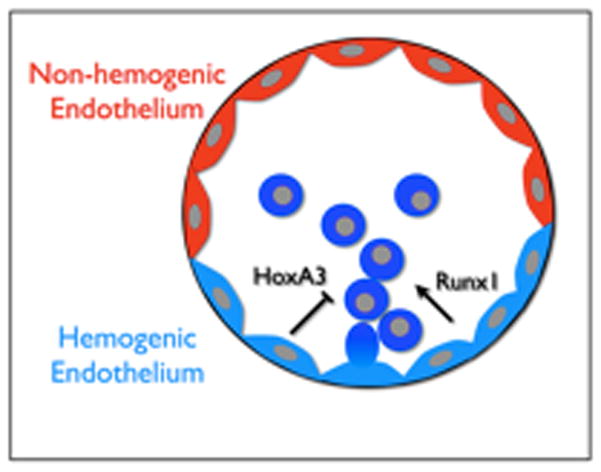
The endothelium of the dorsal aorta is derived from the lateral plate mesoderm. This endothelium, however, contains two functionally distinct regions: the hemogenic endothelium (ventral) and non-hemogenic endothelium (dorsal). HoxA3 expression suppresses the hemogenic capacity of endothelium, while the hematopoietic transcription factor Runx1 induces hematopoiesis.
Coordinated signaling of multiple pathways in endothelial cells
Understanding the regulation of concurrent operational signaling pathways has been a problem challenging to tackle as the integration of signaling inputs and outputs is difficult to trace. Nonetheless, recent advances have shed light on how signaling interplay is able to finely tune cellular output.
Ephrins are transmembrane ligands that interact with Eph receptors and mediate bidirectional signaling: “reverse” signaling via the cytoplasmic PDZ domain of ephrin and “forward” signaling into the Eph receptor tyrosine kinase. EphrinB2 is specific to arterial endothelium and required for developmental angiogenesis, though its mechanism of action has been mysterious. Mutations in the PDZ-domain of ephrinB2 result in a reduction of vascular sprouting and filopodia extensions; while overexpression of ephrinB2 increases filopodia and motility to such an extent that mature vasculature formation is compromised [10, 11]. EphrinB2 protein was found to be concentrated on filopodia of tip cells [11]. This is consistent with the hypothesis that EphrinB2 regulates the directional response to a growth factor gradient, such as VEGF. The effects of ephrinB2 on VEGFR2 and VEGFR3 were assessed in vitro and in vivo. It was found that when ephrinB2 is compromised (by absence or disruption of PDZ signaling) VEGF receptors are not properly endocytosed, which in turn reduces signaling output [10, 11]. These experiments suggest that because of its location, ephrinB2 may be critical in offering precise spatial control of VEGF receptor activity.
Induction of collateral arterial growth remains a critical problem in heart and limb ischemia, particularly since these vessels are poorly responsive to growth factor stimulation. It has been proposed that defective activation of ERK might be the common denominator of this impaired arteriogenesis [**12]. This conclusion was reached by following signaling pathways downstream in a synectin-null mouse model that displays defects in arterial morphogenesis [13]. Lack of synectin is accompanied by a reduced response to VEGF and ERK1/2 activation. Furthermore, it was found that arterial defects due to either synectin inactivation or LDL-R and ApoB48 mutations can be almost completely corrected by enhancing ERK1/2 activation in vitro or in vivo [**12]. This work demonstrates that ERK1/2 signaling is a key regulator of arterial growth, and that growth factor-resistant vessels can instead respond to downstream enhancement of ERK1/2.
Notch-Delta signaling has been shown to coordinate the ratio of tip and stalk cells during vascular sprouting. More recently, VEGFR3 was found to positively-regulate Notch signaling [14]. Endothelial ablation of VEGFR3 resembles a Notch inactivation phenotype that is typified by overabundance of tip-cells [14]. This phenomenon is due to a passive VEGFR3 signaling mechanism, as antibody blockade of VEGFR3 or ligand ablation does not recapitulate the knockout phenotype [14].
The contribution of microRNAs (miRs) to the regulation of vascular development is largely unknown and in fact, the prevalence of miR up-regulation in disease states implies that these molecules are early responders triggered to maintain homeostasis. Nonetheless, novel contributions of miRs during the neo-angiogenic process have been revealed in the past few years [15, 16, 17,18]. More recently, miR-181 was found to regulate Prox1, a transcription factor required for lymphatic endothelial cell specification. miR-181 is highly expressed in the vasculature, but significantly reduced in lymphatic endothelial cells, reciprocally to Prox1 expression [**19]. Forced expression of miR-181 in embryonic lymphatic endothelium reduced lymphatic markers and induced vascular endothelial markers. This data suggests miR-181 is crucial for lymphatic/endothelial lineage divergence [**19]. Although endogenous miR-181 expression was examined in vivo, a miR-181-null mouse model would conclusively reveal the biological relevance of the miR regulation to vascular and lymphatic development.
Lumen Formation
Successful lumenization of vessels requires concurrent changes in endothelial cytoskeleton, cell-cell adhesion and cell-matrix adhesion. Current findings suggest these events hinge upon the formation and maintenance of endothelial cell polarity.
Beta1 integrin has emerged as a novel regulator of endothelial cell polarity and lumen formation in the developing vasculature [*20, 21]. Conditional loss of Beta1 integrin in endothelial cells generated lumenal obstructions in multiple vascular beds of the mouse embryo. Cell tracing experiments revealed that the occlusions consisted of cuboidal endothelial cells that were aberrantly adherent to neighboring cells in all directions. The phenotype ultimately resulted in embryonic lethality between E14.5–17.5 [*19]. Supporting these findings, the cell-polarity protein Par3 was down-regulated in the absence of Beta1 integrin. Multiple adhesion proteins including VE-cadherin, CD99 and PECAM were found to be mis-localized on those endothelial cells lacking the integrin [*19]. The findings are consistent with a previously considered role for Beta1 in the cell polarity of epidermal cells [22, 23].
Further insight on lumen formation was gained from research on the novel endothelium-restrictive small GTPase Rasip1 [24]. Genetic inactivation of Rasip1 results in lethality by E10.5 [**25] with phenotypes that strikingly resemble the polarity defects observed in Beta1 Integrin deletion mutants [*20]. Rasip1 mutants exhibited severe vascular remodeling defects and displayed poor blood flow due to occluded lumens in multiple vascular beds [**25]. Rasip1 and its newly described binding partner Arhgap29 affected the activation, though not levels, of Beta1 integrin [**25]. The data indicates that Rasip1 is upstream the Beta1 integrin pathway and in this way contributes to the regulation of endothelial polarity (Figure 2).
Figure 2. Heterotypic cell-cell interactions in vascular development.
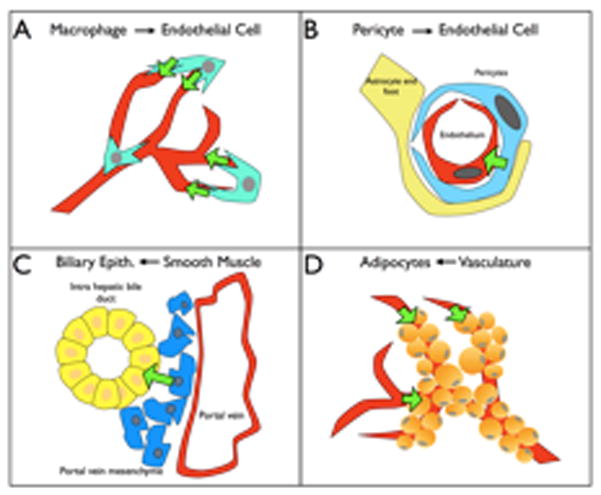
A. Macrophages mediate bridging and connection between tip-cells in the retina and central nervous system.
B. Pericyte contact with endothelial cells in the CNS induces cellular changes that allow proper blood brain barrier function.
C. Deletion of Jag1 in the portal vein mesenchyme disturbs the development of intrahepatic bile ducts. This is an example of instructive signaling of vascular cells to the adjacent parenchyma.
D. Preceding angiogenesis is required for proper adipocyte differentiation.
In addition to molecular signaling events, mechanical forces are important for launching successful lumenization. For a brief period of time before lumenization, cords of endothelial cells form transient apical cell-cell contacts where the lumen eventually emerges. It has been observed that in the time preceding lumenization, the CD34 sialomucin (Podocalyxin/gp135) accumulates on this apical/cell-cell contact face in the mouse aorta [*26]. Because the CD34 sialomucin is decorated with a large number of negatively charged sialic acids, it was hypothesized that lumenization depends on CD34-driven electrostatic repulsion between the adjacent membranes (Figure 2). Enzymatic removal of the negative charge and subsequent whole-embryo culture yield failure of aorta luminization, while restoration of the negative charge by injection of dextran sulfate rescued lumenization [*26]. This study provides a mechanical explanation for the breakdown of cell-cell adhesion in the emerging cord, though it is surprisingly not sufficient for lumenization. Xu et al. observed that, despite failure to lumenize, CD34 is correctly localized between endothelial cells in Rasip1 mutant mice [**25]. This suggests that the electrostatic force generated by CD34 localization is necessary, but not sufficient to drive lumen formation.
The formation of lumenized vessels requires a 3 dimensional matrix that appears to trigger signaling events which initiate the process of tubulogenesis [27]. Furthermore, it has long been recognized that fibroblasts are required in culture to drive vascular lumen formation, however the specific molecules responsible were only identified recently [28]. Fibroblast-secreted factors were shown to significantly stiffen the matrix gel surrounding sprouting endothelial cells; this is predicted to facilitate lumen formation by providing mechanical strength to endothelial cells necessary for adherence [28].
Closely regulating the size and shape (if not directly the genesis) of lumens, Notch has been shown to act cell-autonomously in endothelial cells [29]. Notch was linked to lumen size and vessel patterning in gain- and loss-of-function mutants. Over-expression of activated Notch4 in the endothelium leads to dilated vessels and death at E10.5. Conditional inactivation of endothelial Notch4 or Notch1 results in smaller lumens and mortality at E9.5 [30]. Similar findings were also documented more recently in an endothelial-specific RBPJ knockout model [31].
Molecular basis of vascular diseases that hold developmental roots
Cerebral cavernous malformations (CCMs) are vascular lesions located in the central nervous system and are characterized by an abundant number of vascular tufts with extremely thin walls [32]. Using standard linkage analysis, three genes (CCM1, 2 and 3) were initially identified as hereditary causative components for the disease, albeit functional information on these proteins was absent [33]. More recently, somatic mutations have been shown to cooperate with germline CCM mutations to result in loss of heterozygosity and, ultimately, manifestation of the disease [34, 35]. These discoveries together with the generation of a homozygous mutant mouse and mutant cells have shed light into how CCM proteins function to provide structural stability to the endothelial lining.
Inactivation of both copies of either CCM1 or CCM2 in mice results in early embryonic lethality; though heterozygous animals show no phenotype, possibly because mice do not live long enough to experience a second somatic mutation [36]. To model the two-hit mutation hypothesis, McDonald and colleagues crossed CCM1 heterozygotes with a mismatch repair-deficient background. Imposition of genetic instability was sufficient to facilitate a second hit mutation and resulted in animals that recapitulated most hallmarks of CCM disease [**37]. An alternative mouse model for CCM was created by inducing deletion of CCM at several stages post-birth. Tamoxifen-induced deletion at P1 generates CCM-like lesions in the cerebellum and retina a week post-induction [38]. Interestingly, deletion in adult animals did not yield CCM lesions. This suggests that CCM disease progression may, in fact, be dependent on active angiogenesis, which in humans could occur due to age-related hypoxic events [38].
Although endothelial depletion of CCM proteins has been shown to cause lesions, an endothelial non-autonomous effect of CCM3 deletion was also reported. Deletion of CCM3 in various non-endothelial cell types of the central nervous system yield broadly spread embryonic lethality [39]. Gfap-cre ablation of CCM3 led to an abundance of astrocytes accompanied by dilated, simplified vessels in the brain. About two-thirds of these mice developed CCM-like lesions. Together, the data favors an alternative mechanism of CCM formation based on paracrine signaling.
Another recent development that has contributed to our understanding of hereditary vascular disorders has been in the area of brain arteriovenus malformations (BAVMs). BAVMs are often lethal vascular lesions composed of abnormal arteries and veins. Because BAVMs exhibit no clinical manifestation until adulthood, it is unclear whether they represent a developmental defect or an adult pathology. Abnormal TGF-beta signaling has been associated with BAVMs as mutations in this pathway are correlated with AVMs in multiple organs in human patients [40]. It was found that the TGF-beta-activating integrin Beta8 has reduced expression in human BAVMs [41]. Local deletion of Beta8 in the brain reduces TGF-beta activation and induces enlarged dysplastic capillaries similar to BAVMs. Furthermore, certain Beta8 SNPs in humans were found to correlate with disease progression, suggesting that lack of Beta8 contributes to BAVMs [41].
The Notch pathway has been implicated in a variety of hereditary disorders including Alagille syndrome and patent ductus arteriosus. Alagille syndrome is a multi-symptomatic disorder primarily characterized by a paucity of intrahepatic bile ducts in the liver. This disease has been linked to mutations in Jagged1 (95% of the cases) and in Notch2 (5% of the cases) [42]. The onset and cellular origin of the disease was not well understood until recently. Interestingly, deletion of Jag1 in vascular smooth muscle cells (SMCs) recapitulates Alagille3s paucity of bile ducts [*43]. It was found that biliary progenitors were unable to organize into tubes in mutant animals, and instead remained in a single layer adjacent to the portal vein [*43]. This unexpected finding demonstrates an instructive role of the vasculature in liver/biliary duct morphogenesis, and provided a clear understanding as to the cellular origin of the disease.
Patent ductus arteriosus, the most common congenital heart defect found in humans may also be caused by aberrant Notch signaling. Feng et al. developed a mouse model of patent ductus arteriosus by deletion of the Notch ligand Jag1 in vascular SMCs that results in lethality at P2 [*44]. Immunofluorescence analysis shows that these mice have defects in SMC differentiation, particularly in the ductus arteriosus. The authors found that injection of idomathacin (an anti-inflammatory drug used to inhibit prostanoids) rescues ductus arteriosus closure in approximately 50% of the SMC KO mice [*44]. This study suggests that Jag1 is crucial for SMC differentiation that allows closure of the ductus arteriosus by SMC contraction.
Heterotypic cell interactions in vascular development
Vascular morphogenesis requires the collaborative effort of multiple cell types. Supporting cells from many different lineages have been shown to enhance and direct vessel development in specific organ beds. Individual organs have distinct cell types that impose unique phenotypic characteristics to the endothelium, however the molecular underpinnings are far from clear.
Recent literature highlights the contribution of macrophages to angiogenesis. Specialized macrophages called tumor-associated macrophages (TAMs) and Tie-2 expressing monocytes (TEMs) promote pathological tumor angiogenesis [45]. The discovery of pro-angiogenic macrophages launched interest in the role of these cells during normal developmental angiogenesis. Embryonic Tie2-positive monocytes were observed at active sites of angiogenesis in developing embryos, suggesting an active contribution to angiogenesis [46]. Furthermore, expression profiles of TEMs sorted from mammary tumors were compared to embryonic TEMs, which showed a significant overlap in expression [46].
While the above studies suggested a vague role for macrophages in vascular development, Fantin and colleagues determined that tissue macrophages specifically promote anastamosis. Yolk sac-derived macrophages in the developing hindbrain are found at points of bridging endothelial tip cells. They accumulate at highest numbers when the vasculature is organizing a network at E11.5, suggesting that macrophages aid in the bridging of tip cells [**47] (Figure 3). Absence of yolk-sac macrophages in the PU.1-null mouse results in a reduced number of vascular intersections rather than number of tip-cells. Furthermore, no reduction in VEGF levels was detected. Macrophage depletion shows a phenotype distinct from that of VEGF gradient defects, which reinforces the idea that macrophages aid EC anastamosis rather than promote sprouting [**47].
Figure 3. Molecular regulation of vascular lumen emergence.
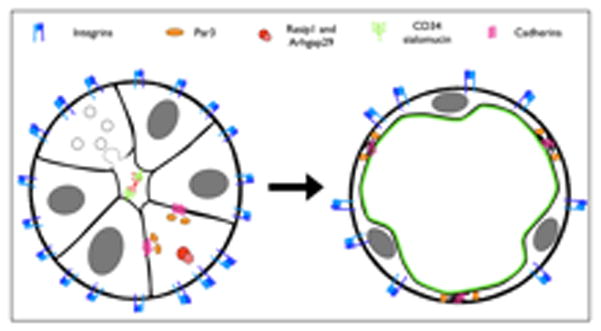
Lumen formation requires the concurrent efforts of many molecules. Beta1 integrin binds to the extracellular matrix and initiates the process of endothelial cell polarization. Recent studies indicate that adequate levels of Beta1 integrin are necessary for expression and localization of the intrinsic polarity protein Par3, and other cell-cell interaction proteins such as VE-cadherin, PECAM-1. Rasip1 is a small GTPase necessary for vascular lumenization. Rasip1 interacts with binding partner Arhgap29 and is needed to activate Beta1 integrin, suggesting an upstream role. Vascular lumenization relies on the concerted exocytosis of large vacuoles which join to become the common lumen, a process disrupted by the deletion of Beta1 integrin. Negatively-charged CD34 sialomucins provides electrostatic repulsion between opposing endothelial faces poised to lumenize. In mature vessels, CD34 is a marker for the apical surface of endothelial cells.
It has been proposed that lymphatic endothelium incorporates trans-differentiated macrophages into the endothelial wall. Gordon and colleagues showed that although intimately localized, a macrophage/lymphatic transition cell was never observed and historical macrophage tracing never yielded lymphatic endothelium that was positive for the tracer. Macrophages do, however, influence lymphatic development. Loss of macrophages results in hyperplastic lymphatic vessels due to overproliferation of dermal lymphatic endothelium [48]. These results indicate a directing role for macrophages in the size, shape and complexity of the developing vasculature.
Endothelial cells vary phenotypically from tissue to tissue due to their interactions with distinct cell types. Strikingly, the blood-brain barrier (BBB) requires endothelial cells to act as the major impediment to the passage of solutes from the blood; a process that depends on specialized, complexed tight junctions and a lower rate of vesicular transport. Armulik and colleagues determined that this endothelial behavior is imposed, in part, by pericytes. Using two pericyte-deficient mouse models, it was found that a lack of pericytes was correlated with increased water content and accumulation of BBB-impermeable dyes in the brain [*49] (Figure 3). The increased BBB permeability was attributed to a defect in transcytosis of the endothelial cells. Analysis of microvascular fragments indicated that in the absence of endothelial-pericyte contact, lower levels of several endothelial BBB markers were noted [*49]. This data indicates for the first time that pericyte contact with endothelial cells induces changes in expression that induce specialized BBB endothelial phenotype.
Relationships between vessels and the organs they inhabit are reciprocal; just as parenchyma informs endothelial development, so too can endothelial cells direct organ differentiation. As previously mentioned, perturbation of vessel function by deletion of Jag1 in vascular smooth muscle cells disrupts the differentiation of bile ducts in the liver [*42]. Correspondingly, adipose tissue development, as studied in the postnatal epididymal adipose tissue (EAT) model, is dependent on preceding angiogenesis. When angiogenesis in EAT is blocked by injection of a VEGF-Trap, adipocytes are of much smaller size and fail to cover the entire vascular network [50] (Figure 3). This research points to an important role held by the vasculature in the regulation of tissue growth and reciprocal instructive interactions.
Conclusion
During the last two years, a significant number of advancements have been achieved using mouse models. In particular, signaling experiments have left the culture dish to find a home in a whole-organism. However, weaving together a multitude of signaling pathways to a point where a complicated input translates to a known cellular output will progressively impose additional challenges. Yet, it is remarkable that using the mouse model, major strides were made towards understanding vascular lumen formation. Future progress in this field also hinges on the development of cell culture systems that are amenable to concurrent live imaging and flow.
Vascular diseases often present no symptoms until adulthood, although the pathology can originate from a developmental defect or genetic incident. Understanding the molecular and cellular origins of vascular diseases will expand the toolbox available for diagnosis and treatment of affected patients. Equally important, this research is critical to our understanding of fundamental developmental processes. In particular, the vasculature appears to hold the ability to impart inductive signals to the adjacent parenchyma and directly contribute to the ultimate 3D architecture of an organ.
Finally, the discovery that certain regions of endothelium have hemogenic potential has opened a pandora3s box: what exactly are the elements that specify hemogenic capacity? Can this capacity be regained? Recent findings suggest a specialized transcription machinery that is distinct to hemogenic endothelium. Reproduction of this capacity ex vivo holds significant therapeutic value.
Key Points.
Hematopoietic stem cells are derived from hemogenic endothelium. Hemogenic potential of the endothelium relies on expression of Runx1 and suppression of HoxA3.
Cross-interactions of distinct signaling pathways such as Ephrin-B2/VEGF or VEGF/Erk1/2, can significantly change cellular output.
Endothelial cell polarity has emerged as a necessary pre-condition for vascular lumen formation.
Vascular diseases that first present in adulthood, such as cerebral cavernous malformations and brain arteriovenus malformations, often have origins in developmental defects.
Heterotypic cell interactions have emerged as critical regulators of vascular morphogenesis and organ specific differentiation.
Acknowledgments
The authors wish to acknowledge funding from the National Institutes of Health RO1CA126935, RO1HL08618 and T32 HL69766.
Footnotes
Conflicts of interest
There are no conflct of interest to disclose.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Ferkowicz MJ, Yoder MC. Blood island formation: longstanding observations and modern interpretations. Exp Hematol. 2005;33:1041–1047. doi: 10.1016/j.exphem.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Cao N, Yao ZX. The hemangioblast: from concept to authentication. Anat Rec (Hoboken) 2011;294:580–588. doi: 10.1002/ar.21360. [DOI] [PubMed] [Google Scholar]
- 3.Speck NA, Iruela-Arispe ML. Conditional Cre/LoxP strategies for the study of hematopoietic stem cell formation. Blood Cells Mol Dis. 2009;43:6–11. doi: 10.1016/j.bcmd.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zovein AC, Hofmann JJ, Lynch M, et al. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell. 2008;3:625–636. doi: 10.1016/j.stem.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oberlin E, El Hafny B, Petit-Cocault L, Souyri M. Definitive human and mouse hematopoiesis originates from the embryonic endothelium: a new class of HSCs based on VE-cadherin expression. Int J Dev Biol. 2010;54:1165–1173. doi: 10.1387/ijdb.103121eo. [DOI] [PubMed] [Google Scholar]
- 6.Lancrin C, Sroczynska P, Stephenson C, et al. The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature. 2009;457:892–895. doi: 10.1038/nature07679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boisset JC, van Cappellen W, Andrieu-Soler C, et al. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010;464:116–120. doi: 10.1038/nature08764. [DOI] [PubMed] [Google Scholar]
- 8*.Zovein AC, Turlo KA, Ponec RM, et al. Vascular remodeling of the vitelline artery initiates extravascular emergence of hematopoietic clusters. Blood. 2010;116:3435–3444. doi: 10.1182/blood-2010-04-279497. This paper demonstrates that hemogenic endothelium has a common lateral plate mesodermal origin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9**.Iacovino M, Chong D, Szatmari I, et al. HoxA3 is an apical regulator of haemogenic endothelium. Nat Cell Biol. 2011;13:72–78. doi: 10.1038/ncb2137. This study is the first to provide a mechanism for the restriction of endothelial hemogenic potential by the transcription factor HoxA3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawamiphak S, Seidel S, Essmann CL, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–491. doi: 10.1038/nature08995. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Nakayama M, Pitulescu ME, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 12**.Ren B, Deng Y, Mukhopadhyay A, et al. ERK1/2-Akt1 crosstalk regulates arteriogenesis in mice and zebrafish. J Clin Invest. 2010;120:1217–1228. doi: 10.1172/JCI39837. This paper demonstrates that manipulation of the ERK pathway downstream of growth factor defects can fully restore arteriogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chittenden TW, Claes F, Lanahan AA, et al. Selective regulation of arterial branching morphogenesis by synectin. Dev Cell. 2006;10:783–795. doi: 10.1016/j.devcel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Tammela T, Zarkada G, Nurmi H, et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol. 2011;13:1202–1213. doi: 10.1038/ncb2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suárez Y, Fernández-Hernando C, Yu J, et al. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc Natl Acad Sci U S A. 2008;105:14082–14087. doi: 10.1073/pnas.0804597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonauer A, Carmona G, Iwasaki M, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 17.Doebele C, Bonauer A, Fischer A, et al. Members of the microRNA-17–92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood. 2010;115:4944–4950. doi: 10.1182/blood-2010-01-264812. [DOI] [PubMed] [Google Scholar]
- 18.Anand S, Majeti BK, Acevedo LM, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010;16:909–914. doi: 10.1038/nm.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19**.Kazenwadel J, Michael MZ, Harvey NL. Prox1 expression is negatively regulated by miR-181 in endothelial cells. Blood. 2010;116:2395–2401. doi: 10.1182/blood-2009-12-256297. Little is known about the role of microRNAs in normal endothelial development. This paper provides evidence that the lymphatic transcription factor Prox1 is regulated by miR-181. [DOI] [PubMed] [Google Scholar]
- 20*.Zovein AC, Luque A, Turlo KA, et al. Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev Cell. 2010;18:39–51. doi: 10.1016/j.devcel.2009.12.006. This paper links Beta1 integrin deficiency to cell polarity, and demonstrates that polarization of the endothelium is essential for lumen formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malan D, Wenzel D, Schmidt A, et al. Endothelial beta1 integrins regulate sprouting and network formation during vascular development. Development. 2010;137:993–1002. doi: 10.1242/dev.045377. [DOI] [PubMed] [Google Scholar]
- 22.Raghavan S, Bauer C, Mundschau G, et al. Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol. 2000;150:1149–1160. doi: 10.1083/jcb.150.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:275–280. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu K, Chong DC, Rankin SA, Zorn AM, Cleaver O. Rasip1 is required for endothelial cell motility, angiogenesis and vessel formation. Dev Biol. 2009;329:269–279. doi: 10.1016/j.ydbio.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Xu K, Sacharidou A, Fu S, et al. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev Cell. 2011;20:526–539. doi: 10.1016/j.devcel.2011.02.010. This paper identifies an endothelial-specific regulator of GTPase that promotes proper cell polarity, cytoskeleton and adhesion molecule changes to allow vascular lumen formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Strilić B, Eglinger J, Krieg M, et al. Electrostatic cell-surface repulsion initiates lumen formation in developing blood vessels. Curr Biol. 2010;20:2003–2009. doi: 10.1016/j.cub.2010.09.061. This study provides the first evidence that the negative charges decorating endothelial cell-surface sialomucins provide force required to initiate lumenization. [DOI] [PubMed] [Google Scholar]
- 27.Davis GE, Koh W, Stratman AN. Mechanisms controlling human endothelial lumen formation and tube assembly in three-dimensional extracellular matrices. Birth Defects Res C Embryo Today. 2007;81:270–285. doi: 10.1002/bdrc.20107. [DOI] [PubMed] [Google Scholar]
- 28.Newman AC, Nakatsu MN, Chou W, et al. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell. 2011;22:3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iruela-Arispe ML, Davis GE. Cellular and molecular mechanisms of vascular lumen formation. Dev Cell. 2009;16:222–231. doi: 10.1016/j.devcel.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uyttendaele H, Ho J, Rossant J, Kitajewski J. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci U S A. 2001;98:5643–5648. doi: 10.1073/pnas.091584598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Copeland JN, Feng Y, Neradugomma NK, et al. Notch signaling regulates remodeling and vessel diameter in the extraembryonic yolk sac. BMC Dev Biol. 2011;11:12. doi: 10.1186/1471-213X-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Del Curling O, Kelly DL, Elster AD, Craven TE. An analysis of the natural history of cavernous angiomas. J Neurosurg. 1991;75:702–708. doi: 10.3171/jns.1991.75.5.0702. [DOI] [PubMed] [Google Scholar]
- 33.Felbor U, Sure U, Grimm T, Bertalanffy H. Genetics of cerebral cavernous angioma. Zentralbl Neurochir. 2006;67:110–116. doi: 10.1055/s-2006-933537. [DOI] [PubMed] [Google Scholar]
- 34.Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18:911–918. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akers AL, Johnson E, Steinberg GK, et al. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet. 2009;18:919–930. doi: 10.1093/hmg/ddn430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitehead KJ, Chan AC, Navankasattusas S, et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15:177–184. doi: 10.1038/nm.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37**.McDonald DA, Shenkar R, Shi C, et al. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum Mol Genet. 2011;20:211–222. doi: 10.1093/hmg/ddq433. It has long been hypothesised that cerebral cavernous malformations are caused by “two hits”, a germline and a somatic mutation. This paper provides proof of principle to this hypothesis using a mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boulday G, Rudini N, Maddaluno L, et al. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J Exp Med. 2011;208:1835–1847. doi: 10.1084/jem.20110571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Louvi A, Chen L, Two AM, et al. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci U S A. 2011;108:3737–3742. doi: 10.1073/pnas.1012617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young WL, Kwok PY, Pawlikowska L, et al. Arteriovenous malformation. J Neurosurg. 2007;106:731–732. doi: 10.3171/jns.2007.106.4.731. author reply 732–733. [DOI] [PubMed] [Google Scholar]
- 41.Su H, Kim H, Pawlikowska L, et al. Reduced expression of integrin alphavbeta8 is associated with brain arteriovenous malformation pathogenesis. Am J Pathol. 2010;176:1018–1027. doi: 10.2353/ajpath.2010.090453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Hofmann JJ, Zovein AC, Koh H, et al. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development. 2010;137:4061–4072. doi: 10.1242/dev.052118. In this study the authors demonstrate that Jag1 in smooth muscle cell progenitors provides an instructive role in the morphogenesis of bile ducts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Feng X, Krebs LT, Gridley T. Patent ductus arteriosus in mice with smooth muscle-specific Jag1 deletion. Development. 2010;137:4191–4199. doi: 10.1242/dev.052043. This paper demonstrates the contribution of Jag1 in the physiological closure of the ductus arteriosus after birth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nucera S, Biziato D, De Palma M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int J Dev Biol. 2011;55:495–503. doi: 10.1387/ijdb.103227sn. [DOI] [PubMed] [Google Scholar]
- 46.Pucci F, Venneri MA, Biziato D, et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood. 2009;114:901–914. doi: 10.1182/blood-2009-01-200931. [DOI] [PubMed] [Google Scholar]
- 47**.Fantin A, Vieira JM, Gestri G, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116:829–840. doi: 10.1182/blood-2009-12-257832. This landmark study demonstrates that macrophages mediate the anastamosis of tip cells during vascular morphogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gordon EJ, Rao S, Pollard JW, et al. Macrophages define dermal lymphatic vessel calibre during development by regulating lymphatic endothelial cell proliferation. Development. 2010;137:3899–3910. doi: 10.1242/dev.050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49*.Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. This manuscript provides molecular information on how heterotypic interactions of EC with pericytes results in unique properties in the organization of the blood-brain barrier. [DOI] [PubMed] [Google Scholar]
- 50.Han J, Lee JE, Jin J, et al. The spatiotemporal development of adipose tissue. Development. 2011;138:5027–5037. doi: 10.1242/dev.067686. [DOI] [PubMed] [Google Scholar]