

Abstract
Pancreatic cancer is an extremely aggressive malignancy with a dismal prognosis. Cancer patients and tumor-bearing mice have multiple immunoregulatory subsets including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSC) that may limit the effectiveness of anti-tumor immunotherapies for pancreatic cancer. It is possible that modulating these subsets will enhance anti-tumor immunity. The goal of this study was to explore depletion of immunoregulatory cells to enhance dendritic cell (DC)-based cancer immunotherapy in a murine model of pancreatic cancer. Flow cytometry results showed an increase in both Tregs and MDSC in untreated pancreatic cancer–bearing mice compared with control. Elimination of Tregs alone or in combination with DC-based vaccination had no effect on pancreatic tumor growth or survival. Gemcitabine (Gem) is a chemotherapeutic drug routinely used for the treatment for pancreatic cancer patients. Treatment with Gem led to a significant decrease in MDSC percentages in the spleens of tumor-bearing mice, but did not enhance overall survival. However, combination therapy with DC vaccination followed by Gem treatment led to a significant delay in tumor growth and improved survival in pancreatic cancer–bearing mice. Increased MDSC were measured in the peripheral blood of patients with pancreatic cancer. Treatment with Gem also led to a decrease of this population in pancreatic cancer patients, suggesting that combination therapy with DC-based cancer vaccination and Gem may lead to improved treatments for patients with pancreatic cancer.
Keywords: Pancreatic cancer, Gemcitabine, Myeloid-derived suppressor cells, Dendritic cells, Immunotherapy, Chemotherapy
Introduction
Pancreatic cancer is the fourth leading cause of cancer deaths in the United States and has the highest cancer fatality rate worldwide [1]. The overwhelming majority of patients with pancreatic adenocarcinoma have locally advanced and/or metastatic disease at the time of presentation [2–4]. Complete tumor resection for pancreatic cancer is the only treatment modality that has been shown to significantly improve survival, but the majority of patients are not eligible for surgical resection at the time of diagnosis [5–9].
Tumor-specific T cells have been identified in patients with pancreatic cancer [10]. Infiltration of pancreatic tumors with CD4+ and CD8+ T cells correlates with an improved prognosis [11]. Dendritic cells (DC) are potent antigen-presenting cells that are highly effective at activating naïve and memory T-cell responses [12]. DC-based vaccinations have been used to induce and expand anti-tumor CD8+ cytotoxic T cells [13–15]. In murine models of pancreatic cancer, DC-based vaccination has led to expansion of anti-tumor T cells and tumor rejection [16]. In clinical trials with pancreatic cancer patients, vaccination with DC has led to positive immunological endpoints, but few cures were achieved [17–19].
There are multiple immunosuppressive populations found in cancer patients that may limit the effectiveness of immunotherapy. Regulatory T cells (Tregs) are characterized as CD4+CD25+FOXP3+ and are increased in the peripheral blood of cancer patients. Tregs have been shown to suppress antigen-specific T-cell responses and are important in the maintenance of tolerance to self-antigens. In murine models, elimination of Tregs leads to enhanced anti-tumor T-cell responses and tumor rejection [20, 21]. In addition to Tregs, myeloid-derived suppressor cells (MDSC) are expanded in cancer patients and tumor-bearing mice [22, 23]. In mice, MDSC are characterized as CD11b+Gr1+ and are comprised of immature macrophages, granulocytes, and DC [24]. In humans, MDSC are characterized as CD11b+CD14−CD33+ or HLA-DRneg/lowLineage−CD33+ [25]. MDSC are able to suppress T-cell responses through various mechanisms. MDSC reduce antigen-specific CD8+ T-cell proliferation, increase T-cell death by apoptosis, foster T-cell tolerance, and change the profile of cytokines secreted by activated T lymphocytes [25, 26]. In addition, MDSCs can indirectly suppress CD8+ T-cell function by inducing the development of Tregs [27]. Elimination of MDSC with drugs such as ATRA, docetaxel, or gemcitabine (Gem) has been shown to enhance anti-tumor immunity and tumor regression in murine models of cancer [28–30]. Both Tregs and MDSC have been shown to be increased in pancreatic cancer patients compared with healthy controls [31, 32].
Combination therapeutic approaches to decrease immune suppression and enhance immunotherapy may be beneficial for combating pancreatic cancer. The purpose of this study was to examine the combination of a DC-based vaccine and depletion of Tregs or MDSC in a murine pancreatic carcinoma model. We report that elimination of Tregs alone or in combination with a DC vaccine has no effect on tumor regression. In contrast, Gem treatment reduces MDSC percentages in tumor-bearing mice and increases survival when combined with DC-based immunotherapy. This study provides evidence that modulating MDSC frequency with Gem therapy improves the efficacy of DC vaccination and may lead to improvements for the treatment of patients with pancreatic cancer.
Materials and methods
Animals
Female C57BL/6 (6–8 weeks old) mice were purchased from Harlan Laboratories, Inc. (Indianapolis, IN, USA). OT-I, OT-II, and pmel mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All mice were maintained in a pathogen-free animal facility for at least 1 week before each experiment. All procedures were approved by the IACUC at the University of South Florida. Mice were humanely euthanatized when tumors exceeded 2.0 cm in diameter, appeared necrotic, or interfered with locomotion.
Patients
Peripheral blood was collected from 5 patients with pancreatic cancer under a protocol approved by the University of South Florida Institutional Review Board. All patients provided written informed consent. Peripheral blood mononuclear cells were collected prior to and 1 week after one cycle of Gem (1,000 mg/m2 i.v.) therapy.
Cell line
Panc02 murine pancreatic adenocarcinoma cell line was established by Corbett et al. [33] by implanting cotton threads impregnated with 3-methylcholanthrene into the pancreas of C57BL/6 mice, which was followed by serial in vivo passage through repeated transplantation. This cell line was maintained in RPMI 1640 medium supplemented with 10 % fetal bovine serum (FBS) (HyClone, Logan, UT, USF), 2 mM l-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin (Gibco BRL, Rockville, MD, USA) at 37 degrees C in 5 % CO2. Cultured cells were tested and found to be negative for mycoplasma and viral contamination.
Bone marrow–derived DCs
Erythrocyte-depleted mouse bone marrow cells were cultured in complete medium supplemented with 20 ng/ml GM-CSF and 10 ng/ml IL-4 (R&D Systems, Minneapolis, MN, USA) as described previously [34]. On day 5, cells were harvested by gentle pipetting and layered onto an Optiprep gradient (Axis-Shield, Oslo, Norway). The low-density cell interface was collected and washed twice. DC were washed 3× in PBS prior to injection. DC (1e6) were directly injected into Panc02 tumor on days 3, 7, and 10 after Panc02 injection.
Regulatory T-cell suppression assay
C57BL/6 mice received 1.5e5 Panc02 cells subcutaneously (s.c.). On day 21 after tumor injection, spleens were collected. Tregs were enriched using a CD4+CD25+ Regulatory T-cell Isolation Kit (Miltenyi Biotec Inc.) and sorted by AutoMACS. OT-I T cells were stimulated with OVA257–264 peptide alone or in the presence of increasing numbers of Tregs for 72 h. T-cell proliferation was measured by 3H incorporation during the final 12 h of the culture. Values are shown as mean ± SEM of triplicate wells.
MDSC suppression assay
Spleens from Panc02 tumor-bearing mice were processed into single cell suspensions. Gr-1+ cells were purified from whole splenocytes by positive selection using anti-Gr-1-PE and anti-PE-magnetic microbeads on an AutoMACS Pro Separator. Sorted MDSC from naïve and tumor-bearing mice were co-incubated at a 1:3 ratio with OT-II-specific (CD4+) T cells primed with DC pulsed with 10 mcg/ml OVA323–339 peptide or with pmel (CD8+) T cells primed with gp10025–33 peptide, for 3 days. T-cell proliferation was measured by 3H incorporation during the final 12 h of the culture. Controls for the assay included T cells incubated alone in complete media (CM).
Treg and MDSC depletion in vivo
C57BL/6 mice received 1.5 × 105 Panc02 cells s.c. in the right flank. For depletion of Tregs, mice were treated intraperitoneally (i.p.) with 12.5 mg/kg PC-61 antibodies starting 3 days after tumor injection. Additional injections were given twice per week for 3 weeks. Depletion of CD25+ cells was verified by flow cytometry (not shown). For MDSC depletion, mice received 120 mg/kg Gem i.p. in 250 mcl PBS starting 10 days after Panc02 injection. Additional injections were given twice per week for 3 weeks. In some experiments, splenocytes were collected after vaccination for in vitro studies.
IFN-gamma ELISA
Briefly, splenocytes were plated at 2 × 106 cells, co-cultured with media alone or with 2 × 105 irradiated B16 or Panc02 cells, and incubated for 48 h. Culture supernatants were analyzed for IFN-γ production using a commercially available ELISA kit (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s protocol.
Flow cytometry
To identify murine Tregs, splenocytes were labeled with anti-mouse CD4+ and CD25+ followed by intracellular staining for FoxP3+. To identify murine MDSC, splenocytes were labeled with anti-mouse CD11b and Gr-1. To identify MDSC subsets, splenocytes were labeled with anti-CD11b, anti-Ly6C, and anti-Ly6G antibodies. Human MDSC were characterized as CD14−CD33+CD11bhigh. For measurement of IFN-γ production, splenocytes were cultured in the presence of PMA (1 ng/ml), ionomycin (0.5 mcg/ml), and brefeldin A (1 mcg/ml) for 6 h. Surface staining was done with anti-mouse CD3 and CD8 antibodies followed by intracellular staining for IFN-γ production. All antibodies were purchased from BD Bioscience PharMingen (San Diego, CA, USA). All analyses were conducted using a FACSCalibur flow cytometer (Becton–Dickinson, Mountain View, CA, USA) and analyzed using FlowJo software.
Statistical analyses
A Mann–Whitney test (unpaired) or a Student’s paired t test was used to compare between two treatment groups. All statistical evaluations of data were performed using GraphPad Prism software. Statistical significance was achieved at p < 0.05.
Results
Expansion of Tregs in Panc02 tumor-bearing mice
First, we examined whether regulatory T cells (Tregs) were expanded in the spleens of tumor-bearing mice. Mice were injected with Panc02 cells, and spleens were collected on day 10. As shown in Fig. 1a, a higher percentage of Tregs within CD4+ cells was measured in tumor-bearing mice (p < 0.05). To determine whether this population suppressed antigen-specific T-cell responses, CD4+CD25+ cells were purified from Panc02-bearing mice. CD4+CD25+ cells were purified from naïve mice as a control. As shown in Fig. 1b, these cells were able to suppress the proliferation of CD8+ OT-I T cells in response to the OVA257–264 peptide.
Fig. 1.

Increased Tregs in the spleens of tumor-bearing mice. a CD4+CD25+Foxp3+ cells in the spleens (n = 4) of naïve and Panc02 tumor-bearing mice were measured by flow cytometry. *p < 0.05. b CD4+CD25+ T cells were purified from the spleens of naïve or Panc02-bearing mice and co-cultured with OT-I T cells stimulated with OVA257-264 peptide. Proliferation was measured after 72 h of co-culture. c Depletion of Tregs does not enhance DC immunotherapy. Mice were injected with Panc02 cells. Starting on day 3 and continuing every 3–4 days after, mice received NrIgG or PC-61 antibodies. Mice received DC injections intratumorally on days 3, 7, and 10. Tumor growth was measured
To test whether elimination of Tregs leads to enhanced anti-tumor immunity, mice were injected with 1.5 × 105 Panc02 cells. Mice received PC-61 antibodies to deplete Tregs or isotype control starting on day 3. PC-61 or isotype control was given every 3–4 days for 3 weeks. As shown in Fig. 1c, PC-61 treatment alone delayed tumor growth, but did not lead to tumor regression. DC vaccination alone or in combination with PC-61 also had no effect on Panc02 growth. Together, these studies indicate that Tregs are increased in Panc02-bearing mice, but depletion of this population does not significantly enhance anti-tumor immunity.
Expansion of MDSC in Panc02 tumor-bearing mice
In order to evaluate MDSC, C57BL/6 mice were injected s.c. with 1.5 × 105 murine Panc02 tumor cells. Splenocytes were harvested on days 0, 14, 20, 24, and 27 after Panc02 injection and stained with anti-Gr1 and anti-CD11b antibodies to measure MDSC percentages in the spleens. Flow cytometry results revealed an increase in the percentages of MDSC in Panc02-bearing mice. On day 0, MDSC made up 2 % of splenocytes. By day 14, this number rose to over 5 %. By day 27, 29 % of the splenocytes displayed the MDSC phenotype (Fig. 2a). To examine infiltration of MDSC into Panc02 tumors, tumors were collected on days 20, 24, and 27. Tumors were excised and processed into a single cell suspension, stained with anti-Gr1 and anti-CD11b antibodies, and analyzed by flow cytometry. As shown in Fig. 2b, the percentage of MDSC within the tumor increased over time.
Fig. 2.

Increased MDSC in the spleens and tumors of Panc02 tumor–bearing mice. a Spleens (n = 4 per group) were collected from naïve and Panc02 tumor–bearing mice on days 14, 20, 24, and 27 after Panc02 injection. b CD11b+Gr1+ MDSC percentages in the tumors of mice on days 20, 24, and 27 after injection of Panc02 tumor. CD11b+Gr1+ cells were measured by flow cytometry
Next, we measured the ability of MDSC derived from Panc02 tumor-bearing mice to suppress antigen-specific immune responses in vitro. To determine whether MDSC could suppress antigen-specific CD8+ T-cell responses, purified MDSC were incubated with pmel T cells and gp10025–33 peptide. As shown in Fig. 3a, a significant decrease in pmel proliferation was measured (p < 0.01 compared with no MDSC). In Fig. 3b, MDSC isolated from Panc02 tumor-bearing mice also suppressed CD4+ T-cell proliferation. A significant inhibition of OT-II T-cell proliferation was measured after co-culture with DC pulsed with OVA323–339 peptide and MDSC (p < 0.01 compared with OT-II cells + DC-OVA323–339 peptide alone). These experiments suggest that modulating MDSC percentages in Panc02 tumor-bearing mice may be beneficial to enhance CD4+ and CD8+ anti-tumor T-cell immune responses.
Fig. 3.

MDSC from Panc02-bearing mice suppress CD8+ and CD4+ T-cell responses. CD11b+Gr1+ cells were purified from the spleens of Panc02-bearing mice. a Pmel T cells were cultured with gp10025-33 peptide alone, or at a 1:3 ratio with purified Gr1− or Gr1+ cells. Proliferation was measured after 72 h. b OT-II T cells were cultured with DC pulsed with OVA323-339 peptide alone, or at a 1:10 ratio with purified Gr1 − or Gr1 + cells. Proliferation was measured after 72 h. *p < 0.01
Depletion of MDSC in Panc02 tumor-bearing mice
Gemcitabine is a chemotherapeutic drug used to treat pancreatic cancer patients and has been shown to decrease MDSC [35, 36]. Therefore, we evaluated whether MDSC percentages are decreased in Panc02-bearing mice after Gem treatment. Starting on day 10, mice received i.p. doses of 120 mg/kg of Gem given 2 times per week. Splenocytes were harvested on day 27. Flow cytometric analysis revealed a significant decrease in the percentage of MDSC in the spleens and tumors of Panc02 tumor-bearing mice treated with Gem (Fig. 4a, b).
Fig. 4.

Gemcitabine reduces MDSC in Panc02-bearing mice. On day 27 after Panc02 injection, a spleens and b tumors were collected from tumor-bearing mice treated with 120 mg/kg of gemcitabine. CD11b+Gr1+ cells were measured in the splenocytes by flow cytometry. *p < 0.001; **p < 0.01
In order to examine the efficacy of Gem therapy in combination with DC-based immunotherapy, mice received 1.5e5 Panc02 cells s.c. and were treated with PBS, Gem alone, DC alone, or combination therapy with DC and Gem. DC vaccinations were given on days 3, 7, and 10. Gem therapy was started on day 10 and given 2 times per week for 3 weeks. As shown in Fig. 5a, Gem therapy alone was able to induce a delay in tumor growth. Combination therapy with Gem and DC vaccination led to a delay in tumor growth, and this combination led to significant survival in Panc02-bearing mice (Fig. 5b, p < 0.05 compared with all other treatment groups). We next determined whether Panc02-specific immunity was enhanced in mice that received DC and Gem therapy. Splenocytes were isolated from mice and restimulated with Panc02 cells for 24 h. As shown in Fig. 5c, there was an increased number of T cells that produced IFN-γ in response to restimulation with Panc02 tumor in mice treated with DC and Gem (p < 0.05 compared with mice treated with Gem alone, DC alone, or PBS). Next, we examined infiltration of T cells into tumors by flow cytometry. A significant increase in CD8+ T-cell infiltration was measured in mice treated with Gem alone or in combination with DC vaccination (Fig. 5d, p < 0.05 compared with PBS- or DC-treated mice). While no difference was measured between these two groups, mice treated with combination Gem and DC also demonstrated enhanced infiltration of CD8+ T cells producing IFN-γ (Fig. 5e, p < 0.05 compared with treatment with Gem alone). Together, these results support the induction of a Panc02-specific T-cell response in mice treated with combination therapy of Gem and DC vaccination.
Fig. 5.
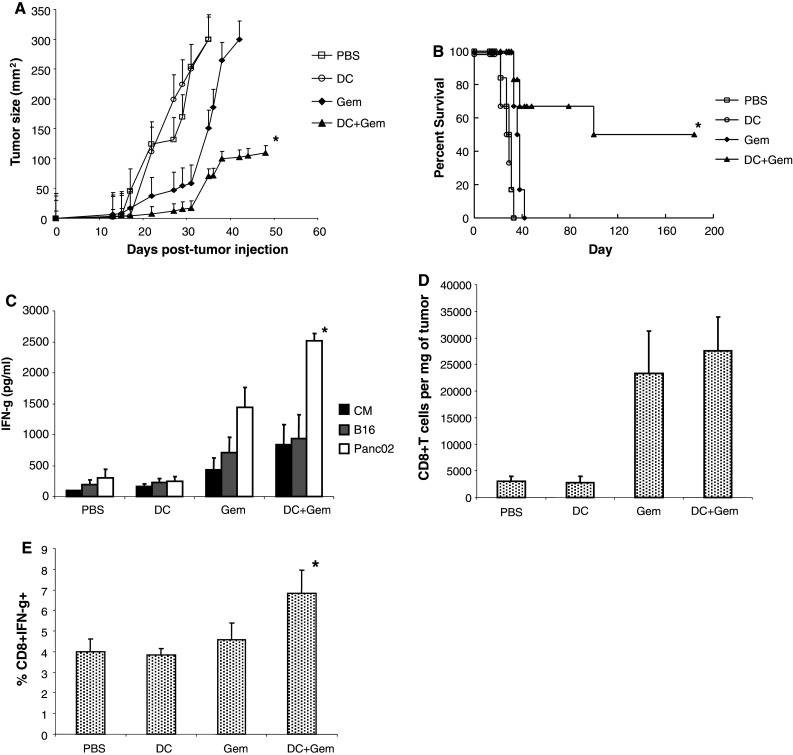
Delay of tumor growth and enhanced survival in mice treated with DC and Gem therapy. Mice were injected s.c. with Panc02 tumor cells (n = 8 mice per group). Mice received 120 mg/kg Gem i.p. starting on day 3 and continuing every 3–4 days until day 42. DC vaccines were injected intratumorally on days 3, 7, and 10. Mice received control (PBS), Gem alone (Gem), DC alone (DC), or combination therapy with DC and Gem (DC + Gem). a Tumor size. b Percent survival. c Splenocytes were collected on day 21, and T cells were restimulated for 48 h with media alone (CM), B16 cells, or Panc02 cells. Supernatants were collected, and IFN-γ production was measured by ELISA. d Tumors were collected on day 21. The number of CD3+CD8+ T cells was calculated per milligram of tumor. e The percentage of CD3+CD8+ T cells producing IFN-gamma was measured. Data show the mean ± SD (n = 3). *p < 0.05
MDSC in patients with pancreatic cancer
To determine whether Gem has an effect on MDSC numbers in patients, we first verified that MDSC were increased in the peripheral blood of patients with pancreatic cancer. As shown in Fig. 6a, patients with pancreatic cancer have an increased percentage of MDSC in peripheral blood (16.1 ± 3.4) compared with healthy donors (5.22 ± 1.8, p < 0.001). In five patients with pancreatic cancer, we measured a decrease in MDSC percentages after one cycle of Gem therapy (15.1 ± 3.2 before Gem therapy, 4.8 ± 2.9 after Gem therapy, p < 0.01, Fig. 6b). These data suggest that utilizing Gem to reduce MDSC may enhance anti-tumor immunity induced by a vaccination strategy and support the design of a clinical trial combining Gem therapy and DC vaccination in patients with pancreatic cancer. An expanded clinical study to further examine the function of MDSC and T-cell subsets in the peripheral blood before and after Gem therapy in pancreatic cancer patients is ongoing.
Fig. 6.

Gemcitabine therapy reduces MDSC in the peripheral blood of patients with pancreatic cancer. a Percentage of CD14−CD11b+CD33+ cells in the PBMC of healthy donors and pancreatic cancer patients as measured by flow cytometry. b Percentage of CD14−CD11b+CD33+ cells in the PBMC of pancreatic cancer patients prior to Gem therapy (pre) and after 1 cycle of 1000 mg/m2 Gem therapy (post-Gem) as measured by flow cytometry. *p < 0.05; **p < 0.01
Discussion
Pancreatic cancer is one of the most difficult human malignancies to treat. Multiple suppressive populations have been measured in patients with pancreatic cancer that may contribute to the aggressive nature of the disease [31]. Using a murine model of pancreatic cancer, we have shown that the growth of pancreatic tumor leads to increased regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSC). Both of these populations have been shown to suppress anti-tumor T-cell responses [21, 23]. In our model, depletion of Tregs had no effect on tumor growth or survival. In contrast, reduction in MDSC with Gem led to a delay in tumor growth and enhanced survival when combined with a dendritic cell vaccination.
Myeloid-derived suppressor cells are able to suppress T-cell responses through various mechanisms, and depletion of MDSC has been shown to enhance immunity in tumor-bearing mice [35, 37, 38]. Gem is a deoxycytidine analogue used as a monotherapy for the treatment for advanced pancreatic cancer [39]. Approved by the FDA for its beneficial effects on pain and quality of life, Gem has little effect on prolonging survival in pancreatic patients [39, 40]. While Gem has little effect on T-cell subsets, this study and others support the use of Gem to modulate MDSC percentages [36, 41]. It has been shown that Gem may directly induce necrosis and apoptosis of MDSC in tumor-bearing mice [42]. This may lead to a reduction in tumor pro-inflammatory soluble factors as macrophages can interact with tumor-induced MDSC, leading to a significant reduction in IL-12 cytokine production, critical for DC maturation and proper antigen presentation required for induction of anti-tumor immune responses [43]. In addition, MDSC associated with macrophages produce significant amounts of IL-10, which can lead to the down-regulation of MHC class I and class II molecules to halt antigen presentation and hinder maturation of DC. Targeting MDSC with Gem may prevent interaction with macrophages that halt the production of pro-inflammatory factors in the microenvironment that promote tumor progression.
Several populations of MDSC have been shown to be important in suppressing T-cell function. The granulocytic subset characterized by expression of CD11b and Ly6C has been shown to suppress by secreting high levels of ROS and little NO. The monocytic subset is characterized by expression of CD11b and Ly6G and mediates suppression primarily through the production of NO with little ROS production [44]. In our model, we evaluated which of these subsets were affected by Gem treatment. We observed a significant reduction in the granulocytic MDSC subset in mice that received Gem alone or in combination with DC vaccination. There was no significant difference in the monocytic MDSC between the study groups (data not shown). These data suggest that Gem specifically targets granulocytic MDSC in mice bearing Panc02 tumors.
Dendritic cell–based vaccination has been shown to be effective in murine models of pancreatic cancer. Treatment of tumor-bearing mice with DC has led to increased tumor-specific lysis by CD8+ T cells, expansion of IFN-γ-secreting T cells, and tumor regression [45–48]. Targeting primary or metastatic lesions with intratumoral DC has been shown to be effective in murine pancreatic cancer models and may be beneficial as most patients are unresectable at the time of diagnosis [49]. In a clinical trial, intratumoral injection of DC led to enhanced immunity and regression of tumor in several patients with pancreatic cancer [18].
Our studies support combination therapy with intratumoral DC vaccination and Gem treatment to reduce the number of MDSC and enhance survival in mice bearing pancreatic cancer. Further studies are ongoing to determine the mechanisms of enhanced anti-tumor responses in mice treated with DC vaccination and Gem. As we have measured a reduction in MDSC in the peripheral blood of pancreatic cancer patients treated with Gem, this research has the potential to lead to better therapeutic approaches for patients with pancreatic cancer.
Acknowledgments
We would like to acknowledge and thank The Moffitt Cancer Center and University of South Florida (USF) Fred Wright Jr. Flow Cytometry facilities for their support. We like to thank Dr. Maya Jerald, Jeffrey Nagle, and Nadine Nelson for their technical assistance. This work was supported, in part, by grants from NIH/NCI R00CA128825-01 (SPT), Frederick A. Coller Surgical Society Resident Research Award (NV), and an Institutional Research Grant 93-032-13 from the American Cancer Society (TG).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Tomar Ghansah and Nasreen Vohra: equally contributed.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 2.Henne-Bruns D, Vogel I, Luttges J, Kloppel G, Kremer B. Ductal adenocarcinoma of the pancreas head: survival after regional versus extended lymphadenectomy. Hepatogastroenterology. 1998;45:855–866. [PubMed] [Google Scholar]
- 3.Johnson CD, Schwall G, Flechtenmacher J, Trede M. Resection for adenocarcinoma of the body and tail of the pancreas. Br J Surg. 1993;80:1177–1179. doi: 10.1002/bjs.1800800937. [DOI] [PubMed] [Google Scholar]
- 4.Mukaiya M, Hirata K, Satoh T, Kimura M, Yamashiro K, et al. Lack of survival benefit of extended lymph node dissection for ductal adenocarcinoma of the head of the pancreas: retrospective multi-institutional analysis in Japan. World J Surg. 1998;22:248–252. doi: 10.1007/s002689900378. [DOI] [PubMed] [Google Scholar]
- 5.Cleary SP, Gryfe R, Guindi M, Greig P, Smith L, et al. Prognostic factors in resected pancreatic adenocarcinoma: analysis of actual 5-year survivors. J Am Coll Surg. 2004;198:722–731. doi: 10.1016/j.jamcollsurg.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Helm JF, Centeno BA, Coppola D, Druta M, Park JY, et al. Outcomes following resection of pancreatic adenocarcinoma: 20-year experience at a single institution. Cancer Control. 2008;15:288–294. doi: 10.1177/107327480801500403. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez J, Mullinax J, Clark W, Toomey P, Villadolid D, et al. Survival after pancreaticoduodenectomy is not improved by extending resections to achieve negative margins. Ann Surg. 2009;250:76–80. doi: 10.1097/SLA.0b013e3181ad655e. [DOI] [PubMed] [Google Scholar]
- 8.Raut CP, Tseng JF, Sun CC, Wang H, Wolff RA, et al. Impact of resection status on pattern of failure and survival after pancreaticoduodenectomy for pancreatic adenocarcinoma. Ann Surg. 2007;246:52–60. doi: 10.1097/01.sla.0000259391.84304.2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toomey P, Hernandez J, Morton C, Duce L, Farrior T, et al. Resection of portovenous structures to obtain microscopically negative margins during pancreaticoduodenectomy for pancreatic adenocarcinoma is worthwhile. Am Surg. 2009;75:804–809. doi: 10.1177/000313480907500911. [DOI] [PubMed] [Google Scholar]
- 10.Schmitz-Winnenthal FH, Volk C, Z’Graggen K, Galindo L, Nummer D, et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005;65:10079–10087. doi: 10.1158/0008-5472.CAN-05-1098. [DOI] [PubMed] [Google Scholar]
- 11.Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–e31. doi: 10.1097/00006676-200401000-00023. [DOI] [PubMed] [Google Scholar]
- 12.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 13.Banchereau J, Ueno H, Dhodapkar M, Connolly J, Finholt JP, et al. Immune and clinical outcomes in patients with stage IV melanoma vaccinated with peptide-pulsed dendritic cells derived from CD34+ progenitors and activated with type I interferon. J Immunother. 2005;28:505–516. doi: 10.1097/01.cji.0000171292.79663.cb. [DOI] [PubMed] [Google Scholar]
- 14.Chang AE, Redman BG, Whitfield JR, Nickoloff BJ, Braun TM, et al. A phase I trial of tumor lysate-pulsed dendritic cells in the treatment of advanced cancer. Clin Cancer Res. 2002;8:1021–1032. [PubMed] [Google Scholar]
- 15.Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna EA, Yanik GA, et al. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001;61:8513–8519. [PubMed] [Google Scholar]
- 16.Bauer C, Bauernfeind F, Sterzik A, Orban M, Schnurr M, et al. Dendritic cell-based vaccination combined with gemcitabine increases survival in a murine pancreatic carcinoma model. Gut. 2007;56:1275–1282. doi: 10.1136/gut.2006.108621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lepisto AJ, Moser AJ, Zeh H, Lee K, Bartlett D, et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008;6:955–964. [PMC free article] [PubMed] [Google Scholar]
- 18.Mule JJ. Dendritic cell-based vaccines for pancreatic cancer and melanoma. Ann N Y Acad Sci. 2009;1174:33–40. doi: 10.1111/j.1749-6632.2009.04936.x. [DOI] [PubMed] [Google Scholar]
- 19.Pecher G, Haring A, Kaiser L, Thiel E. Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunol Immunother. 2002;51:669–673. doi: 10.1007/s00262-002-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsushita N, Pilon-Thomas SA, Martin LM, Riker AI. Comparative methodologies of regulatory T cell depletion in a murine melanoma model. J Immunol Methods. 2008;333:167–179. doi: 10.1016/j.jim.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, et al. Depletion of CD4+ CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. 2006;13:1252–1258. doi: 10.1245/s10434-006-9015-y. [DOI] [PubMed] [Google Scholar]
- 22.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pilon-Thomas S, Nelson N, Vohra N, Jerald M, Pendleton L, et al. Murine pancreatic adenocarcinoma dampens SHIP-1 expression and alters MDSC homeostasis and function. PLoS ONE. 2011;6:e27729. doi: 10.1371/journal.pone.0027729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 25.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;16:348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 26.Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184:3106–3116. doi: 10.4049/jimmunol.0902661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–243. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 28.Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, et al. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16:4583–4594. doi: 10.1158/1078-0432.CCR-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 30.Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, et al. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010;120:1111–1124. doi: 10.1172/JCI40269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goedegebuure P, Mitchem JB, Porembka MR, Tan MC, Belt BA, et al. Myeloid-derived suppressor cells: general characteristics and relevance to clinical management of pancreatic cancer. Curr Cancer Drug Targets. 2011;11:734–751. doi: 10.2174/156800911796191024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44:717–726. [PubMed] [Google Scholar]
- 34.Pilon-Thomas S, Li W, Briggs JJ, Djeu J, Mule JJ, et al. Immunostimulatory effects of CpG-ODN upon dendritic cell-based immunotherapy in a murine melanoma model. J Immunother. 2006;29:381–387. doi: 10.1097/01.cji.0000199199.20717.67. [DOI] [PubMed] [Google Scholar]
- 35.Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010;16:1812–1823. doi: 10.1158/1078-0432.CCR-09-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le HK, Graham L, Cha E, Morales JK, Manjili MH, et al. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9:900–909. doi: 10.1016/j.intimp.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 38.Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol. 2012;189:5147–5154. doi: 10.4049/jimmunol.1200274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 40.Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100,313 patients diagnosed from 1985–1995, using the National Cancer Database. J Am Coll Surg. 1999;189:1–7. doi: 10.1016/S1072-7515(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 41.Plate JM, Plate AE, Shott S, Bograd S, Harris JE. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol Immunother. 2005;54:915–925. doi: 10.1007/s00262-004-0638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 43.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 44.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dauer M, Herten J, Bauer C, Renner F, Schad K, et al. Chemosensitization of pancreatic carcinoma cells to enhance T cell-mediated cytotoxicity induced by tumor lysate-pulsed dendritic cells. J Immunother. 2005;28:332–342. doi: 10.1097/01.cji.0000164038.41104.f5. [DOI] [PubMed] [Google Scholar]
- 46.Kim HS, Choo YS, Koo T, Bang S, Oh TY, et al. Enhancement of antitumor immunity of dendritic cells pulsed with heat-treated tumor lysate in murine pancreatic cancer. Immunol Lett. 2006;103:142–148. doi: 10.1016/j.imlet.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 47.Nagaraj S, Ziske C, Strehl J, Messmer D, Sauerbruch T, et al. Dendritic cells pulsed with alpha-galactosylceramide induce anti-tumor immunity against pancreatic cancer in vivo. Int Immunol. 2006;18:1279–1283. doi: 10.1093/intimm/dxl059. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt T, Ziske C, Marten A, Endres S, Tiemann K, et al. Intratumoral immunization with tumor RNA-pulsed dendritic cells confers antitumor immunity in a C57BL/6 pancreatic murine tumor model. Cancer Res. 2003;63:8962–8967. [PubMed] [Google Scholar]
- 49.Nair SK, Hull S, Coleman D, Gilboa E, Lyerly HK, et al. Induction of carcinoembryonic antigen (CEA)-specific cytotoxic T-lymphocyte responses in vitro using autologous dendritic cells loaded with CEA peptide or CEA RNA in patients with metastatic malignancies expressing CEA. Int J Cancer. 1999;82:121–124. doi: 10.1002/(SICI)1097-0215(19990702)82:1<121::AID-IJC20>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]