

Abstract
To probe the importance of the heterocyclic core of estrogen receptor (ER) ligands, we prepared a series of thiophene-core ligands by Suzuki cross-coupling of aryl boronic acids with bromo-thiophenes, and we assessed their receptor binding and cell biological activities. The disposition of the phenol substituents on the thiophene core, at alternate or adjacent sites, and the nature of substituents on these phenols all contribute to binding affinity and subtype selectivity. Most of the bis(hydroxyphenyl)-thiophenes were ERβ selective, whereas the tris(hydroxyphenyl)-thiophenes were ERα selective; analogous furan-core compounds generally have lower affinity and less selectivity. Some diarylthiophenes show distinct superagonist activity in reporter gene assays, giving maximal activities 2–3 times that of estradiol, and modeling suggests that these ligands have a different interaction with a hydrogen-bonding residue in helix-11. Ligand-core modification may be a new strategy for developing ER ligands whose selectivity is based on having transcriptional activity greater than that of estradiol.
INTRODUCTION
The beneficial physiological actions of estrogens on development and functional maintenance of many target tissues (reproductive system, bone,1 vasculature,2 and brain,3 etc.4) are well recognized, as are the pathological roles that they play in promoting breast and uterine cancers.5–8 This spectrum of effects in diverse tissues provides an intriguing opportunity for the creation of tissue-selective estrogens that on balance have net desirable vs. undesirable activities, such as ones that would provide bone and cardiovascular protection without stimulation of breast or uterus. The challenge of creating such selective estrogens has, over time, been approached in different ways.
The actions of estrogens are mediated by two members of the Nuclear Receptor superfamily,9 the estrogen receptor (ER) subtypes, ERα and the more recently identified ERβ.10 These subtypes have distinct tissue distributions (ERα mainly in the female reproductive system and ERβ mainly in prostate, colon, cardiovascular and central nervous systems11), and even when both ER subtypes are present in the same cells, estrogen action through these receptors leads to differential gene regulation, resulting in distinct physiological outcomes.12, 13 Thus, much effort has been made to develop ER subtype-selective ligands having tissue- and gene-selective biological activities. A major focus has been on obtaining ERβ-selective compounds, because this ER subtype is considered more likely to mediate desirable estrogen action without the increased risk of cancer from stimulation of the breast and uterus.14–18 Despite the similarity of their ligand-binding pockets (LBPs),19–21 considerable success has been achieved in obtaining ER ligands having affinity or potency preferences for one or the other ER subtype (Figure 1),14 but thus far preferential efficacy or intrinsic activity has been only in favor of ERα.22 The medical utility of ERβ subtype-selective estrogens, however, is less clear.18
Figure 1.

Estradiol and examples of some ERα (left) and ERβ-selective (right) ligands and a SERM
Prior to the discovery of ERβ, efforts to obtain tissue-selective estrogens focused on compounds, termed SERMs (for selective estrogen receptor modulators), that showed different levels of partial agonist/antagonist activity (i.e., intrinsic activity) in different target tissues. The selectivity of SERMs was believed to arise from their differential engagement of coregulator proteins in a target cell- and gene-specific manner,23 and compounds such as tamoxifen (Figure 1), as well as raloxifene, lasofoxifene, and basedoxifene, were found to be largely agonistic in bone and antagonistic in breast, but having varying activity in the uterus.
In the development of both SERMs and subtype-selective ligands, extensive investigation has been made of non-steroidal compounds having heterocyclic cores. In fact, while the peripheral substituents—typically phenols, simple alkyl groups, and basic and polar phenyl substituents—have generally remained the same, a wide variety of heterocyclic cores has been explored. We and others have examined ER ligand cores consisting of five-membered ring heterocycles5, 24–37 and to a lesser extent six-membered ring heterocycles.38 Some examples of the 5-membered ring heterocycle series are presented below (Figure 2).
Figure 2.

Structure and binding affinity data of the most promising pyrazole, furan and imidazole and the title thiophene compounds. Binding, determined by competitive radiometric binding assays, is expressed as Relative Binding Affinity (RBA) values, with estradiol (E2) as 100. α/β is the ratio of ERα RBA to ERβ RBA
We noted that the highest affinity ligands had 5-membered heterocyclic cores of low polarity (furan > pyrazole > imidazole), presumably reflecting both the penalty for desolvation to remove the ligand from water to bind to ER, as well as an intolerance of the receptor ligand binding pockets for core units having large dipole moments.22 Also, in the pyrazole and furan series, we mapped quite extensively the requirements for high ERα binding affinity, and concluded that tri and even tetra substitution was required.24, 30 Thiophenes, by contrast, have been relatively unexplored, and the only two prepared in our prior studies had lower levels of substitution and quite low affinities for ER.30 Hartmann and co-workers have reported on certain disubstituted thiophenes as inhibitors of 17β-hydroxysteroid dehydrogenase type1 (17β-HSD1), but they also showed very low ER binding affinity,39, 40 consistent with our prior observations.
In connection with recent work on a series of very different ER ligands, based on an inherently three-dimensional 7-thia-bicyclo[2.2.1]hept-5-ene-7-oxide core, we prepared a series of 3,4-disubstituted thiophene precursors. These thiophenes were used as diene components in an in situ oxidative Diels-Alder reaction with various dienophiles to give the sulfoxide-bridged bicyclic system.41 As part of that work, we tested the binding affinity of some of the diaryl-substituted thiophene Diels-Alder diene precursors (4a–d), and we were pleased to find that they showed significant ER binding affinities, in some cases with modest ERβ selectivity.
To pursue this lead further, we have now extended our investigation of thiophene-core ER ligands by making systematic modifications of the number, nature, and orientation of substituents on these ligands, examining their binding affinity and cellular potencies for ERα and ERβ; some congeneric furan-core ligands were also prepared for comparison. We have been able to map structure-activity relationships (SARs) for these compounds, and most intriguingly, we have identified among these thiophene-core ligands, compounds that have striking superagonist activity. Superagonism, which is intrinsic activity greater than that of a paradigmatic agonist, in this case the endogenous hormone estradiol (E2), has only scant precedent in the nuclear receptor field (see Discussion). The enhanced intrinsic activity of ligands showing superagonism might provide a novel mechanism for the development of estrogens having desirable selective activity that is complementary to the reduced intrinsic activity of SERMs.42 In addition, molecular modeling of these novel ligands complexed to ER offers a framework for understanding the structural basis for their superagonist activity, and might reveal a general strategy for obtaining superagonists for other members of the nuclear receptor superfamily.
RESULTS
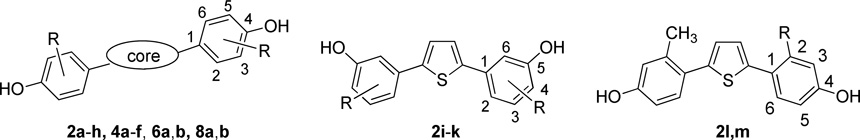
(We have deviated somewhat from the IUPAC rules in specifying the locant positions of the hydroxyl, methyl, and halogen substituents on the phenyl groups attached to the core heterocycle (i.e., thiophene or furan): Strict application of IUPAC rules is awkward because it causes locant positions on the phenyl groups to change when the nature of the substituent changes from –OMe (the thiophene or furan is the root) to –OH (the phenol becomes the root). Thus, to facilitate comparisons across compound series and discuss structure-activity relationships in a more understandable way, we have opted to use consistently the heterocyclic core (thiophene or furan) as the root name, regardless of the nature of the substituents on the phenyl groups. We also display these locant positions clearly in indicator structures in the schemes and tables. In addition, for clarity, the locant positions at which the aryl groups are attached to the thiophene or furan core rings are given in italic typeface.)
Chemical Synthesis
Thiophenes
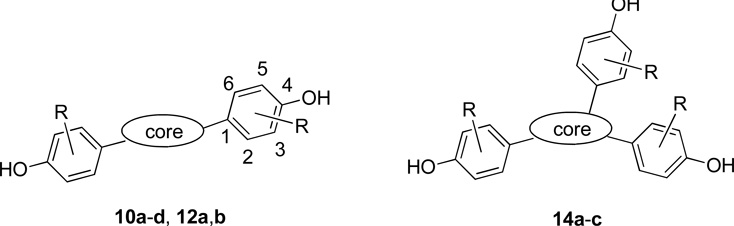
In the synthesis of compounds 2a–m (Scheme 1), key intermediates 1a–m were obtained by a one-step double Suzuki cross-coupling reaction.43, 44 Compounds 1a–d and 1g–k were easily obtained using Pd(OAc)2/PPh3 as catalyst. This reaction failed, however, with compounds 1e and 1f, and attempts to employ the two-step Suzuki coupling using Pd(OAc)2/PPh3 also gave very low overall yields of 1e and 1f, accompanied by a large amount of homo coupled diaryl byproducts. In a brief catalyst screen, we found that while Pd2(dba)2, Pd(PPh3)Cl2, and Pd(CN)2Cl2 did not give improved yields, Pd(dppf)Cl2 gave the products 1e and 1f in 45% and 62% yields, respectively, with little by products (Scheme 1A). We presume that this improvement is due to the increased rate of reductive elimination resulting from the increased bite angle of the dppf ligand.45 Unsymmetrical compounds 1l and 1m were obtained by a two-step Suzuki coupling (Scheme 1B). In the first step, 1 equivalent of commercially available 2,5-dibromothiophene reacted with 2 equivalents of aryl boronic acid under standard conditions to give the monosubstituted thiophene 19, and a second cross-coupling with 2 equivalents of aryl boronic acid gave the intermediates 1l–m. Finally, treatment of the compounds 1a–m with boron tribromide gave the final 2,5-disubstituted diphenolic thiophenes 2a–m in good yields.
Scheme 1.


Synthesis of 2,5-substituted series compounds 2a–m.a
Reagents and conditions: (a) [Pd] catalyst, Na2CO3, toluene/water (1:1), reflux, 24h; (b) BBr3, CH2Cl2, −20 °C to r.t., 4h. a To simplify comparisons between compounds in closely related series, we designate locant positions of the substituents on the phenyl groups with respect to the thiophene core; locant positions on the thiophene core itself are given by numbers in italics.
In the synthesis of 3,4-, 2,4-, 2,3-disubstituted thiophene derivatives 4a–f, 6a–b and 8a–b (Scheme 2), we used Pd(OAc)2/PPh3 for the preparation of 3a–d but again turned to Pd(dppf)Cl2 as catalyst for the preparation of compounds 3e–f, 5a–b and 7a–b, which were obtained in moderate to good yields from the appropriate dibromothiophene precursor. Ether cleavage gave final products 4a–f, 6a–b and 8a–b in 60–80% yields.
Scheme 2.

Synthesis of 3,4-, 2,4-, 2,3-substituted thiophenes 4a–f, 6a–b and 8a–b. a
Reagents and conditions: (a) [Pd] catalyst, Na2CO3, toluene/water (1:1), reflux, 24h; (b) BBr3, CH2Cl2, −20 °C to r.t., 4h. a See footnote a in Scheme 1.
To investigate the effect of a third substituent on the thiophene core, we synthesized a series of trisubstituted thiophene compounds 10a–d, 12a–b and 14a–c (Scheme 3). 3-Methyl-2,5-dibromothiophene (20) was first synthesized by bromination of 3-methylthiophene using NBS.46 A one-step Suzuki coupling with Pd(dppf)Cl2 afforded 9a–d, which gave the products 10a–d in good yield after ether cleavage (Scheme 3A). 3-Phenyl bisphenol thiophenes 12a–b were obtained by demethylation of 11a–b, prepared by two successive cross-coupling reactions (Scheme 3B). In the first step, 1 equivalent of commercially available 2,3,5-tribromothiophene was reacted with 4 equivalents of aryl boronic acid using standard conditions, and the resulting 2,5-bis-substituted thiophenes 21 and 22 were subsequently submitted to a second cross-coupling reaction with 2 equivalents of phenyl boronic acid to yield the intermediates 11a–b.
Scheme 3.


Synthesis of tri-substituted compounds 10a–d, 12a–b and 14a–c. a
Reagents and conditions: (a) [Pd] catalyst, Na2CO3, toluene/water (1:1), reflux, 24h; (b) BBr3, CH2Cl2, −20 °C to r.t., 4h; (c) NBS, THF/AcOH (1:1), r. t., 12h. a See footnote a in Scheme 1.
In our previous work on furan-core ER ligands,30 we found that triaryl, especially trisphenol, furans showed higher binding affinity and subtype selectivity than the corresponding bisphenol analogues. Thus, we also wanted to introduce a third phenol ring into the thiophene-core compounds. Key intermediates 13a–c were prepared using 1 equivalent of 2,3,5-tribromothiophene with 5 equivalents of aryl boronic acid using standard conditions; ether cleavage then gave compounds 14a–c in very good yields (Scheme 3C).
Furans
For comparison with selected thiophenes, we prepared their furan-core counterparts (16a–d and 18a–d, Scheme 4). 2,5-Dibromofuran (23) was synthesized by bromination of furan using DMF as solvent with dropwise addition of Br2 (Scheme 4A).47 3,4-Dibromofuran (24) was synthesized by oxidation of commercially available (E)-2,3-dibromo-2-butane-1,4-diol using chromosulfuric acid (Scheme 4B).48 Because 3,4-dibromofuran decomposes under the reaction conditions, it was continuously removed from the reaction mixture by steam distillation and stored below 0 °C as a white solid. We first used Suzuki coupling reaction conditions similar to those used for the thiophenes, but we failed to get to corresponding furans. Considering the lower aromaticity of furan compared to thiophene, we then switched to Pd(PPh3)4 as Pd(0) catalyst.49 The dibromofurans were coupled with 5 equivalents of the aryl boronic acid and then deprotected with boron tribromide to give the final products 16a–d and 18a–d in moderate to good yields.
Scheme 4.

Synthesis of corresponding furan-core compounds 16a–d and 18a–d.a
Reagents and conditions: (a) Na2CO3, THF/water (1:1), Pd(PPh3)4, reflux, 12h; (b)BBr3, CH2Cl2, −20 °C to r.t., 4h; (c) K2Cr2O7, H2SO4, H2O, steam distillation; (d) Br2, DMF, 30–35 °C, overnight. a See footnote a in Scheme 1.
The identity of all final compounds were confirmed by 1H NMR, 13C NMR and mass spectroscopy (see experimental section).
Biological Results
(See comment on nomenclature at the start of the Results Section)
Relative Binding Affinities
The relative binding affinities were determined by a competitive radiometric receptor-binding assay and are reported in Tables 1–3. These affinities are presented as relative binding affinity (RBA) values, where estradiol has an affinity of 100. For reference, the KD of estradiol is 0.2 nM for ERα and 0.5 nM for ERβ. (The affinities for a subset of compounds representing the different modes of diaryl core substitution are compared schematically in Figure 3.)
Table 1.
ERα and ERβ relative binding affinity (RBAs) of compounds 2a–m, 4a–f, 6a–b and 8a–b.a
 | ||||||
|---|---|---|---|---|---|---|
| 2a–h, 4a–f, 6a,b, 8a,b | 2i–k | 2l,m | ||||
| entry | cmpd | core | R | ERαb | ERβb | β/α |
| 1 | 2a | H | 0.040 ± 0.004 | 0.79 ± 0.11 | 19.8 | |
| 2 | 2b | 2-Me | 1.43 ± 0.19 | 1.74 ± 0.26 | 1.2 | |
| 3 | 2c | 3-Me | 0.004 ± 0.001 | 0.002 | 0.5 | |
| 4 | 2d | 3,5-diMe | 0.003 ± 0.001 | 0.005 ± 0.001 | 1.7 | |
| 5 | 2e | 2-F | 2.03 ± 0.19 | 33.1 ± 5.8 | 16 | |
| 6 | 2f | 2-Cl | 6.7 ± 1.3 | 10.0 ± 2.4 | 1.5 | |
| 7 | 2g | 3-F | 0.013 ± 0.004 | 0.142 ± 0.004 | 11 | |
| 8 | 2h | 3-Cl | 0.009 ± 0.001 | 0.036 | 4.0 | |
| 9 | 2i | 2-F | 0.008 ± 0.002 | 0.066 ± 0.066 | 8.3 | |
| 10 | 2j | 3-F | 0.009 ± 0.006 | 0.041 ± 0.004 | 0.4 | |
| 11 | 2k | 4-F | ~0.001 | 0.010 ± 0.002 | 10 | |
| 12 | 2l | F | 0.97 ± 0.01 | 0.91 ± 0.26 | 0.94 | |
| 13 | 2m | Cl | 4.29 ± 0.64 | 5.89 ± 0.21 | 1.4 | |
| 14 | 4a | 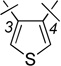 |
H | 0.57 ± 0.12 | 1.69 ± 0.45 | 2.9 |
| 15 | 4b | 2-Me | 2.16 ± 0.54 | 4.9 ± 1.3 | 2.25 | |
| 16 | 4c | 3-Me | 0.830 | 0.297 | 0.36 | |
| 17 | 4d | 3,5-diMe | 0.039 ± 0.003 | 0.004 ± 0.001 | 0.10 | |
| 18 | 4e | 2-F | 3.03 ± 0.85 | 9.1 ± 2.2 | 3.0 | |
| 19 | 4f | 2-Cl | 13.07 ± 1.1 | 17.5 ± 2.1 | 1.3 | |
| 20 | 6a | 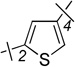 |
2-F | 1.22 ± 0.32 | 10.4 ± 1.2 | 8.5 |
| 21 | 6b | 2-Cl | 4.21 ± 0.54 | 25.6 ± 5.7 | 6.1 | |
| 22 | 8a | 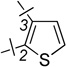 |
2-F | 1.22 ± 0.35 | 2.54 ± 0.76 | 2.1 |
| 23 | 8b | 2-Cl | 6.50 ± 0.61 | 7.73 ± 2.1 | 1.2 | |
To simplify comparisons between compounds in closely related series, we designate locant positions of the substituents on the phenyl groups with respect to the thiophene core; locant positions within the thiophene core itself are given by numbers in italics.
Relative binding affinity (RBA) values are determined by competitive radiometric binding assays and are expressed as IC50estradiol/IC50compound×100 ± the range or standard deviation (RBA, estradiol = 100%). In these assays, the KD for estradiol is 0.2 nM on ERα and 0.5 nM on ERβ.51 For details, see the Experimental Section.
Table 3.
ERα and ERβ relative binding affinity (RBAs) of furan-core compounds 16a–d and 18a–b.
 | ||||||
|---|---|---|---|---|---|---|
| 16a–d, 18a–d | ||||||
| entry | cmpd. | core | R | ERαb | ERβb | β/α |
| 1 | 16a |  |
H | 0.082 ± 0.007 | 0.34 ± 0.10 | 4.2 |
| 2 | 16b | Me | 0.78 ± 0.12 | 0.24 ± 0.02 | 0.31 | |
| 3 | 16c | F | 6.92 ± 0.73 | 32.2 ± 1.0 | 4.7 | |
| 4 | 16d | Cl | 2.73 ± 0.41 | 6.9 ± 1.7 | 2.5 | |
| 5 | 18a | 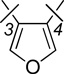 |
H | 0.22 ± 0.06 | 0.69 ± 0.07 | 3.1 |
| 6 | 18b | Me | 0.79 ± 0.21 | 2.28 ± 0.34 | 2.9 | |
| 7 | 18c | F | 1.19 ± 0.20 | 5.41 ± 0.49 | 4.5 | |
| 8 | 18d | Cl | 2.99 ± 0.70 | 5.44 ± 0.19 | 1.8 | |
Figure 3.

Comparisons of ERα and ERβ binding affinities of congeneric bisphenolic thiophene and furan-core ER ligands. In each table, the RBA values are given for binding to ERα or [ERβ] for the various biaryl thiophenes and furans we have studied. The top tables are for the alternate-substituted systems and the bottom tables for the adjacent-substituted systems. The disposition of the aryl groups with respect to the heteroatom (S or O) is indicated by the numbers in parentheses, which are color coded to enable reference to be made between the structures and the appropriate columns in the tables.
A number of the bis(hydroxyphenyl)thiophenes (Table 1) show moderate levels of ERβ selectivity, which is a general characteristic of rather slender ligands.14 In terms of absolute affinity, however, substituents on the phenol rings have major effects on their binding affinity and selectivity. Among the 2,5-bisphenol thiophenes (Series 2), those substituted at C2 (meta to the phenol) bound better than the corresponding non-substituted compound 2a. The highest affinity ERβ ligand, 2,5-bis(2-fluoro-4-hydroxyphenyl)thiophene (2e), binds to ERβ with an affinity of 33 and a 16-fold ERβ binding affinity preference (Table 1, entry 5). The highest affinity ERα ligand in the Series 2 was the 2-chloro analogue 2f, with an RBA of 6.7, but it still showed ERβ selectivity (RBA of 10 for ERβ, Table 1, entry 6).
The thiophenes having substituents at the C3 position (i.e., ortho to the phenol) all show dramatically decreased binding affinities compared to the corresponding ones substituted at C2 (i.e., meta to the phenol). Their affinities are even lower than that of the corresponding unsubstituted compound 2a (Table 1, entry 1), with the tetra ortho methyl compound 2d binding particularly poorly (Table 1, entry 4). This is very likely due to steric occlusion of the hydrogen bonding capacity of the phenols by ortho substitution, which is known to greatly reduce affinity in both steroidal and other non-steroidal estrogens.50
The position of the hydroxyl group is also of great importance, and when this group is moved from the para to the meta position of the phenyl ring (e.g., 2i–k), binding affinities are remarkably lowered (Table 1, entries 9–11), again consistent with known trends in other ligand series. Unsymmetrically substituted compounds (e.g., 2l and 2m) also had somewhat lower binding affinities than their corresponding symmetrical counterparts 2e and 2f (Table 1, entries 5 and 6).
Like of 2,5-disubstituted compounds (Series 2), similar trends were also observed for the 3,4-disubstituted (Series 4), 2,4-disubstituted (Series 6), and 2,3-disubstituted compounds (Series 8). The highest affinities were those substituted at C2, meta to the phenol, and the lowest, those with C3 (ortho) substitution or no substitution. Chlorine substitution gave the highest affinity for both ERs (4f and 6b, Table 1, entries 19 and 21). Other comparisons among the disubstituted thiophenes (and furans) can be viewed more easily in Figure 3 (see below).
To investigate the influence of an additional substituent on the thiophene core, trisubstituted thiophenes 10a–d, 12a–b and 14a–c were prepared (Table 2). Introduction of a methyl group at the 3-position of the thiophene core (Table 2, 10a–d) did not cause a significant change in RBA but appeared to cause a decrease in selectivity for ERβ (10a vs. 2a, 10b vs. 2b, 10c vs. 2e, 10d vs. 2f). When the methyl group was replaced by a phenyl group, as with compounds 12a–b, there was a further decrease in binding for ERβ (RBA of ERβ 12a 2.1 vs. 2e 33, 12b 3.0 vs. 2f 10), and when the third substituent is replaced by a phenol moiety, the subtype selectivity was actually reversed. Thus, compounds 14a–c show ERα-selectivity, with 14c having an affinity for ERα of 86 and a selectivity of 14, which is similar to that reported earlier for the triphenolic furans and pyrazoles.24, 30 This reversal of ER subtype binding preference as additional substituents are added to the heterocycle core likely reflects the fact that the ligand-binding pocket of ERα is larger than in ERβ.24, 30 Also, because members of the tris-phenol series (Series 14) bound better than the phenyl bis-phenols (Series 12), we inferred that the third phenolic hydroxyl group might be involved in a hydrogen bond with another residue in the ligand binding pocket, most likely T347 in ERα. Similar enhancement in binding with a third phenolic hydroxyl group can be found in the furan and pyrazole systems that we have previously investigated.24, 30
Table 2.
ERα and ERβ relative binding affinity (RBAs) of compounds 10a–d, 12a–b and 14a–c.a
 | ||||||
|---|---|---|---|---|---|---|
| 10a–d, 12a,b | 14a–c | |||||
| entry | cmpd. | core | R | ERαb | ERβb | β/α |
| 1 | 10a | 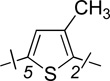 |
H | 0.95 ± 0.16 | 1.6 ± 0.2 | 1.7 |
| 2 | 10b | 2-Me | 5.15 ± 0.44 | 4.8 ± 0.6 | 0.92 | |
| 3 | 10c | 2-F | 9.3 ± 1.1 | 20.6 ± 4.1 | 2.2 | |
| 4 | 10d | 2-Cl | 10.4 ± 1.2 | 5.41 ± 0.81 | 0.52 | |
| 5 | 12a | 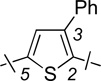 |
2-F | 1.88 ± 0.28 | 2.02 ± 0.57 | 1.1 |
| 6 | 12b | 2-Cl | 3.65 ± 0.41 | 3.01 ± 0.74 | 0.83 | |
| 7 | 14a | 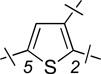 |
H | 1.79 ± 0.23 | 0.47 ± 0.07 | 0.26 |
| 8 | 14b | 2-F | 6.35 ± 0.26 | 0.98 ± 0.02 | 0.15 | |
| 9 | 14c | 2-Cl | 85.7 ± 2.5 | 6.0 ± 1.6 | 0.07 | |
Finally, to compare the affinity of thiophene-core ligands with furan-core ligands, we prepared a limited series of bis(hydroxylphenyl)furans (16a–d and 18a–d, Table 3). Gratifyingly, most of these disubstituted furans retained the ERβ selectivity of the corresponding thiophenes. The best furan, compound 16c, had an RBA of 6.9 and 32 for ERα and ERβ, respectively, giving a 4.7-fold ERβ selectivity (Table 3, entry 3). Where direct comparisons can be made, however, the furans (except for compound 16c) have lower affinity and poorer selectivity than their corresponding thiophenes. This might be due to the higher electron negativity of the oxygen atom, making the furan core more polar than the thiophene core, and/or the longer length of the C-S vs. C-O bond, which somewhat alters the geometric disposition of the substituents.
Global comparisons of structure-affinity relationships among the disubstituted thiophenes and furans can better be made by reference to Figure 3, which displays an overlay of the two core substitution patterns that unite congeneric (positionally isomeric) compounds, an “alternate” pattern, with the phenols attached to non-adjacent positions (2,5- and 2,4- patterns of substitution), and an “adjacent” pattern, with phenols on neighboring positions (2,3- and 3,4-patterns of substitution). RBA values for ERα and [ERβ] are shown to the right. In all cases, the importance of C2 substitution on enhancing affinity is evident. This has been noted in our prior work on ER ligands with bicyclic cores41, 52, 53 and in a recent report by Gust and colleagues on pyrroles.29 Beyond this, the halogens, Cl and F, bind better than the proteo and Me analogs, with Cl being better than F in most cases. Curiously, despite their considerably different shapes, no clear pattern emerges in comparing the alternate vs. adjacent substitution patterns (upper vs. lower table), the two types of alternate or adjacent substitutions (blue vs. red in the thiophene tables), nor are there systemic differences between the furans and the congeneric thiophenes (right vs. left). In addition to the comparisons illustrated in this figure, other trends noted above were the increased affinity and ERα preference that comes from adding a third substituent, especially a phenol, onto the thiophene and furan cores.
We will return to this type of comparative analysis where we discuss the results of the cellular activity of these compounds and when we present examples of docking representative members into the ERα ligand-binding pocket and examine ligand volumes, geometries, and conformations.
Transcriptional Activation Assays
The thiophene- and furan-core compounds that have significant affinity for ERα or ERβ were characterized for activation of an estrogen response element (ERE)-driven luciferase reporter in HepG2 liver cells cotransfected with ERα or ERβ; this is considered to be a versatile cell system for characterizing the efficacy of ER ligands.54 Dose-response curves were generated, and when full curves were evident, potency (EC50) values and efficacy (Eff, % of E2) values were determined. The results are summarized in Table 4, and dose-response curves for representative ligands, are shown in Figure 4.
Table 4.
Potency and Intrinsic Activity of Thiophene and Furan Core Ligandsa
| HepG2 | Ishikawa | |||||||
|---|---|---|---|---|---|---|---|---|
| ERα | ERβ | ERα | ||||||
| cmpd | core | R | EC50 (µM) | Eff (% E2) | EC50 (µM) | Eff (% E2) | Eff (% E2) | |
| THIOPHENES | ||||||||
| 2a | 2,5- | 4-OH | H | 2.35 | 125 ± 4 | 1.54 | 198 ± 4 | 111 ± 32 |
| 2b | 4-OH | 2-Me | - | 78 ± 4 | 6.60 | 87 ± 15 | 148 ± 23 | |
| 2e | 4-OH | 2-F | - | 181 ± 14 | - | 336 ± 63 | 315 ± 125 | |
| 2f | 4-OH | 2-Cl | 2.18 | 111 ± 5 | 1.94 | 169 ± 18 | 169 ± 14 | |
| 2l | 4-OH | 2-Me, 2'-F | 0.47 | 117 ± 8 | 0.61 | 277 ± 15 | 8 ± 5 | |
| 2m | 4-OH | 2,Me, 2'-Cl | 1.91 | 78 ± 3 | 1.92 | 111 ± 7 | 102 ± 7 | |
| 4a | 3,4- | H | 2.25 | 69 ± 4 | 1.27 | 73 ± 2 | 46 ± 4 | |
| 4b | 2-Me | 3.15 | 97 ± 11 | 1.87 | 67 ± 4 | 120 ± 14 | ||
| 4c | 3-Me | - | 73 ± 5 | - | 70 ± 4 | 4 ± 0 | ||
| 4e | 2-F | 0.12 | 79 ± 3 | 0.780 | 29 ± 4 | 63 ± 5 | ||
| 4f | 2-Cl | 0.045 | 90 ± 7 | 0.203 | 57 ± 3 | 90 ± 3 | ||
| 6a | 2,4- | 2-F | 1.06 | 123 ± 6 | 1.34 | 177 ± 17 | 201 ± 13 | |
| 6b | 2-Cl | 0.563 | 87 ± 5 | 0.660 | 117 ± 2 | 81 ± 5 | ||
| 8a | 2,3- | 2-F | 65.9 | 69 ± 3 | - | 33 ± 10 | 67 ± 6 | |
| 8b | 2-Cl | 1.16 | 82 ± 8 | 5.70 | 116 ± 14 | 114 ± 6 | ||
| 10a | 2,5- | 3-Me | H | 1.52 | 116 ± 5 | 1.23 | 200 ± 10 | 225 ± 27 |
| 10b | 3-Me | 2-Me | 0.439 | 70 ± 5 | 2.46 | 106 ± 23 | 56 ± 4 | |
| 10c | 3-Me | 2-F | 0.447 | 149 ± 1 | 10.8 | 284 ± 44 | 260 ± 53 | |
| 10d | 3-Me | 2-Cl | 0.101 | 82 ± 1 | 0.295 | 113 ± 4 | 72 ± 8 | |
| 12a | 2,5- | 3-Ph | 2-F | 0.615 | 70 ± 1 | - | 39 ± 11 | 91 ± 15 |
| 12b | 3-Ph | 2-Cl | 0.066 | 72 ± 2 | - | 1 ± 1 | 92 ± 4 | |
| 14a | 2,3,5- | H | 0.485 | 82 ± 6 | 2.23 | −22 ± 1 | 145 ± 9 | |
| 14b | 2-F | 0.387 | 84 ± 5 | - | −1 ± 4 | 63 ± 2 | ||
| 14c | 2-Cl | 0.009 | 52 ± 5 | - | −13 ± 2 | 35 ± 1 | ||
| FURANS | ||||||||
| 16a | 2,5- | H | - | 62 ± 3 | - | 94 ± 26 | 91 ± 21 | |
| 16b | Me | 3.79 | 36 ± 4 | - | 8 ± 6 | 58 ± 8 | ||
| 16c | F | - | 154 ± 7 | - | 159 ± 13 | 61 ± 48 | ||
| 16d | Cl | 3.89 | 82 ± 7 | - | 106 ± 3 | 118 ± 11 | ||
| 18a | 3,4- | H | 10.6 | 81 ± 2 | - | 37 ± 5 | 82 ± 25 | |
| 18b | Me | 0.944 | 97 ± 5 | 5.11 | 56 ± 3 | 104 ± 6 | ||
| 18c | F | 3.64 | 73 ± 2 | - | 85 ± 16 | 85 ± 11 | ||
| 18d | Cl | 0.266 | 86 ± 6 | 17.5 | 54 ± 4 | 116 ± 9 | ||
See footnote a in Table 1.
Values indicating pronounced superagonism (i.e., Eff values ≥120) are indicated in boldface type.
Figure 4. Activity profile of ERα selective compounds.

HepG2 cells were transfected with 3xERE-luc and expression plasmids for ERα or ERβ, and then dispensed in 384-well plates. The next day cells were treated with the indicated compounds for 24 hr and assayed for luciferase activity. Compound with ERα (red solid line), with ERβ (blue solid line); estradiol with ERα (red dashed line, panel B only), with ERβ (blue dashed line, panel B only); ICI with ERβ (black line, panel C only)
Alternate-Position Aryl-Disubstituted Thiophenes (2,5-diaryl Series 2 and 2,4-diaryl Series 6)
The thiophenes in these two regioisomeric series are congeneric except for the position of the sulfur atom in the thiophene ring. In the reporter gene assays, the Series 2 (2,5-bisphenol-substituted) thiophenes mostly profiled as superagonists on both ERα and ERβ, showing efficacies in ERβ 2–3 times greater than that of estradiol (e.g., see compound 2l, Figure 4B). To test whether the superagonist activity was cell-type specific, compounds were also tested at a single, high dose (10 µM) for activation of the ERE-luciferase reporter in Ishikawa endometrial cells that contain native ERα, and here again most of the Series 2 tested profiled as superagonists (Table 4).
Although these compounds have affinities in the low nanomolar range, most of their potencies in the HepG2 assays were in the micromolar range, reflecting the fact that cellular potency involves the affinities of the receptor both for the ligand and for the recruited coregulators that provide the essential signal for activation of transcription.55 In addition, the liver-derived HepG2 cells are known to metabolize small molecules, which sometimes reduces their cellular potency.56,57
With respect to the effects of substitutions on the phenolic groups, the 2-methyl analog 2b displayed lower potency than 2a, despite its higher affinity. Thus, the lack of superagonist activity for 2b in the HepG2 cells may reflect the fact that maximal activity (i.e., saturation) was not reached at the doses we tested. While the 2-Cl substitution in 2f had little effect on activity, the 2-F containing analog 2e displayed even greater super-agonist activity. The other alternate-position thiophene regioisomers, Series 6, differ only in the location of the sulfur atom in the thiophene core from Series 2. Nevertheless, the superagonist activity for the 2-F compound 6a was attenuated with respect to the Series 2 regioisomers, with the 2-Cl substituted 6b having only a full agonist profile. Further interpretation of the structural basis of superagonism is presented in the Discussion.
The congeneric 2,5-disubstituted furan Series 16 displayed less agonist activity, but this may reflect the lower potency and lack of curve saturation at the highest dose tested (10 µM). Again, these differences in potency were not related to affinity, as was the case with the corresponding compounds in Series 2.
Adjacent-Position Aryl-Disubstituted Thiophenes (3,4-diaryl Series 4 and 2,3-diaryl 8) and Trisubstituted Thiophenes
Thiophenes in these series contain substitutions on adjacent positions on the thiophene, drastically changing the overall shape of these ligands compared to those of the alternate-position ligand. Also, compared to the planar ground-state conformation of the alternate-position ligands, steric interaction of the adjacent-disposed aryl groups also causes substantial (ca. 30°) twisting of the phenols out of the plane of the thiophene ring. These compounds displayed variable partial to full agonist activity profiles, with little evidence of superagonist activity. Surprisingly, 4e and 4f displayed >25 fold better EC50 values on ERα and ERβ than the corresponding 8a and 8b, despite affinity differences of only ~2 fold.
The effects of adding a third substituent onto the core were very dramatic. For the methyl-substituted Series 10, the efficacies were similar to Series 2, but with substantial increases in potency for ERα. The phenyl 12 or phenol 14 series as well displayed selective activation of ERα. In fact, compounds 12b and 14c (Figure 4A and D) displayed low nM potency for activating ERα. All of these changes in potency reflect the trends in binding affinity. Notably, while 12b had no activity on ERβ up to 10 µM, 14a and 14c displayed inverse agonist activity on ERβ (Figure 4C and D), representing to the best of our knowledge first-in-class ligands. Even the full antagonist ICI 182,780 (Fulvestrant) does not have inverse agonist activity on ERβ (Figure 4C, black line).
A summary of efficacy values for the alternate and adjacent di-substituted thiophenes and furans are presented in Figure 5, according to the same layout as were the binding affinities (Figure 3); efficacy values above 120 are noted in boldface. The predominance of superagonism in the alternate compared to the adjacent mode thiophenes, and in the thiophenes compared to the furans is very evident. To ascertain that the effects of these compounds are ER mediated, we confirmed their activity in a mammalian 2 hybrid assay of ER-coactivator binding and also by the induction of a canonical ER target gene, GREB1 (See supporting information). A proposed structural model for superagonist activity is presented in the Discussion.
Figure 5.

Comparisons of ERα and ERβ intrinsic activities (Eff % of E2) of congeneric bisphenolic thiophene and furan-core ER ligands. Maximal activities above 120 are indicated in boldface. In each table, the %Eff. values are given for the reporter gene assays of the various biaryl thiophenes and furans we have studied with ERα or [ERβ]. The top tables are for the alternate-substituted systems and the bottom tables for the adjacent-substituted systems. The disposition of the aryl groups with respect to the heteroatom (S or O) is indicated by the numbers in parentheses, which are color coded to enable reference to be made between the structures and the appropriate columns in the tables.
Discussion
The X-ray structures of many ER-ligand complexes show a pocket volume that is considerably larger than the ligand volume, with a ligand such as E2 appearing to be “suspended” by close contacts and hydrogen bonding at its ends (the A and D rings) but having relatively few close contacts at its middle (B and C rings). The ER can form a variety of pocket shapes upon binding structurally different ligands, and these shapes relate to the overall conformation of the ER-ligand complex, defining its interaction with coregulators, and ultimately determining its biological output. To understand more fully the connections between ligand shape, ligand-binding pocket shape, and biological output of the ER, we have examined the respective contributions that the core vs. the peripheral substituents of an ER ligand makes to its affinity, potency, efficacy, and ER-subtype selectivity in a number of systems.
Reviving Thiophene-Core Ligands
In this work, we have extended our examination of ER ligands having 5-membered ring heterocyclic cores such as isoxazoles,58 imidazoles,58 pyrazoles,23, 41 and furans,30 to thiophenes. Previously, we found that these 5-membered, as well as some related 6-membered ring heterocyclic cores, afforded high affinity when the core had low polarity (or a small dipole moment) and when there were a sufficient number of peripheral substituents. Larger ligands showed distinct preferential affinity and potency for ERα and smaller ligands for ERβ. However, our initial efforts to prepare thiophene-core ligands from the furan 1,4-diones precursors in the presence of thiolating agents were largely thwarted by the strong tendency of the highly substituted diones to form the furans in preference to the thiophenes.30
Our interest in the thiophenes was revived when we prepared some 3,4-diarylthiophenes to use as dienes for the preparation of 7-thia-bicyclo[2.2.1]hept-5-ene-7-oxides through a Diels-Alder reaction (using their corresponding monoxides).41 This alternative approach of preparing the di- and tri-substituted thiophenes from di- and tribromothiophene precursors using palladium-mediated cross coupling with aryl boronic acids, described here, works very well, and it enabled us to prepare the larger series of thiophenes (as well as related furans) that are examined herein.
Structural Trends in Binding Affinities and Estrogen Receptor Subtype Selectivity in the Thiophenes and Furans
From the binding data in Tables 1–3 and the summary of the diaryl congeners (Figure 3), we can make some novel observations and note a number of familiar trends.
With earlier biaryl-substituted heterocycle-core ER ligands, we generally used simple 4-hydroxyphenyl (phenol) substituents and enhanced affinities by adding a third phenol and even a fourth (alkyl) group. Here, we have enhanced affinities by adding halogens (F, Cl) and small alkyl groups (Me) at C3, meta to the phenolic OH. The boost in affinity can be quite remarkable (>100 fold on ERα for compounds 2f vs. 2a, and >10 fold for many others) and presumably arises from a more complete filling of the ligand-binding pocket, such that high affinity binding can be obtained even with thiophenes and furans containing only two substituted phenols. By contrast, as has long been known in many ligand series, substitution at C3, ortho to the phenol, greatly reduces affinity, presumably by interfering with critical hydrogen bonding.20,50 It is of note that in a series of B-seco or A-CD steroidal estrogens studied earlier, a beneficial effect of meta but not ortho substitution was also found.59, 60 Similar observations of the beneficial effect of meta substitution were made with a number of our bicyclo[2.2.1]heptane core ligands,41, 52, 53 and have been noted recently by Gust and colleagues in a series of pyrroles.29 Thus, filling voids in ligand volume without interfering with critical receptor interactions is an emerging strategy to generate high affinity ER ligands. It was also surprising that in the bisaryl-substituted thiophenes and furans, there are no great differences in the affinity between members of the alternate-position vs. adjacent-position series, despite marked differences in ligand length and shape, as well as phenol-heterocycle core torsional angles.
Other general trends are well known. The ERβ preference of smaller ligands (bisphenols or methyl-bisphenols) vs. larger ligands (trisphenols or phenyl bisphenols) is known from the pyrazoles and furans,24,30 and doubtless reflects the generally smaller pockets that form around ERβ ligands.21 Bisphenol C3-substituted ligands have high affinity and high ERβ selectivity; the best bisphenolic compound is 2e, with an ERβ affinity of 33 and selectivity of 16; the best tris(hydroxyphenyl) thiophene is compound 14c, with an ERα affinity of 85 and selectivity of 14. Also, where they can be directly compared (2a/16a, 2b/16b, 2e/16c, 2f/16d, 4a/18a, 4b/18b, 4e/16c, and 4f/18d), the thiophenes generally bind somewhat better than the furans (though not by a large factor), perhaps because the thiophene core is less polar than the furan core. This trend was also evident in comparisons of the more polar core imidazoles with the corresponding pyrazoles.24, 58 The greater electron density and higher polarizability of the sulfur atom as well as the geometric changes due to the longer C-S bond vs. C-O bond could also be factors that enhance the affinity of the thiophenes (see below).
Models for Thiophenes Binding in the Estrogen Receptor α Ligand Binding Pocket and a Possible Model for Superagonism
The most unusual finding in this study is the pronounced superagonism shown by a number of the alternate-substituted thiophenes. Ligands exhibiting superagonistic activity have been described in the nuclear receptor field. Gustafsson and others have reported that some phytoestrogens gave a somewhat higher reporter gene maximal transcriptional output than estradiol, especially on ERβ.61–64 Because the maximum transcriptional output from ERβ at saturating concentrations of both expression plasmid and E2 ligand is only about 25–40% of that from ERα,62 one could speculate that other ligands might be able to drive ERβ into a yet more active conformation. Also, there is evidence that steroidal ligands other than E2, notably 3β-adiol 65 and Δ5-3β-adiol,66 are likely the endogenous ligands for ERβ in some tissue contexts. Nevertheless, we find that certain of the thiophenes can drive superagonism from ERα as well as from ERβ; so, superagonism is not exclusively the domain of ERβ.
We have used molecular modeling to examine potential modes of interaction of the thiophenes with ER and to explore what might be the molecular basis for superagonism, which is seen predominantly with the alternate-position thiophenes. Crystal structures of ERα-ligand complexes show that the A-ring phenolic hydroxyl of estradiol has hydrogen bonding interactions with helix 3 residues Glu-353, Arg-394, and an ordered water molecule, while the D-ring hydroxyl hydrogen bonds with the helix 11 residue, His-524 (Figure 7A). This stabilizes helices 3 and 11, and allows helix 12 to dock across them. In this so-called active conformation, helix 12 forms one side of a hydrophobic groove on the surface where transcriptional coactivators bind. Hydrogen bonding to helix 3 via an A-ring mimetic has been seen in every ligand-bound ER crystal structure to date, while hydrogen bonding to helix 11 is more variable.
Figure 7. Model of thiophene ligands bound to ERα and comparisons with estradiol and OBHS.

A portion of the ligand binding pocket is rendered as a ribbon, with selected amino acids shown as sticks. (A) Structure of E2 bound ERα. The A ring of E2 forms hydrogen bonds with E353 and R394 while the D ring bonds to H524 (PDB ID: 1ERE). (B) Structure of Oxabicyclic heptane sulfonate (OBHS) bound ERα.52 OBHS H-bonds to both the conserved R394/E353 pair, and also T347 on helix 3. The phenyl sulfonate binds extends between helices 8 and 11. (C) Model of 2e bound to ERα with the conserved H-bonding to R394/E353, and the other phenol extending between helices 8 and 11. (D) Model of 4e with H-bonds to E353 and T347.
Molecular modeling of complexes with ERα suggests that thiophenes in the two alternate-substituted series bind in a different manner compared to a typical steroidal agonist. It seems reasonable to assume that the thiophenes in the 2,5- and 2,4-alternate-substituted series (2 and 6) would bind to ER in a congruent fashion. The only expected difference would be in the placement of the thiophene sulfur atom towards opposite sides of the pocket, where it might engage in different interactions with residues in the ligand-binding pocket. However, there seem to be no pronounced differences between the 2,5- and 2,4-bisphenolic thiophenes and furans in terms of affinity and intrinsic activity, except the more pronounced superagonism exhibited by the thiophenes compared to the furans. The most likely polar interactions for the alternate-pattern bisphenols, then are with Glu-353 and Arg-394 on one end and His-524 on the other (See Figure 7). While it is a speculation, superagonism might be the consequence of the increased O-O distance between the two phenols in the thiophenes compared to other ligands. This distance is 10.9 Å in E2 and 11.8 Å in diethylstilbestrol; however, in both alternate thiophene bisphenols it is 12.8 Å, that is, 1–2 Å longer. The O-O distance in the thiophenes is also somewhat longer than in the alternate-position furan bisphenols, due to the greater length of the C-S bonds (1.47 Å) vs. the C-O bonds (1.24 Å) in the heterocyclic core. (This bond length difference also affects to a lesser degree, the display angle of the two phenols on the heterocyclic core.) Thus, the alternate-position bisphenol thiophenes might be too long to form adequate hydrogen bonds with His-524.
We recently published on a series of oxabicyclic compounds, exemplified by OBHS, where the side group extends between helices 8 and 11 (Figure 7B).52 Modeling predicts that the thiophene 2e will bind similarly between helices 8 and 11 (Figure 7C), but this does not explain their superagonist activity. We have found that ligand-specific “mis-positioning” of helix 11 can interfere with the agonist conformation of helix 12;52 so, we suggest that the superagonist activity may stem from “improved” positioning of helix 11 that stabilizes helix 12 in the agonist conformation to an even greater degree than does E2. This issue is being examined further by X-ray crystallography.
The binding of thiophenes with adjacent phenols (Series 4 and 8) was modeled with compound 4e, based on the assumption that the two hydroxyls will form hydrogen bonds similar to the two phenols in OBHS by wrapping around helix 3. In this model (Figure 7D), the ligand makes no contacts with helix 11, which would be a novel form of binding for an ER ligand, but one likely utilized by members of the cyclofenil class of ligands. Changing the position of the sulfur (i.e., 2,3- vs. 3,4- substitution) could provide subtle shifts in helix 3, which comprises part of the coactivator-binding site on the external surface. The adjacent phenol thiophenes do not show superagonism, and from this model, there is no apparent reason why they should. All of these issues are being examined further by X-ray crystallography.
Conclusion
In this paper, we present the synthesis of a novel series of thiophenes and an investigation of their ER binding and biological activity. Ligands of this series can be easily prepared by Suzuki coupling, an approach that may prove convenient for further library synthesis. These compounds show an interesting binding affinity pattern: Small substituents placed meta to the phenols cause marked enhancements of binding affinity, and in general, most of the bis(hydroxylphenyl) thiophenes were selective for ERβ, whereas the tris(hydroxyphenyl) thiophenes were selective for ERα. Reporter gene assays reflect these trends, but also reveal that a number of the alternate-pattern thiophene bisphenols have marked superagonistic activity, more so than the adjacent-pattern bisphenols or the corresponding furans from either series. Molecular modeling suggests that the 1–2 Å increased O-O distance in the alternate-mode thiophenes might be responsible for this superagonism. Further studies of the cellular and in vivo biological activities of key members of this new class of ER ligands, as well as X-ray crystallographic analyses, which are underway, will be reported in due course.
The generation of this new series of ER ligands provides important insight into the diversity of structures that can function as ligands for both ER subtype, and our findings may open new strategies for the design of both ERα- and ERβ-selective ligands. They also illustrate that changes to the ligand core structure, though it is remote from close contacts with residues in the ligand-binding pocket, can have important effects on activity, in this case increased efficacy leading to superagonism. When better understood at the level of molecular interactions, superagonism itself might prove to be a new mode for developing ER ligands having selective activities.42
Experimental section
General
Starting materials were purchased from Aldrich, Acros, Aladin-reagent, Alfa-Asar and were used without purification. Tetrahydrofuran was freshly distilled from sodium/benzophenone under Argon. Toluene was freshly distilled from sodium and dichloromethane was distilled from anhydrous CaH2. Glassware was oven-dried, assembled while hot, and cooled under an inert atmosphere. Unless otherwise noted, all reactions were conducted in an inert atmosphere. All reactions were performed under an Ar atmosphere unless otherwise specified. Reaction progress was monitored by analytical thin-layer chromatography (TLC). Visualization was achieved by UV light (254 nm).
1H NMR and 13C NMR spectra were measured on a Bruker Biospin AV400 (400 MHz) instrument. Chemical shifts are reported in ppm (parts per million) and are referenced to either tetramethylsilane or the solvent. Melting points were determined on a X-4 Beijing Tech melting point apparatus and the data were uncorrected. The purity of all compounds for biological testing was determined by HPLC method (see Supporting Information), confirming >95% purity.
General Procedure for Suzuki Coupling
Under Ar atmosphere, a mixture of bromothiophene (1 equiv), arylboronic acid (2 equivalents for mono-substituted, 4 equivalents for di-substituted, 5 equivalents for tri-substituted thiophenes), Pd catalyst, sodium carbonate (2 equiv.) in an oxygen-free toluene/water (1:1) solution was stirred at 120 °C for 24 hour. After the reaction mixture was cooled to room temperature. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous MgSO4/Na2SO4, then filtered and concentrated on vacuum. The product was purified by column chromatography (CC).
General Procedure for Ether Cleavage
Under argon atmosphere, To a solution of methoxyphenyl derivative (1 equiv.) in dry dichloromethane at −20 °C, boron tribromide (3 equivalents per methoxy function) was added dropwise. The reaction mixture was stirred at room temperature. After 4h, water was added to quench the reaction, and ethyl acetate was used to extract the aqueous layer. The combined organic layers were washed with brine, dried over anhydrous MgSO4/Na2SO4, then filtered and concentrated on vacuum. The product was purified by column chromatography (CC).
2,5-Bis(4-methoxyphenyl)thiophene (1a)
Compound 1a was prepared by reaction of 2,5-dibromothiophene and (4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 94–98 °C). 1H NMR (400 MHz, CDCl3) δ 7.57 – 7.51 (m, 4H), 7.15 (s, 2H), 6.95 – 6.89 (m, 4H), 3.84 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 159.12, 144.51,126.84, 122.90, 114.31, 55.38.
2,5-Bis(4-methoxy-2-methylphenyl)thiophene (1b)
Compound 1b was prepared by reaction of 2,5-dibromothiophene and (4-methoxy-2-methylphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 72–74 °C). 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.4 Hz, 2H), 6.86 (s, 2H), 6.72 (d, J = 2.5 Hz, 2H), 6.68 (dd, J = 8.4, 2.6 Hz, 2H), 3.72 (s, 6H), 2.37 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 159.11, 142.33, 137.55, 131.49, 126.87, 126.04, 116.24, 111.29, 55.29, 21.56.
2,5-Bis(4-methoxy-3-methylphenyl)thiophene (1c)
Compound 1c was prepared by reaction of 2,5-dibromothiophene and (4-methoxy-3-methylphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 78–81 °C). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 8.4 Hz, 2H), 6.89 (s, 2H), 6.72 (d, J = 2.5 Hz, 2H), 6.58 (dd, J = 8.4, 2.6 Hz, 2H), 3.82 (s, 6H), 2.29 (s, 6H)..
2,5-Bis(4-methoxy-3,5-dimethylphenyl)thiophene (1d)
Compound 1d was prepared by reaction of 2,5-dibromothiophene and (4-methoxy-3,5-dimethylphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 99–101 °C). 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 4H), 7.16 (s, 2H), 3.74 (s, 6H), 2.32 (s, 12H). 13C NMR (101 MHz, CDCl3) δ 156.63, 142.91, 131.36, 130.04, 126.07, 123.35, 59.83, 16.20.
2,5-Bis(2-fluoro-4-methoxyphenyl)thiophene (1e)
Compound 1e was prepared by reaction of 2,5-dibromothiophene and (2-fluoro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow solid (mp 94–96 °C). 1H NMR (400 MHz, CDCl3) δ 7.55 (t, J = 8.9 Hz, 2H), 7.34 (s, 2H), 6.78 – 6.67 (m, 4H), 3.83 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 160.96, 158.47, 136.41, 129.07, 125.66, 114.84, 110.54, 102.30, 55.68.
2,5-Bis(2-chloro-4-methoxyphenyl)thiophene (1f)
Compound 1f was prepared by reaction of 2,5-dibromothiophene and (2-chloro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as green solid (mp 107–110 °C). 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 8.7 Hz, 2H), 7.26 (s, 2H), 7.03 (d, J = 2.6 Hz, 2H), 6.86 (dd, J = 8.7, 2.6 Hz, 2H), 3.84 (s, 6H).
2,5-Bis(3-fluoro-4-methoxyphenyl)thiophene (1g)
Compound 1g was prepared by reaction of 2,5-dibromothiophene and (3-fluoro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as green solid (mp 92–97 °C). 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 4H), 7.17 (s, 2H), 7.00 (d, J = 2.6 Hz, 2H), 3.95 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 154.46, 141.69, 127.95, 127.38, 124.96, 123.65, 122.95, 112.31, 56.29.
2,5-Bis(3-chloro-4-methoxyphenyl)thiophene (1h)
Compound 1h was prepared by reaction of 2,5-dibromothiophene and (3-chloro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as green solid (mp 108–110°C). 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 2.3 Hz, 2H), 7.46 (dd, J = 8.5, 2.3 Hz, 2H), 7.16 (s, 2H), 6.94 (d, J = 8.6 Hz, 2H), 3.94 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 147.35, 142.37, 142.17, 138.80, 123.65, 121.47, 113.80, 113.26, 56.38.
2,5-Bis(2-fluoro-5-methoxyphenyl)thiophene (1i)
Compound 1i was prepared by reaction of 2,5-dibromothiophene and (2-fluoro-5-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow solid (mp 102–104 °C). 1H NMR (400 MHz, CDCl3) δ 7.38 (s, 2H), 7.06 (dd, J = 6.1, 3.0 Hz, 2H), 7.01 – 6.94 (m, 2H), 6.69 (dt, J = 8.9, 3.4 Hz, 2H), 3.74 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 155.82, 154.92, 152.51, 137.44, 127.16, 122.36, 117.04, 113.94, 55.84.
2,5-Bis(3-fluoro-5-methoxyphenyl)thiophene (1j)
Compound 1j was prepared by reaction of 2,5-dibromothiophene and (3-fluoro-5-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow solid (mp 89–90 °C). 1H NMR (400 MHz, CDCl3) δ 7.18 (s, 2H), 6.84 (dd, J = 7.9, 1.9 Hz, 4H), 6.51 – 6.45 (m, 2H), 3.76 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 159.86, 157.43, 155.99, 137.63, 131.12, 119.54, 102.13, 99.89 (s, 3H), 99.66, 95.60, 50.40.
2,5-Bis(4-fluoro-3-methoxyphenyl)thiophene (1k)
Compound 1k was prepared by reaction of 2,5-dibromothiophene and (4-fluoro-5-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow solid (mp 105–108 °C). 1H NMR (400 MHz, CDCl3) δ 7.19 (m, 3H), 7.17 – 7.12 (m, 3H), 7.08 (dd, J = 10.7, 8.4 Hz, 2H), 3.96 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 153.40, 150.94, 147.91, 142.78, 130.87, 124.06, 118.29, 116.49, 56.35.
2-(2-Fluoro-4-methoxyphenyl)-5-(4-methoxy-2-methylphenyl)thiophene (1l)
Compound 1l was prepared by reaction of compound 19 and (2-fluoro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.54 (t, J = 8.9 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.32 (d, J = 2.5 Hz, 1H), 6.97 (d, J = 3.6 Hz, 1H), 6.82 (d, J = 2.4 Hz, 1H), 6.75 (ddd, J = 20.7, 10.9, 2.5 Hz, 3H), 3.83 (s, 6H), 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.92, 159.89, 159.20, 158.44, 142.34, 137.62, 136.51, 131.48, 129.08, 126.63, 125.17, 116.22, 115.03, 111.28, 110.51, 102.31, 102.05, 55.65, 55.28, 21.46.
2-(2-Chloro-4-methoxyphenyl)-5-(4-methoxy-2-methylphenyl)thiophene (1m)
Compound 1m was prepared by reaction of compound 19 and (2-chloro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.6 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 3.6 Hz, 1H), 6.91 (d, J = 2.5 Hz, 1H), 6.86 (d, J = 3.6 Hz, 1H), 6.72 (dd, J = 8.8, 2.5 Hz, 2H), 6.66 (dd, J = 8.4, 2.5 Hz, 1H), 3.70 (d, J = 2.5 Hz, 6H), 2.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 158.22, 142.21, 138.25, 136.52, 131.78, 130.82, 130.45, 125.95, 125.58, 124.96, 124.71, 115.19, 114.59, 112.22, 110.23, 54.51, 54.18.
2,5-Bis-(4-hydroxyphenyl) thiophene (2a)
Compound 2a was prepared by 2,5-bis-(4-methoxyphenyl) thiophene (1a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as white solid (mp 168–170 °C). 1H NMR (400 MHz, acetone-d6) δ 8.50 (s, 1H), 7.42 – 7.33 (m, 4H), 7.08 (s, 2H), 6.81 – 6.71 (m, 4H). 13C NMR (101 MHz, acetone-d6) δ 158.18, 143.20, 127.51, 126.88, 123.52, 116.67. HRMS (MALDI/DHB) calcd for C16H12O2S (M+H+) 268.0556, found 268.05525.
2,5-Bis(4-hydroxy-2-methylphenyl)thiophene (2b)
Compound 2b was prepared by 2,5-bis(4-methoxy-2-methylphenyl)thiophene (1b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as white solid (mp 172–175 °C). 1H NMR (400 MHz, acetone-d6) δ 7.17 (t, J = 4.6 Hz, 2H), 6.92 (s, 2H), 6.74 (d, J = 12.5 Hz, 2H), 6.69 – 6.60 (m, 2H), 2.06 (d, J = 5.8 Hz, 6H). 13C NMR (101 MHz, acetone-d6) δ 144.04, 136.74, 132.19, 131.69, 128.41, 126.86, 118.76, 115.40, 21.00. HRMS (MALDI/DHB) calcd for C18H16O2S (M+H+) 296.0875, found 296.08655.
2,5-Bis(4-hydroxy-3-methylphenyl)thiophene (2c)
Compound 2b was prepared by 2,5-bis(4- methoxy-3-methylphenyl)thiophene (1c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp187–189 °C). 1H NMR (400 MHz, DMSO-d6) δ 7.38 (s, 2H), 6.95 (d, J = 1.2 Hz, 2H), 6.70 (dd, J = 8.2, 1.8 Hz, 2H), 6.64 (d, J = 8.2 Hz, 2H), 2.06 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 149.05, 142.28, 137.54, 131.46, 126.84, 125.99, 116.18, 111.24, 21.53. HRMS (MALDI/DHB) calcd for C18H16O2S (M+H+) 296.0875, found 296.08655.
2,5-Bis(4-hydroxy-3,5-dimethylphenyl)thiophene (2d)
Compound 2d was prepared by 2,5-bis-(4-methoxy-3,5-dimethylphenyl)thiophene (1d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 201–203 °C). 1H NMR (400 MHz, acetone-d6) δ 7.27 (s, 4H), 7.18 (s, 2H), 2.27 (s, 12H).13C NMR (101 MHz, acetone-d6) δ 153.99, 143.32, 126.96, 126.36, 125.41, 123.27, 16.71. HRMS (MALDI/DHB) calcd for C20H20O2S (M+H+) 324.1184, found 324.11785.
2,5-Bis(2-fluoro-4-hydroxyphenyl)thiophene (2e)
Compound 2e was prepared by 2,5-bis(2-fluoro-4-methoxyphenyl)thiophene (1e) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 178–180 °C). 1H NMR (400 MHz, acetone-d6) δ 7.59 (t, J = 8.9 Hz, 2H), 7.37 (s, 2H), 6.76 (ddd, J = 15.3, 10.8, 2.4 Hz, 4H). 13C NMR (101 MHz, acetone-d6) δ 161.72, 159.29, 137.16, 130.04, 126.03, 114.17, 113.13, 104.20. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0369, found 304.03641.
2,5-Bis(2-chloro-4-hydroxyphenyl)thiophene (2f)
Compound 2f was prepared by 2,5-bis(2-chloro-4-methoxyphenyl)thiophene (1f) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 194–196 °C). 1H NMR (400 MHz, acetone-d6) δ 7.50 (d, J = 8.5 Hz, 2H), 7.28 (s, 2H), 7.04 (d, J = 2.5 Hz, 2H), 6.91 (dd, J = 8.5, 2.5 Hz, 2H).13C NMR (101 MHz, acetone-d6) δ 141.00, 133.08, 133.06, 127.75, 124.89, 117.89, 117.81, 115.81, 115.73. HRMS (MALDI/DHB) calcd for C16H10O2Cl2S (M+H+) 335.9788, found 335.97731.
2,5-Bis(3-fluoro-4-hydroxyphenyl)thiophene (2g)
Compound 2g was prepared by 2,5-Bis(3-fluoro-4-methoxyphenyl)thiophene (1g) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as green solid (mp 187–190 °C). 1H NMR (400 MHz, acetone-d6) δ 7.47 – 7.37 (m, 1H), 7.37 – 7.31 (m, 1H), 7.28 (d, J = 8.3 Hz, 1H), 7.05 (t, J = 8.8 Hz, 1H). 13C NMR (101 MHz, acetone-d6) δ 133.04, 127.60, 124.87, 123.08, 122.51, 118.89, 114.81, 113.50. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0361, found 304.03641.
2,5-Bis(3-chloro-4-hydroxyphenyl)thiophene (2h)
Compound 2h was prepared by 2,5-Bis(3- chloro-4-methoxyphenyl)thiophene (1h) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as green solid (mp 192–195 °C). 1H NMR (400 MHz, DMSO-d6) δ 7.65 (s, 2H), 7.45 (d, J = 8.5 Hz, 2H), 7.39 (s, 2H), 7.02 (d, J = 8.7 Hz, 2H).13C NMR (101 MHz, DMSO-d6) δ 141.03, 138.97, 126.75, 126.48, 125.55, 124.53, 120.76, 117.50. HRMS (MALDI/DHB) calcd for C16H10O2Cl2S (M+H+) 335.9774, found 335.97731.
2,5-Bis(2-fluoro-5-hydroxyphenyl)thiophene (2i)
Compound 2i was prepared by 2,5-bis(2-fluoro-5-methoxyphenyl)thiophene (1i) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as yellow solid (mp 176–179 °C). 1H NMR (400 MHz, acetone-d6) δ 7.39 (s, 2H), 7.07 (dd, J = 6.1, 2.8 Hz, 2H), 7.01 – 6.90 (m, 2H), 6.78 – 6.61 (m, 2H). 13C NMR (101 MHz, acetone-d6) δ 154.81, 152.50, 138.16, 127.77, 122.81, 117.92, 117.68, 114.74. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0361, found 304.03641.
2,5-Bis(3-fluoro-5-hyroxyphenyl)thiophene (2j)
Compound 2j was prepared by 2,5-bis(3-fluoro-5-methoxyphenyl)thiophene (1j) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as yellow solid (mp 212–214 °C). 1H NMR (400 MHz, DMSO-d6) δ 7.55 (s, 2H), 7.01 (d, J = 9.9 Hz, 2H), 6.90 (s, 2H), 6.55 (d, J = 10.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.57, 162.16, 159.35, 141.79, 135.74, 125.81, 108.29, 102.82, 102.59, 102.04, 101.81. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0361, found 304.03641.
2,5-Bis(4-fluoro-3-hydroxyphenyl)thiophene (2k)
Compound 2k was prepared by 2,5-bis(4-fluoro-3-methoxyphenyl)thiophene (1k) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as yellow solid (mp 169–172 °C). 1H NMR (400 MHz, acetone-d6) δ 8.74 (s, 2H), 7.16 (s, 4H), 6.98 (d, J = 5.3 Hz, 4H). 13C NMR (101 MHz, acetone-d6) δ 153.33, 150.93, 146.15, 143.19, 131.96, 125.22, 117.99, 115.71. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0361, found 304.03641.
2-(2-Fluoro-4-hydroxyphenyl)-5-(4-hydroxy-2-methylphenyl)thiophene (2l)
Compound 2l was prepared by 2-(2-fluoro-4-methoxyphenyl)-5-(4-methoxy-2-methylphenyl) thiophene (1l) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as yellow solid (mp 151–153 °C). 1H NMR (400 MHz, acetone-d6) δ 7.57 (t, J = 8.9 Hz, 1H), 7.34 (d, J = 3.7 Hz, 1H), 7.27 (d, J = 8.3 Hz, 1H), 7.02 (d, J = 3.7 Hz, 1H), 6.84 – 6.68 (m, 4H), 2.38 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 161.67, 159.21, 158.11, 143.23, 138.12, 137.10, 132.17, 130.05, 127.22, 126.67 – 125.17, 118.32, 113.89, 113.04, 104.25, 104.00, 21.42. HRMS (MALDI/DHB) calcd for C17H13O2FS (M+H+) 300.0607, found 300.06148.
2-(2-Chloro-4-hydroxyphenyl)-5-(4-hydroxy-2-methylphenyl)thiophene (2m)
Compound 2m was prepared by 2-(2-chloro-4-methoxyphenyl)-5-(4-methoxy-2-methylphenyl) thiophene (1m) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 167–169 °C). 1H NMR (400 MHz, acetone-d6) δ 7.49 (d, J = 8.5 Hz, 1H), 7.27 (dd, J = 7.4, 6.2 Hz, 2H), 7.02 (d, J = 3.5 Hz, 2H), 6.90 (dd, J = 8.5, 2.5 Hz, 1H), 6.81 (d, J = 1.7 Hz, 1H), 6.75 (d, J = 8.3 Hz, 1H), 2.39 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 158.65, 158.19, 144.22, 139.94, 138.12, 133.02, 132.20, 127.74, 126.74, 125.99, 125.17, 124.44, 118.36, 117.83, 115.73, 113.93, 21.44. HRMS (MALDI/DHB) calcd for C17H13O2ClS (M + H+) 316.0325, found 316.03193.
3,4-Bis(4-methoxyphenyl)thiophene (3a)
Compound 3a was prepared by reaction of 3,4- dibromothiophene and (4-methoxyphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 105–106 °C). 1H NMR (400 MHz, CDCl3) δ 7.23 (s, 2H), 7.12 (d, J = 8.8 Hz, 4H), 6.80 (d, J = 8.8 Hz, 4H), 3.80 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 158.57, 141.32, 130.10, 129.22, 123.08, 113.57, 55.22.
3,4-Bis(4-methoxy-2-methylphenyl)thiophene (3b)
Compound 3b was prepared by reaction of 3,4-dibromothiophene and (4-methoxy-2-methylphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 72–76 °C). 1H NMR (400 MHz, CDCl3) δ 7.15 (s, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.64–6.58 (m, 4H), 3.75 (s, 6H), 2.03 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 158.46, 141.98, 137.67, 131.61, 128.98, 123.26, 115.26, 110.62, 55.08, 20.52.
3,4-Bis(4-methoxy-3-methylphenyl)thiophene (3c)
Compound 3c was prepared by reaction of 3,4-dibromothiophene and (4-methoxy-3-methylphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 77–78 °C). 1H NMR (400 MHz, CDCl3) δ 7.21 (s, 2H), 7.05 (s, 2H), 6.93 (dd, J = 8.4, 2.1 Hz, 2H), 6.69 (d, J = 8.4 Hz, 2H), 3.81 (s, 6H), 2.16 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 154.56, 139.23, 129.02, 125.14, 124.46, 120.58, 107.17, 53.00, 13.98.
3,4-Bis(4-methoxy-3,5-dimethylphenyl)thiophene (3d)
Compound 3d was prepared by reaction of 3,4-dibromothiophene and (4-methoxy-3,5-dimethylphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 92–96 °C). 1H NMR (400 MHz, CDCl3) δ 7.15 (s, 2H), 6.76 (s, 4H), 3.64 (s, 6H), 2.12 (s, 12H). 13C NMR (101 MHz, CDCl3) δ 156.01, 141.45, 132.13, 130.21, 129.49, 123.08, 59.76, 16.01.
3,4-Bis(2-fluoro-4-methoxyphenyl)thiophene (3e)
Compound 3e was prepared by reaction of 3,4-dibromothiophene and (2-fluoro-4-methoxyphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 88–90 °C). 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 2H), 6.94 (t, J = 8.7 Hz, 2H), 6.54–6.47 (m, 4H), 3.71 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 161.47, 159.01, 135.52, 131.57, 124.49, 109.65, 101.81, 101.55, 55.49.
3,4-Bis(2-chloro-4-methoxyphenyl)thiophene (3f)
Compound 3f was prepared by reaction of 3,4-dibromothiophene and (2-chloro-4-methoxyphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 121–125 °C). 1H NMR (400 MHz, CDCl3) δ 7.24 (s, 2H), 6.92 (d, J = 8.5 Hz, 2H), 6.80 (d, J = 2.6 Hz, 2H), 6.59 (dd, J = 8.6, 2.6 Hz, 2H), 3.69 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 159.15, 139.18, 134.06, 132.37, 127.60, 124.54, 114.66, 112.49, 55.43.
3,4-Bis(4-hydroxyphenyl)thiophene (4a)
Compound 4a was prepared by 3,4-bis(4-methoxyphenyl) thiophene (3a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as white solid (mp 198–201 °C). 1H NMR (400 MHz, acetone-d6) δ 8.49 (s, 2H), 7.33 (s, 2H), 7.03 (d, J = 8.2 Hz, 4H), 6.75 (d, J = 8.1 Hz, 4H). 13C NMR (101 MHz, acetone-d6) δ 157.45, 142.46, 130.87, 129.04, 123.63, 115.86. HRMS (MALDI/DHB) calcd for C16H12O2S (M+H+) 268.0563, found 268.05525.
3,4-Bis(4-hydroxy-2-methylphenyl)thiophene (4b)
Compound 4b was prepared by 3,4-bis(4- methoxy-2-methylphenyl)thiophene (3b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as white solid (mp 214–215 °C). 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 2H), 7.41 (s, 2H), 6.80 (d, J = 8.2 Hz, 2H), 6.57 (s, 2H), 6.50 (d, J = 8.2 Hz, 2H), 1.98 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 156.09, 141.68, 136.75, 131.19, 126.97, 123.24, 116.38, 112.20, 20.00. HRMS (MALDI/DHB) calcd for C18H16O2S (M+H+) 296.0875, found 296.08655.
3,4-Bis(4-hydroxy-3-methylphenyl)thiophene (4c)
Compound 4c was prepared by 3,4-bis(4- methoxy-3-methylphenyl)thiophene (3c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as white solid (mp 183–184 °C). 1H NMR (400 MHz, acetone-d6) δ 8.38 (s, 1H), 7.30 (s, 2H), 7.01 (s, 2H), 6.78 (d, J = 8.1 Hz, 2H), 6.70 (d, J = 8.2 Hz, 2H), 2.13 (s, 6H). 13C NMR (101 MHz, acetone-d6) δ 155.40, 142.64, 132.15, 129.07, 128.18, 124.67, 123.35, 115.02, 16.23. HRMS (MALDI/DHB) calcd for C18H16O2S (M+H+) 296.0875, found 296.08655.
3,4-Bis(4-hydroxy-3,5-dimethylphenyl)thiophene (4d)
Compound 4d was prepared by 3,4-bis(4- methoxy-3,5-dimethylphenyl)thiophene (3d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 193–194 °C). 1H NMR (400 MHz, acetone-d6) δ 7.29 (s, 2H), 6.82 (s, 4H), 2.14 (s, 12H). HRMS (MALDI/DHB) calcd for C20H20O2S (M+H+) 324.1184, found 324.11785.
3,4-Bis(2-fluoro-4-hydroxyphenyl)thiophene (4e)
Compound 4e was prepared by 3,4-bis(2-fluoro-4-methoxyphenyl)thiophene (3e) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 4:1) gave the title compound as white solid (mp 207–209 °C). 1H NMR (400 MHz, acetone-d6) δ 8.91 (s, 1H), 7.45 (s, 2H), 6.97 (t, J = 8.6 Hz, 2H), 6.57 (ddd, J = 14.1, 10.0, 2.4 Hz, 4H). 13C NMR (101 MHz, acetone-d6) δ 162.32, 159.09, 136.91, 132.62, 125.16, 116.35, 111.98, 103.59. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0378, found 304.03641.
3,4-Bis(2-chloro-4-hydroxyphenyl)thiophene (4f)
Compound 4f was prepared by 3,4-bis(2-chloro-4-methoxyphenyl)thiophene (3f) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 4:1) gave the title compound as white solid (mp 179–183 °C). 1H NMR (400 MHz, acetone-d6) δ 7.42 (s, 2H), 6.98 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 2.5 Hz, 2H), 6.69 (dd, J = 8.4, 2.5 Hz, 2H). 13C NMR (101 MHz, acetone-d6) δ 158.20, 140.43, 134.40, 133.54, 127.38, 125.26, 116.85, 114.65. HRMS (MALDI/DHB) calcd for C16H10O2Cl2S (M+H+) 335.9861, found 335.98513.
2,4-Bis(2-fluoro-4-methoxyphenyl)thiophene (5a)
Compound 5a was prepared by reaction of 2,4- dibromothiophene and (2-fluro-4-methoxyphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 88–90 °C). 1H NMR (400 MHz, CDCl3) δ 7.54 – 7.34 (m, 4H), 6.68 – 6.57 (m, 4H), 3.72 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 161.64, 161.01, 160.06, 159.17, 158.52, 137.12, 136.01, 129.78, 129.30, 125.32, 121.45, 116.33, 114.82, 110.55, 110.27, 102.30, 55.64, 55.59.
2,4-Bis(2-chloro-4-methoxyphenyl)thiophene (5b)
Compound 5b was prepared by reaction of 2,4-dibromothiophene and (2-chloro-4-methoxyphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 135–138 °C). 1H NMR (400 MHz, CDCl3) δ 7.47 (dd, J = 5.0, 3.6 Hz, 2H), 7.38 (dd, J = 5.0, 3.6 Hz, 2H), 7.02 (dd, J = 4.3, 2.6 Hz, 2H), 6.85 (ddd, J = 8.7, 2.6, 0.6 Hz, 2H), 3.83 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 159.50, 159.26, 139.40, 139.02, 133.04, 132.93, 132.06, 131.64, 128.93, 127.81, 125.57, 123.51, 115.57, 115.34, 113.29, 113.15, 55.64, 55.61.
2,4-Bis(2-fluoro-4-hydroxyphenyl)thiophene (6a)
Compound 6a was prepared by 2,4-bis (2-fluoro-4-methoxyphenyl)thiophene (5a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 190–194 °C). 1H NMR (400 MHz, acetone-d6) δ 9.26 (s, 1H), 9.12 (s, 1H), 7.62 – 7.60 (m, 4H), 6.75 – 6.73 (m, 4H). 13C NMR (101 MHz, acetone-d6) δ 161.79, 159.99, 159.33, 139.61, 138.21, 137.06, 131.00, 130.37, 125.81, 121.85, 114.25, 113.08, 112.74, 110.89, 110.09, 103.99. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0373, found 304.03641.
2,4-Bis(2-chloro-4-hydroxyphenyl)thiophene (6b)
Compound 6b was prepared by 2,4-bis(2-chloro-4-methoxyphenyl)thiophene (5b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 163–165 °C). 1H NMR (400 MHz, acetone-d6) δ 7.56 – 7.47 (m, 3H), 7.41 (d, J = 8.5 Hz, 1H), 7.03 (dd, J = 11.6, 2.5 Hz, 2H), 6.90 (td, J = 8.3, 2.5 Hz, 2H). 13C NMR (101 MHz, acetone-d6) δ 158.83, 158.72, 158.45, 158.34, 140.33, 140.08, 133.15, 132.88, 129.55, 127.36, 125.07, 124.20, 117.85, 117.55, 115.64, 115.48. HRMS (MALDI/DHB) calcd for C16H10O2Cl2S (M+H+) 335.9784, found 335.97731.
2,3-Bis(2-fluoro-4-methoxyphenyl)thiophene (7a)
Compound 7a was prepared by reaction of 2,3-dibromothiophene and (2-fluoro-4-methoxyphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 76–78 °C). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 5.2 Hz, 1H), 7.11 – 7.04 (m, 2H), 6.95 (t, J = 8.5 Hz, 1H), 6.58 – 6.49 (m, 4H), 3.72 (s, 3H), 3.71 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.79, 159.15, 137.60, 136.71, 135.01, 133.94, 133.58, 132.40, 129.86, 127.62, 125.01, 124.18, 114.80, 112.72, 55.46.
2,3-Bis(2-chloro-4-methoxyphenyl)thiophene (7b)
Compound 7b was prepared by reaction of 2,3-dibromothiophene and (2-chloro-4-methoxyphenyl)boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 115–117 °C). 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 5.2 Hz, 1H), 7.08 (dd, J = 6.9, 1.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 1H), 6.83 (dd, J = 11.1, 2.6 Hz, 2H), 6.63 (dd, J = 8.6, 2.6 Hz, 1H), 6.57 (dd, J = 8.6, 2.6 Hz, 1H), 3.70 (s, 3H), 3.69 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.42, 140.20, 132.90, 131.97, 127.02, 125.52, 115.61, 113.30, 55.63.
2,3-Bis(2-fluoro-4-hydroxyphenyl)thiophene (8a)
Compound 8a was prepared by 2,3-bis (2-fluoro-4-methoxyphenyl)thiophene (7a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 158–162 °C). 1H NMR (400 MHz, acetone-d6) δ 7.54 (d, J = 5.2 Hz, 1H), 7.15 (dd, J = 5.2, 1.7 Hz, 1H), 7.09 (t, J = 8.6 Hz, 1H), 6.98 (t, J = 8.5 Hz, 1H), 6.67 – 6.54 (m, 4H). 13C NMR (101 MHz, acetone-d6) 166.73, 162.32, 160.05, 134.63, 133.66, 133.40, 132.91, 132.53, 131.23, 130.84, 130.16, 128.90, 125.51, 112.42, 103.96. HRMS (MALDI/DHB) calcd for C16H10O2F2S (M+H+) 304.0377, found 304.03641.
2,3-Bis(2-chloro-4-hydroxyphenyl)thiophene (8b)
Compound 8b was prepared by 2,3-bis (2-chloro-4-methoxyphenyl)thiophene (7b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as white solid (mp 160–163 °C). 1H NMR (400 MHz, acetone-d6) δ 9.16 (s, 1H), 8.98 (s, 1H), 7.53 (d, J = 5.2 Hz, 1H), 7.19 – 7.09 (m, 2H), 6.98 – 6.86 (m, 3H), 6.74 (dd, J = 8.4, 2.4 Hz, 1H), 6.66 (dd, J = 8.4, 2.4 Hz, 1H). 13C NMR (101 MHz, acetone-d6) δ159.09, 158.33, 135.31, 134.71, 134.29, 133.47, 130.69, 125.10, 124.58, 117.30,116.86, 114.95. HRMS (MALDI/DHB) calcd for C16H10O2Cl2S (M+H+) 335.9769, found 335.97731.
2,5-bis(4-methoxyphenyl)-3-methylthiophene (9a)
Compound 9a was prepared by reaction of 3-methyl-2,5-dibromothiophene and 4-methoxy-phenyl boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 117–120 °C). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.41 (m, 2H), 7.36 – 7.31 (m, 2H), 6.94 (s, 1H), 6.89 – 6.85 (m, 2H), 6.84 – 6.80 (m, 2H), 3.76 (s, 3H), 3.74 (s, 3H), 2.22 (s, 3H).
2,5-bis(4-methoxy-2-methylphenyl)-3-methylthiophene (9b)
Compound 9b was prepared by reaction of 3-methyl-2,5-dibromothiophene and (2-methyl-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 112–114 °C). 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.4 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 6.78 – 6.66 (m, 5H), 3.77 (s, 3H), 3.75 (s, 3H), 2.39 (s, 3H), 2.18 (s, 3H), 1.97 (s, 3H).
2,5-Bis(2-fluoro-4-methoxyphenyl)-3-methylthiophene (9c)
Compound 9c was prepared by reaction of 3-methyl-2,5-dibromothiophene and (2-fluoro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 90–94 °C). 1H NMR (400 MHz, CDCl3) δ 7.49 (t, J = 8.9 Hz, 1H), 7.30 (t, J = 8.7 Hz, 1H), 7.19 (d, J = 1.1 Hz, 1H), 6.78 – 6.64 (m, 4H), 3.80 (d, J = 8.5 Hz, 6H), 2.18 (d, J = 1.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.64, 161.04, 159.94, 159.17, 158.46, 136.06, 132.61, 130.08, 129.02, 128.38, 114.86, 114.11, 110.49, 110.05, 102.08, 55.64, 55.62, 14.89.
2,5-Bis(2-chloro-4-methoxyphenyl)-3-methylthiophene (9d)
Compound 9d was prepared by reaction of 3-methyl-2,5-dibromothiophene and (2-chloro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 122–124 °C). 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.7 Hz, 1H), 7.32 (d, J = 8.5 Hz, 1H), 7.13 (s, 1H), 7.03 (d, J = 2.6 Hz, 1H), 7.00 (d, J = 2.6 Hz, 1H), 6.85 (dd, J = 8.1, 2.6 Hz, 2H), 3.83 (s, 3H), 3.81 (s, 3H), 2.11 (s, 3H).
2,5-Bis(4-hydroxyphenyl)-3-methylthiophene (10a)
Compound 10a was prepared by 2,5-bis (4-methoxyphenyl)-3-methylthiophene (9a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as white solid (mp 160–163 °C). 1H NMR (400 MHz, acetone-d6) δ 8.69 (s, 1H), 8.67 (s, 1H), 7.52 – 7.46 (m, 2H), 7.38 – 7.32 (m, 2H), 7.10 (s, 1H), 6.89 (dt, J = 4.8, 2.9 Hz, 4H), 2.27 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 157.79, 141.78, 136.80, 133.86, 130.76, 128.23, 116.77, 15.25. HRMS (MALDI/DHB) calcd for C17H14O2S (M+H+) 282.1027, found 282.10260.
2,5-Bis(4-hydroxy-2-methylphenyl)-3-methylthiophene (10b)
Compound 10b was prepared by 2,5-bis (4-methoxy-2-methylphenyl)-3-methylthiophene (9b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate =3:1) gave the title compound as brown solid (mp 158–163 °C). 1H NMR (400 MHz, acetone-d6) δ 8.68 (d, J = 9.1 Hz, 1H), 8.46 (s, 1H), 7.53–7.32 (m, 4H), 7.10 (s, 1H), 6.99–6.84 (m, 4H), 2.40 (s, 3H), 2.18 (s, 3H), 2.02 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 158.34, 157.77, 141.05, 139.24, 135.80, 137.90, 34.94, 133.15, 132.04, 129.23, 126.42, 124.59, 15.59, 117.66, 117.27, 113.97, 113.63, 29.91, 20.29, 14.49, 14.58. HRMS (MALDI/DHB) calcd for C19H18O2S (M+H+) 310.1027, found 310.10220.
2,5-Bis(2-fluoro-4-hydroxyphenyl)-3-methylthiophene (10c)
Compound 10c was prepared by 2,5-bis(2-fluoro-4-methoxyphenyl)-3-methylthiophene (9c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 162–166 °C). 1H NMR (400 MHz, acetone-d6) δ 9.20 (d, J = 9.8 Hz, 2H), 7.55 (t, J = 8.9 Hz, 1H), 7.27 (dd, J = 19.0, 10.4 Hz, 2H), 6.84–6.67 (m, 4H), 2.16 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 162.46, 161.70, 160.00, 159.43, 159.07, 136.57, 136.31, 133.56, 130.86, 129.97, 128.71, 114.22, 113.53, 112.59, 104.42, 103.46, 14.89. HRMS (MALDI/DHB) calcd for C17H12O2SF2 (M+H+) 318.0527, found 318.05206.
2,5-Bis(2-chloro-4-hydroxyphenyl)-3-methylthiophene (10d)
Compound 10d was prepared by 2,5-bis(2-chloro-4-methoxyphenyl)-3-methylthiophene (9d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as yellow solid (mp 112–116 °C). 1H NMR (400 MHz, acetone-d6) δ 9.13 (s, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.16 (s, 1H), 7.03 (dd, J = 11.0, 2.5 Hz, 2H), 6.90 (ddd, J = 8.7, 6.5, 2.5 Hz, 2H), 2.09 (s, 3H). 13C NMR (101 MHz, acetone-d6) δ 159.34, 158.60, 139.44, 136.23, 135.54, 135.26, 134.49, 133.32, 132.89, 130.22, 125.08, 124.40, 117.89, 117.25, 115.76, 115.25, 14.775. HRMS (MALDI/DHB) calcd for C17H12O2SCl2 (M+H+) 349.9938, found 349.99296.
2,5-Bis(2-fluoro-4-methoxyphenyl)-3-phenylthiophene (11a)
Compound 11a was prepared by reaction of 3-bromo-2,5-bis(2-fluoro-4-methoxyphenyl)thiophene (21) and phenylboronic acid according to the general procedure for Suzuki coupling using Pd(dppf)Cl2. Purification by CC (petroleum ether: ethyl acetate = 95:5) gave the title compound as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.48 (t, J = 8.9 Hz, 1H), 7.38 (s, 1H), 7.25 – 7.09 (m, 6H), 6.67 (d, J = 7.1 Hz, 2H), 6.55 (t, J = 9.7 Hz, 2H), 3.76 (s, 3H), 3.72 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.91, 159.22, 157.52, 139.37, 135.70, 131.79, 128.03, 127.31, 126.38, 125.84, 109.54, 109.09, 101.22, 101.11, 101.13, 54.58.
2,5-Bis(2-chloro-4-methoxyphenyl)-3-phenylthiophene (11b)
Compound 11b was prepared by reaction of 3-bromo-2,5-bis(2-chloro-4-methoxyphenyl)thiophene (22) and phenylboronic acid according to the general procedure for Suzuki coupling using Pd(dppf)Cl2. Purification by CC (petroleum ether: ethyl acetate = 95:5) gave the title compound as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.6 Hz, 1H), 7.34 (s, 1H), 7.15 (td, J = 12.6, 6.4 Hz, 6H), 6.94 (d, J = 2.5 Hz, 1H), 6.87 (d, J = 2.4 Hz, 1H), 6.76 (dd, J = 8.6, 2.5 Hz, 1H), 6.67 (dd, J = 8.6, 2.4 Hz, 1H), 3.73 (s, 3H), 3.70 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.13, 159.58, 140.12, 139.53, 136.55, 135.31, 134.83, 133.78, 132.92, 131.93, 128.60, 128.34, 127.06, 126.81, 125.37, 115.77, 115.23, 113.37, 113.01, 55.62.
2,5-Bis(2-fluoro-4-hydroxyphenyl)-3-phenylthiophene (12a)
Compound 12a was 2,5-bis(2-fluoro-4-methoxyphenyl)-3-phenylthiophene (11a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 2:1) gave the title compound as green solid (mp 142–145 °C). 1H NMR (400 MHz, acetone-d6) δ 9.22 (s, 1H), 7.67 (t, J = 8.9 Hz, 1H), 7.53 (s, 1H), 7.36 – 7.16 (m, 6H), 6.83 – 6.67 (m, 3H), 6.62 (dd, J = 11.6, 2.4 Hz, 1H). 13C NMR (101 MHz, acetone-d6) δ 159.19, 158.81, 158.32, 139.33, 138.51, 137.36, 135.25, 134.74, 134.27, 133.51, 132.95, 130.34, 127.25, 124.77, 124.33, 117.96, 117.16, 115.86, 115.02. HRMS (MALDI/DHB) calcd for C22H14O2SF2 (M+H+) 380.0676, found 380.06771.
2,5-Bis(2-chloro-4-hydroxyphenyl)-3-phenylthiophene (12b)
Compound 12b was 2,5-bis(2- chloro-4-methoxyphenyl)-3-phenylthiophene (11b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 2:1) gave the title compound as yellow solid (mp 152–154 °C). 1H NMR (400 MHz, acetone-d6) δ 7.58 (d, J = 8.5 Hz, 1H), 7.48 (s, 1H), 7.32 – 7.21 (m, 6H), 7.06 (d, J = 2.4 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.92 (dd, J = 8.7, 2.4 Hz, 1H), 6.84 (dd, J = 8.4, 2.4 Hz, 1H). 13C NMR (101 MHz, acetone-d6) δ158.83, 140.77, 135.61, 134.79, 133.17,132.89, 129.29,128.85, 127.72, 124.63, 117.87, 117.31, 115.78, 115.34. HRMS (MALDI/DHB) calcd for C22H14O2SCl2 (M+H+) 412.0103, found 412.00861.
2,3,5-Tris(4-methoxyphenyl)thiophene (13a)
Compound 13a was prepared by reaction of 2,3,5-tribromothiophene and 4-methoxy-phenyl boronic acid according to the general procedure for Suzuki coupling using Pd(dppf)Cl2. Purification by CC (petroleum ether: ethyl acetate = 95:5) gave the title compound as red solid (mp 142–145 °C). 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 8.7 Hz, 2H), 7.18 (dd, J = 8.1, 2.1 Hz, 2H), 7.13 (s, 1H), 6.85 (d, J = 8.7 Hz, 2H), 6.75 (dd, J = 11.4, 8.7 Hz, 4H), 3.77 (s, 3H), 3.74 (s, 3H), 3.73 (s, 3H).
2,3,5-Tris(2-fluoro-4-methoxyphenyl)thiophene (13b)
Compound 13b was prepared by reaction of 2,3,5-tribromothiophene and (2-fluoro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(dppf)Cl2. Purification by CC (petroleum ether: ethyl acetate = 95:5) gave the title compound as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.65 (dd, J = 9.3, 4.1 Hz, 1H), 7.34 (s, 1H), 7.10 (t, J = 8.7 Hz, 1H), 7.01 (t, J = 8.8 Hz, 1H), 6.71 – 6.61 (m, 2H), 6.54 (td, J = 10.0, 2.5 Hz, 4H), 3.76 (s, 3H), 3.72 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 161.52, 161.01, 160.66, 160.05, 159.05, 158.53, 136.25, 133.87, 132.29, 131.61, 129.08, 127.92, 116.81, 114.57, 114.08, 110.57, 109.96, 102.31, 101.85, 55.80 – 55.40.
2,3,5-Tris(2-chloro-4-methoxyphenyl)thiophene (13c)
Compound 13c was prepared by reaction of 2,3,5-tribromothiophene and (2-chloro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(dppf)Cl2. Purification by CC (petroleum ether: ethyl acetate = 95:5) gave the title compound as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 8.7 Hz, 1H), 7.37 (s, 1H), 7.19 (d, J = 8.6 Hz, 1H), 7.05 – 6.98 (m, 2H), 6.91 (dd, J = 11.9, 2.5 Hz, 2H), 6.85 (dd, J = 8.7, 2.6 Hz, 1H), 6.70 (dd, J = 8.6, 2.5 Hz, 1H), 6.64 (dd, J = 8.6, 2.5 Hz, 1H), 3.82 (s, 3H), 3.76 (d, J = 3.3 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.84, 159.44, 159.22, 138.59, 137.42, 136.63, 134.95, 133.97, 133.62, 132.84, 132.44, 131.82, 129.69, 127.76, 125.42, 124.94, 115.71, 114.94, 113.34, 112.74, 55.71.
2,3,5-Tris(4-hydroxyphenyl)thiophene (14a)
Compound 14a was prepared by 2,3,5-tris (4-methoxyphenyl)thiophene (13a) and boron tribromide according to the general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 2:1) gave the title compound as brown solid (mp 285–288 °C). 1H NMR (400 MHz, acetone-d6) δ 8.84 (s, 1H), 8.69 (s, 1H), 8.62 (s, 1H), 7.60 – 7.54 (m, 2H), 7.15 – 7.05 (m, 2H), 7.01 – 6.95 (m, 2H), 6.91 – 6.85 (m, 2H), 6.78 – 6.73 (m, 2H). 13C NMR (101 MHz, acetone-d6) δ 158.17, 138.99, 136.74, 136.43, 132.74, 131.35, 130.77, 128.07, 125.90, 125.39, 125.30, 116.66, 116.44, 116.19, 115.62, 111.18. HRMS (MALDI/DHB) calcd for C22H16O3S (M+H+) 360.0819, found 360.08147.
2,3,5-Tris(2-fluoro-4-hydroxyphenyl)thiophene (14b)
Compound 14b was prepared by 2,3,5-tris(2-fluoro-4-methoxyphenyl)thiophene (13b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 2:1) gave the title compound as brown solid (mp 232–238 °C). 1H NMR (400 MHz, acetone-d6) δ 9.21 (s, 1H), 9.14 (s, 1H), 8.98 (s, 1H), 7.63 (t, J = 8.9 Hz, 1H), 7.41 (s, 1H), 7.14 (t, J = 8.6 Hz, 1H), 7.05 (t, J = 8.7 Hz, 1H), 6.77 (ddd, J = 15.3, 10.7, 2.3 Hz, 2H), 6.70 – 6.57 (m, 4H). 13C NMR (101 MHz, acetone-d6) δ 162.36, 159.92, 158.23, 136.85, 135.35, 135.01, 134.97, 133.32, 132.58, 130.04, 128.31, 125,57, 124.45, 120.12, 113.10, 112.56, 112.50, 112.33, 104.28, 104.04. HRMS (MALDI/DHB) calcd for C22H13O3SF3 (M+H+) 414.0541, found 414.05320.
2,3,5-Tris(2-chloro-4-hydroxyphenyl)thiophene (14c)
Compound 14c was prepared by 2,3,5-tris(2-chloro-4-methoxyphenyl)thiophene (13c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 2:1) gave the title compound as yellow solid (mp 185–187 °C). 1H NMR (400 MHz, acetone-d6) δ 9.17 (s, 1H), 9.09 (s, 1H), 8.91 (s, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.36 (s, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 2.5 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.95 – 6.89 (m, 3H), 6.77 (dd, J = 8.4, 2.4 Hz, 1H), 6.68 (dd, J = 8.4, 2.4 Hz, 1H). 13C NMR (101 MHz, acetone-d6) δ 159.19, 159.07, 158.81, 158.44, 158.32, 138.51, 135.25, 134.27, 132.95, 127.25, 124.33, 118.00, 117.91, 117.25, 117.16, 117.07, 115.90, 115.86, 115.16, 115.07, 114.97, 114.88. HRMS (MALDI/DHB) calcd for C22H13O3SCl3 (M+H+) 461.9662, found 461.96455.
2,5-Bis(4-methoxyphenyl)furan (15a)
Compound 15a was prepared by reaction of 2,5-dibromofuran and 4-methoxy-phenyl boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 113–114 °C). 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 8.8 Hz, 4H), 6.86 (t, J = 5.8 Hz, 4H), 6.50 (s, 2H), 3.77 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 158.9, 152.8, 125.0, 124.0, 114.1, 105.6, 55.3.
2,5-Bis(4-methoxy-2-methylphenyl)furan (15b)
Compound 15b was prepared by reaction of 2,5-dibromofuran and (4-methoxy-2-methylphenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 106–108 °C). 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.4 Hz, 2H), 6.74 (d, J = 9.6 Hz, 4H), 6.45 (s, 2H), 3.76 (s, 2H), 2.46 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 158.73, 152.21, 136.15, 128.32, 123.43, 116.50, 111.48, 108.98, 55.26, 22.21.
2,5-Bis(2-fluoro-4-methoxyphenyl)furan (15c)
Compound 15c was prepared by reaction of 2,5-dibromofuran and (2-fluoro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 95–97 °C). 1H NMR (400 MHz, CDCl3) δ 7.71 (t, J = 8.8 Hz, 2H), 6.74 – 6.65 (m, 2H), 6.61 (dd, J = 12.9, 2.4 Hz, 2H), 3.74 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.66, 159.79, 158.17, 146.90, 126.53, 112.06, 110.37, 102.19, 101.94, 55.63.
2,5-Bis(2-chloro-4-methoxyphenyl)furan (15d)
Compound 15d was prepared by reaction of 3,4-dibromofuran and (2-chloro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 112–114 °C). 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.8 Hz, 2H), 7.07 (s, 2H), 7.00 (d, J = 2.6 Hz, 2H), 6.90 (dd, J = 8.8, 2.6 Hz, 2H), 3.84 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 158.99, 149.23, 131.01, 128.84, 122.17, 115.72, 113.46, 111.26, 55.59.
2,5-Bis(4-hydroxyphenyl)furan (16a)
Compound 16a was prepared by 2,5-bis(4-methoxyphenyl)furan (15a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as brown solid (mp 176–177 °C). 1H NMR (400 MHz, acetone-d6) δ 8.64 (s, 2H), 7.64 (d, J = 8.6 Hz, 4H), 6.90 (d, J = 8.7 Hz, 4H), 6.69 (s, 2H). 13C NMR (101 MHz, acetone-d6) δ 159.18, 145.20, 128.61, 126.88, 124.32, 116.68. HRMS (MALDI/DHB) calcd for C16H12O3 (M+H+) 252.0783, found 252.07810.
2,5-Bis(4-hydroxy-2-methylphenyl)furan (16b)
Compound 16b was prepared by 2,5-Bis(4- methoxy-2-methylphenyl)furan (15b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 4:1) gave the title compound as brown solid (mp 184–187 °C). 1H NMR (400 MHz, acetone-d6) δ 6.86 – 6.78 (m, 4H), 6.65 (d, J = 2.2 Hz, 2H), 6.55 (dd, J = 8.4, 2.4 Hz, 2H), 2.10 (s, 6H). 13C NMR (101 MHz, acetone-d6) δ 159.04, 146.02, 138.60, 132.18, 128.93, 123.75, 117.98, 113.29, 20.40. HRMS (MALDI/DHB) calcd for C18H16O3 (M+H+) 280.1091, found 280.10940.
2,5-Bis(2-fluoro-4-hydroxyphenyl)furan (16c)
Compound 16c was prepared by 2,5-bis(2-fluoro-4-methoxyphenyl)furan (15c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 4:1) gave the title compound as brown solid (mp 219–222 °C). 1H NMR (400 MHz, acetone-d6) δ 9.16 (s, 1H), 7.81 (t, J = 8.8 Hz, 2H), 6.81 (dd, J = 8.6, 2.3 Hz, 2H), 6.75 (dd, J = 5.8, 2.5 Hz, 3H), 6.71 (d, J = 2.3 Hz, 1H). 13C NMR (101 MHz, acetone-d6) δ 161.52, 159.43, 147.91, 127.66, 112.87, 111.38, 110.53, 104.23, 103.99. HRMS (MALDI/DHB) calcd for C16H16O3F2 (M+H+) 288.0595, found 288.05925.
2,5-Bis(2-chloro-4-hydroxyphenyl)furan (16d)
Compound 16d was prepared by 2,5-bis(2-chloro-4-methoxyphenyl)furan (15d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as yellow solid (mp 194–198 °C). 1H NMR (400 MHz, acetone-d6) δ 7.70 (d, J = 8.7 Hz, 2H), 6.91 (s, 2H), 6.88 (d, J = 2.5 Hz, 2H), 6.80 (dd, J = 8.7, 2.5 Hz, 2H). 13C NMR (101 MHz, acetone-d6) δ 158.36, 150.23, 131.34, 130.08, 121.62, 118.03, 115.77, 111.54. HRMS (MALDI/DHB) calcd for C16H16O3Cl2 (M+H+) 320.0003, found 320.00015.
3,4-Bis(4-methoxyphenyl)furan (17a)
Compound 17a was prepared by reaction of 3,4- dibromofuran and 4-methoxy-phenyl boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate =98:2) gave the title compound as white solid (mp 144–148 °C).1H NMR (400 MHz, CDCl3) δ 7.42 (s, 0H), 7.08 (d, J = 8.8 Hz, 1H), 6.76 (d, J = 8.8 Hz, 1H), 3.73 (s, 6H).
3,4-Bis(4-methoxy-2-methylphenyl)furan (17b)
Compound 17b was prepared by reaction of 3,4- dibromofuran and (4-methoxy-2-methylphenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 102–106 °C).1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 1.7 Hz, 1H), 7.33 (d, J = 1.7 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 6.82 (d, J = 2.4 Hz, 2H), 6.77 (dd, J = 8.4, 2.5 Hz, 2H), 3.82 (s, 6H), 2.24 (s, 5H).
3,4-Bis(2-fluoro-4-methoxyphenyl)furan (17c)
Compound 17c was prepared by reaction of 3,4-dibromofuran and (2-fluoro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 117–119 °C). 1H NMR (400 MHz, acetone-d6) δ 7.75 (s, 2H), 7.10 (t, J = 8.8 Hz, 2H), 6.78 – 6.68 (m, 4H), 3.82 (s, 6H). 13C NMR (101 MHz, acetone-d6) δ 162.57, 161.43, 160.13, 142.53, 132.02, 120.55, 110.92, 102.63, 102.37, 56.00.
3,4-Bis(2-chloro-4-methoxyphenyl)furan (17d)
Compound 17d was prepared by reaction of 3,4-dibromofuran and (2-chloro-4-methoxy-phenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(PPh)4. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as white solid (mp 137–140 °C). 1H NMR (400 MHz, acetone-d6) δ 7.84 (d, J = 8.8 Hz, 2H), 7.07 (s, 2H), 7.00 (d, J = 2.6 Hz, 2H), 6.90 (dd, J = 8.8, 2.6 Hz, 2H), 3.84 (s, 6H). 13C NMR (101 MHz, acetone-d6) δ 160.47, 140.17, 134.63, 133.47, 128.47, 125.58, 115.38, 113.44, 55.91.
4,4'-(furan-3,4-diyl)diphenol (18a)
Compound 18a was prepared by 3,4-Bis(4-methoxyphenyl) furan (17a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate =3:1) gave the title compound as brown solid (mp 172–174 °C). 1H NMR (400 MHz, acetone-d6) δ 7.63 (s, 2H), 7.10 – 7.05 (m, 4H), 6.78 (dd, J = 11.2, 4.5 Hz, 4H). 13C NMR (101 MHz, acetone-d6) δ 157.53, 140.98, 130.45, 126.60, 124.21, 116.06. HRMS (MALDI/DHB) calcd for C16H12O3 (M+H+) 252.0782, found 252.07810.
3,4-Bis(4-hydroxy-2-methylphenyl)furan (18b)
Compound 18b was prepared by 3,4-bis (4-methoxy-2-methylphenyl)furan (17b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate =3:1) gave the title compound as brown solid (mp 170 °C). 1H NMR (400 MHz, acetone-d6) δ 8.28 (s, 2H), 7.53 (s, 2H), 6.88 (d, J = 8.3 Hz, 2H), 6.64 (d, J = 2.3 Hz, 2H), 6.57 (dd, J = 8.2, 2.5 Hz, 2H), 2.00 (s, 6H). 13C NMR (101 MHz, acetone-d6) δ 157.52, 141.30, 138.59, 132.41, 127.12, 124.25, 117.68, 113.31, 20.67. HRMS (MALDI/DHB) calcd for C18H16O3 (M+H+) 280.1091, found 280.10940.
3,4-Bis(2-fluoro-4-hydroxyphenyl)furan (18c)
Compound 18c was prepared by 3,4-bis(2-fluoro-4-methoxyphenyl)furan (17c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as gray solid (mp 209–211 °C). 1H NMR (400 MHz, acetone-d6) δ 9.03 (s, 1H), 7.71 (s, 2H), 7.00 (t, J = 8.6 Hz, 2H), 6.63 – 6.58 (m, 4H). 13C NMR (101 MHz, acetone-d6) δ 162.09, 159.70, 137.53, 130.41, 126.40, 114.54, 113.54, 104.61. HRMS (MALDI/DHB) calcd for C16H16O3F2 (M+H+) 288.0595, found 288.05925.
3,4-Bis(2-chloro-4-hydroxyphenyl)furan (18d)
Compound 18d was prepared by 3,4-bis(2-chloro-4-methoxyphenyl)furan (17d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether: ethyl acetate = 3:1) gave the title compound as gray solid (mp 179–183 °C).1H NMR (400 MHz, acetone-d6) δ 7.68 (s, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 2.5 Hz, 2H), 6.72 (dd, J = 8.4, 2.4 Hz, 2H). 13C NMR (101 MHz, acetone-d6) δ 160.47, 140.17, 134.63, 133.47, 128.47, 125.58, 115.38, 113.44. HRMS (MALDI/DHB) calcd for C16H16O3Cl2 (M+H+) 320.0003, found 320.00015.
2-Bromo-5-(4-methoxy-2-methylphenyl)thiophene (19)
Compound 19 was prepared by reaction of 2,5-dibromofuran and (4-methoxy-2-methylphenyl) boronic acid according to the general procedure for Suzuki coupling using Pd(OAc)2. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 3.7 Hz, 1H), 6.73 (m, 3H), 3.78 (s, 3H), 2.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.57, 144.81, 137.75, 133.82, 131.56, 129.96, 126.39, 125.98, 116.28, 111.38, 55.32, 21.28.
2,5-Dibromo-3-methylthiophene (20)
Compound 20 was prepared by bromination of 3-methyl thiophene using NBS. Distillation gave the title compound as yellow oil. 1H NMR (400 MHz, CDCl3) δ 6.74 (s, 1H), 2.14 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 138.07, 131.90, 110.23, 108.47, 15.23.
3-Bromo-2,5-bis(2-fluoro-4-methoxyphenyl)thiophene (21)
Compound 21 was prepared by reaction of 2,3,5-dibromofuran and (2-fluoro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.44 – 7.33 (m, 2H), 7.23 (s, 1H), 6.73 – 6.60 (m, 4H), 3.77 (s, 3H), 3.75 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.51, 161.07, 160.62, 159.18, 158.58, 132.65, 128.79, 128.18, 110.70, 110.06, 102.37, 102.13, 101.90, 55.68.
3-Bromo-2,5-bis(2-chloro-4-methoxyphenyl)thiophene (22)
Compound 21 was prepared by reaction of 2,3,5-dibromofuran and (2-chloro-4-methoxyphenyl) boronic acid according to the general procedure for Suzuki coupling. Purification by CC (petroleum ether: ethyl acetate = 98:2) gave the title compound as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 8.7 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 7.15 (s, 1H), 6.93 (dd, J = 13.3, 2.5 Hz, 2H), 6.76 (ddd, J = 12.4, 8.6, 2.6 Hz, 2H), 3.74 (s, 3H), 3.72 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.63, 159.87, 140.52, 135.28, 133.48, 132.94, 131.68, 129.42, 124.35, 123.61, 115.79, 115.18, 113.39, 112.81, 110.66, 55.63.
2,5-Dibromofuran (23)
Compound 23 was prepared by bromination of furan using Br2 as brown oil. 1H NMR (400 MHz, CDCl3) δ 6.29 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 121.88, 114.24, 77.36, 77.04, 76.72.
3,4-Dibromofuran (24)
Compound 24 was prepared through oxidation of (E)-2,3-dibromo-2-3-butane-1,4-diol using chromosulfuric acid by steam distillation as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.45 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 141.60, 103.97.
Estrogen Receptor Binding Affinity
Relative binding affinities were determined by a competitive radiometric binding assay as previously described,51, 67 using 2 nM [3H]estradiol as tracer ([2,4,6,7-3H]-estra-1,3,5(10)-triene-3,17β-diol, 70–115 Ci/mmol, Perkin Elmer, Waltham, MA), and purified full-length human ERα and ERβ were purchased from PanVera/Invitrogen (Carlsbad, CA). Incubations were for 18–24 h at 0 °C. Hydroxyapatite (BioRad, Hercules, CA) was used to absorb the receptor-ligand complexes, and free ligand was washed away. The binding affinities are expressed as relative binding affinity (RBA) values with the RBA of estradiol set to 100%. The values given are the average ± range or SD of two to three independent determinations. Estradiol binds to ERα with a Kd of 0.2 nM and to ERβ with a Kd of 0.5 nM; these values were determined by Scatchard analysis using the binding assay protocol described previously.51
Gene Transcriptional Activity
Assays were performed as previously described.52 HepG2 and Ishikawa cells cultured in Dulbecco’s minimum essential medium (DMEM) (Cellgro by Mediatech, Inc. Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Hyclone by Thermo Scientific, South Logan, UT), and 1% non-essential amino acids (Cellgro), Penicillin-Streptomycin-Neomycin antibiotic mixture and Glutamax (Gibco by Invitrogen Corp. Carlsbad, CA), were maintained at 37 °C and 5% CO2. HepG2 cells were transfected with 10 µg of 3X ERE-luciferase reporter plus 1.6 µg of ERα or ERβ expression vector per 10 cm dish using FugeneHD reagent (Roche Applied Sciences, Indianapolis IN). The next day, the cells were transferred to phenol red-free growth media supplemented with 10% charcoal-dextran sulfate-stripped FBS at a density of 20,000 cells/well, incubated in 384-well plates overnight at 37 °C and 5% CO2, and stimulated with various concentrations of compounds in triplicate. Ishikawa cells grown in phenol red-free growth media were transfected with 10 ug of 3X ERE-luciferase reporter per 10 cm dish using FugeneHD reagent. The next day, cells were plated at a density of 20,000 cells/well, incubated in 384-well plates overnight at 37 °C and 5% CO2, and treated with compounds dissolved in DMSO using a robotic pin-tool dispenser. Compounds were assayed in dose curves ranging from 10 µM to 100 pM for ERE luciferase assays in HepG2 cells and at a single dose of 10 µM for ERE luciferase assays in Ishikawa cells. Luciferase activity was measured after 24 hours using BriteLite reagent (Perkin-Elmer Inc., Shelton, CT) according to manufacturer’s protocol. Raw data measured as relative light units were normalized for each plate using the average of DMSO treated samples as 0% and the average of top of the E2 curve as 100%.
Molecular Modeling
The thiophenes 2e or 4e were superpositioned using68 COOT with OBHS in the OBHS-bound ER crystal structure52 so that the A-ring mimetic formed the canonical hydrogen-bonding pattern with R394 and E353. For 2e the other phenol was superposed with the phenyl-sulfonate substituent of OBHS, and for 4e, the second phenol was positioned to hydrogen bond with T347. The models were transferred to CCP4MG for presentation.69
Supplementary Material
Acknowledgment
Supported by grants from NSFC (20872116, 81172935, 91017005), the Program for New Century Excellent Talents in University (NCET-10-0625), Key Project of Ministry of Education of China (313040), the Research Fund for the Doctoral Program of Higher Education of China (20100141110021) and the Fundamental Research Funds for the Central Universities. Research support from the National Institutes of Health (PHS 5R37 DK015556 to J.A.K. and R01 DK077085 to K.W.N.) is gratefully acknowledged. S.S. is supported by the Frenchman’s Creek Women for Cancer Research. We are grateful to Teresa Martin to help in the binding assays.
Abbreviations
- ER
estrogen receptor
- CC
column chromatography
- DCM
dicholoromethane
- E2
estradiol
- HEC-1
human endometrial cancer cells
- HepG2
human hepatoma cells
- LBD
ligand-binding domain
- LBP
ligand-binding pocket
- RBA
relative binding affinity
- SAR
structure-activity relationship
- THF
tetrahydrofuran
- TLC
thin layer chromatography
Footnotes
Supporting Information Available: HPLC results, HPLC spectra for final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wang CL, Tang XY, Chen WQ, Su YX, Zhang CX, Chen YM. Association of estrogen receptor alpha gene polymorphisms with bone mineral density in Chinese women: a meta-analysis. Osteoporos. Int. 2007;18:295–305. doi: 10.1007/s00198-006-0239-2. [DOI] [PubMed] [Google Scholar]
- 2.Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308:1583–1587. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 3.Behl C, Manthey D. Neuroprotective activities of estrogen: An update. J. Neurocytol. 2000;29:351–358. doi: 10.1023/a:1007109222673. [DOI] [PubMed] [Google Scholar]
- 4.Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation Estrogen receptor alpha and estrogen receptor beta: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai ZL, Gust R. Breast Cancer, Estrogen Receptor and Ligands. Arch. Pharm. (Weinheim) 2009;342:133–149. doi: 10.1002/ardp.200800174. [DOI] [PubMed] [Google Scholar]
- 6.Deroo BJ, Korach KS. Estrogen receptors and human disease. J. Clin. Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fox EM, Davis RJ, Shupnik MA. ER beta in breast cancer - Onlooker, passive player, or active protector? Steroids. 2008;73:1039–1051. doi: 10.1016/j.steroids.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osipo C, Liu H, Meeke K, Jordan VC. The consequences of exhaustive antiestrogen therapy in breast cancer: Estrogen-induced tumor cell death. Exp. Biol. Med. 2004;229:722–731. doi: 10.1177/153537020422900804. [DOI] [PubMed] [Google Scholar]
- 9.Robinson-Rechavi M, Garcia HE, Laudet V. The nuclear receptor superfamily. J. Cell Sci. 2003;116:585–586. doi: 10.1242/jcs.00247. [DOI] [PubMed] [Google Scholar]
- 10.Mueller SO, Korach KS. Estrogen receptors and endocrine diseases: lessons from estrogen receptor knockout mice. Curr. Opin. Pharmacol. 2001;1:613–619. doi: 10.1016/s1471-4892(01)00105-9. [DOI] [PubMed] [Google Scholar]
- 11.Swedenborg E, Power KA, Cai W, Pongratz I, Ruegg J. Regulation of estrogen receptor beta activity and implications in health and disease. Cell. Mol. Life Sci. 2009;66:3873–3894. doi: 10.1007/s00018-009-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang EC, Charn TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen Receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol. Endocrinol. 2008;22:1032–1043. doi: 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 14.Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor β ligands: Recent advances and biomedical applications. Med. Res. Rev. 2011;31:364–442. doi: 10.1002/med.20186. [DOI] [PubMed] [Google Scholar]
- 15.Harris HA. Preclinical characterization of selective estrogen receptor beta agonists: new insights into their therapeutic potential; Ernst Schering Foundation symposium proceedings; 2006. pp. 149–161. [DOI] [PubMed] [Google Scholar]
- 16.Nilsson S, Gustafsson JA. Estrogen receptors: therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 2011;89:44–55. doi: 10.1038/clpt.2010.226. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson S, Koehler KF, Gustafsson JA. Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 2011;10:778–792. doi: 10.1038/nrd3551. [DOI] [PubMed] [Google Scholar]
- 18.Harris HA. Estrogen receptor-beta: recent lessons from in vivo studies. Mol. Endocrinol. 2007;21:1–13. doi: 10.1210/me.2005-0459. [DOI] [PubMed] [Google Scholar]
- 19.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. U.S. A. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson J-A, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 21.Pike ACW, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JK, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katzenellenbogen JA. The 2010 Philip S. Portoghese Medicinal Chemistry Lectureship: addressing the "core issue" in the design of estrogen receptor ligands. J. Med. Chem. 2011;54:5271–5282. doi: 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonnell DP, Wardell SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr. Opin. Pharmacol. 2010;10:620–628. doi: 10.1016/j.coph.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: Structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J. Med. Chem. 2000;43:4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 25.Naoum F, Kasiotis KM, Magiatis P, Haroutounian SA. Synthesis of novel nitro-substituted triaryl pyrazole derivatives as potential estrogen receptor ligands. Molecules. 2007;12:1259–1273. doi: 10.3390/12071259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou HB, Carlson KE, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA. Analogs of methyl-piperidinopyrazole (MPP): Antiestrogens with estrogen receptor alpha selective activity. Bioorg. Med. Chem. Lett. 2009;19:108–110. doi: 10.1016/j.bmcl.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schafer A, Wellner A, Gust R. Synthesis and investigations on the oxidative degradation of C3/C5-alkyl-1,2,4-triarylpyrroles as ligands for the estrogen receptor. Chemmedchem. 2011;6:794–803. doi: 10.1002/cmdc.201000537. [DOI] [PubMed] [Google Scholar]
- 28.Schafer A, Wellner A, Strauss M, Wolber G, Gust R. Development of 2,3,5-triaryl-1H-pyrroles as estrogen receptor alpha selective ligands. Chemmedchem. 2011;6:2055–2062. doi: 10.1002/cmdc.201100283. [DOI] [PubMed] [Google Scholar]
- 29.Schafer A, Wellner A, Strauss M, Wolber G, Gust R. Influence of Chlorine or Fluorine Substitution on the Estrogenic Properties of 1-Alkyl-2,3,5-tris(4-hydroxyphenyl)-1H-pyrroles. J. Med. Chem. 2012;55:9607–9618. doi: 10.1021/jm300860j. [DOI] [PubMed] [Google Scholar]
- 30.Mortensen DS, Rodriguez AL, Carlson KE, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Synthesis and biological evaluation of a novel series of furans: Ligands selective for estrogen receptor alpha. J. Med. Chem. 2001;44:3838–3848. doi: 10.1021/jm010211u. [DOI] [PubMed] [Google Scholar]
- 31.Zimmermann J, Liebl R, von Angerer E. 2,5-Diphenylfuran-based pure antiestrogens with selectivity for the estrogen receptor alpha. J. Steroid Biochem. Mol. Biol. 2005;94:57–66. doi: 10.1016/j.jsbmb.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 32.Zimmermann J, von Angerer E. Estrogenic and antiestrogenic activities of 2,4-diphenylfuran-based ligands of estrogen receptors alpha and beta. J. Steroid Biochem. Mol. Biol. 2007;104:259–268. doi: 10.1016/j.jsbmb.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 33.Haddad T, Gershman R, Dilis R, Labaree D, Hochberg RB, Hanson RN. Synthesis and evaluation of 4-(substituted styryl/alkenyl)-3,5-bis(4-hydroxyphenyl)-isoxazoles as ligands for the estrogen receptor. Bioorg. Med. Chem. Lett. 2012;22:5999–6003. doi: 10.1016/j.bmcl.2012.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gust R, Keilitz R, Schmidt K. Investigations of new lead structures for the design of selective estrogen receptor modulators. J. Med. Chem. 2001;44:1963–1970. doi: 10.1021/jm001131d. [DOI] [PubMed] [Google Scholar]
- 35.Wiglenda T, Ott I, Kircher B, Schumacher P, Schuster D, Langer T, Gust R. Synthesis and pharmacological evaluation of 1H-imidazoles as ligands for the estrogen receptor and cytotoxic inhibitors of the cyclooxygenase. J. Med. Chem. 2005;48:6516–6521. doi: 10.1021/jm050190u. [DOI] [PubMed] [Google Scholar]
- 36.Wiglenda T, Gust R. Structure-activity relationship study to understand the estrogen receptor-dependent gene activation of aryl- and alkyl-substituted 1H-imidazoles. J. Med. Chem. 2007;50:1475–1484. doi: 10.1021/jm061106t. [DOI] [PubMed] [Google Scholar]
- 37.Schlenk M, Ott I, Gust R. Cobalt-alkyne complexes with imidazoline ligands as estrogenic carriers: synthesis and pharmacological investigations. J. Med. Chem. 2008;51:7318–7322. doi: 10.1021/jm8008376. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh U, Ganessunker D, Sattigeri VJ, Carlson KE, Mortensen DJ, Katzenellenbogen BS, Katzenellenbogen JA. Estrogenic diazenes: heterocyclic non-steroidal estrogens of unusual structure with selectivity for estrogen receptor subtypes. Bioorg. Med. Chem. 2003;11:629–657. doi: 10.1016/s0968-0896(02)00309-7. [DOI] [PubMed] [Google Scholar]
- 39.Bey E, Marchais-Oberwinkler S, Werth R, Negri M, Al-Soud YA, Kruchten P, Oster A, Frotscher M, Birk B, Hartmann RW. Design, synthesis, biological evaluation and pharmacokinetics of bis(hydroxyphenyl) substituted azoles, thiophenes, benzenes, and aza-benzenes as potent and selective nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD1) J. Med. Chem. 2008;51:6725–6739. doi: 10.1021/jm8006917. [DOI] [PubMed] [Google Scholar]
- 40.Bey E, Marchais-Oberwinkler S, Negri M, Kruchten P, Oster A, Klein T, Spadaro A, Werth R, Frotscher M, Birk B, Hartmann RW. New insights into the SAR and binding modes of bis(hydroxyphenyl)thiophenes and -benzenes: influence of additional substituents on 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD1) inhibitory activity and selectivity. J. Med. Chem. 2009;52:6724–6743. doi: 10.1021/jm901195w. [DOI] [PubMed] [Google Scholar]
- 41.Wang P, Min J, Nwachukwu JC, Cavett V, Carlson KE, Guo P, Zhu M, Zheng Y, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. Identification and structure-activity relationships of a novel series of estrogen receptor ligands based on 7-thiabicyclo[2.2.1]hept-2-ene-7-oxide. J. Med. Chem. 2012;55:2324–2341. doi: 10.1021/jm201556r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riggs BL, Hartmann LC. Selective estrogen-receptor modulators -- mechanisms of action and application to clinical practice. N. Engl. J. Med. 2003;348:618–629. doi: 10.1056/NEJMra022219. [DOI] [PubMed] [Google Scholar]
- 43.Amb CM, Rasmussen SC. Sterics versus electronics: Regioselective cross-coupling of polybrominated thiophenes. Eur. J. Org. Chem. 2008:801–804. [Google Scholar]
- 44.Tung DT, Tuan DT, Rasool N, Villinger A, Reinke H, Fischer C, Langer P. Regioselective Palladium(0)-Catalyzed Cross-Coupling Reactions and Metal-Halide Exchange Reactions of Tetrabromothiophene: Optimization, Scope and Limitations. Adv. Synth. Catal. 2009;351:1595–1609. [Google Scholar]
- 45.van Leeuwen P, Kamer PCJ, Reek JNH, Dierkes P. Ligand bite angle effects in metal-catalyzed C-C bond formation. Chem. Rev. 2000;100:2741–2769. doi: 10.1021/cr9902704. [DOI] [PubMed] [Google Scholar]
- 46.Hou JH, Yang CH, He C, Li YF. Poly 3-(5-octyl-thienylene-vinyl)-thiophene : A side-chain conjugated polymer with very broad absorption band. Chem. Commun. 2006:871–873. doi: 10.1039/b516576h. [DOI] [PubMed] [Google Scholar]
- 47.Ramalingan C, Lee I-S, Kwak Y-W. Novel Furanylarylene Arylsulfonylindolesulfonamides: Synthesis and Their Antibacterial Evaluation. Chem. Pharm. Bull. 2009;57:591–596. doi: 10.1248/cpb.57.591. [DOI] [PubMed] [Google Scholar]
- 48.Mee SPH, Lee V, Baldwin JE, Cowley A. Total synthesis of 5,5',6,6'-tetrahydroxy-3,3'-biindolyl, the proposed structure of a potent antioxidant found in beetroot (Beta vulgaris) Tetrahedron. 2004;60:3695–3712. [Google Scholar]
- 49.Hussain M, Khera RA, Nguyen TH, Langer P. Site-selective Suzuki-Miyaura cross-coupling reactions of 2,3,4,5-tetrabromofuran. Org. Biomol. Chem. 2011;9:370–373. doi: 10.1039/c0ob00695e. [DOI] [PubMed] [Google Scholar]
- 50.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 51.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 52.Zheng Y, Zhu M, Srinivasan S, Nwachukwu JC, Cavett V, Min J, Carlson KE, Wang P, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. Development of selective estrogen receptor modulator (SERM)-like activity through an indirect mechanism of estrogen receptor antagonism: defining the binding mode of 7-oxabicyclo[2.2.1]hept-5-ene scaffold core ligands. Chemmedchem. 2012;7:1094–1100. doi: 10.1002/cmdc.201200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu M, Zhang C, Nwachukwu JC, Srinivasan S, Cavett V, Zheng Y, Carlson KE, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. Bicyclic core estrogens as full antagonists: synthesis, biological evaluation and structure-activity relationships of estrogen receptor ligands based on bridged oxabicyclic core arylsulfonamides. Org. Biomol. Chem. 2012;10:8692–8700. doi: 10.1039/c2ob26531a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gould JC, Leonard LS, Maness SC, Wagner BL, Conner K, Zacharewski T, Safe S, McDonnell DP, Gaido KW. Bisphenol A interacts with the estrogen receptor alpha in a distinct manner from estradiol. Mol. Cell. Endocrinol. 1998;142:203–214. doi: 10.1016/s0303-7207(98)00084-7. [DOI] [PubMed] [Google Scholar]
- 55.Jeyakumar M, Carlson KE, Gunther JR, Katzenellenbogen JA. Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J. Biol. Chem. 2011;286:12971–12982. doi: 10.1074/jbc.M110.205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Li Y, Lavigne JA, Trush MA, Yager JD. Increased mitochondrial superoxide production in rat liver mitochondria, rat hepatocytes, and HepG2 cells following ethinyl estradiol treatment. Toxicol. Sci. 1999;51:224–235. doi: 10.1093/toxsci/51.2.224. [DOI] [PubMed] [Google Scholar]
- 57.Dumont C, Perdu E, de Sousa G, Debrauwer L, Rahmani R, Cravedi JP, Chagnon MC. Bis(hydroxyphenyl)methane-bisphenol F-metabolism by the HepG2 human hepatoma cell line and cryopreserved human hepatocytes. Drug Chem. Toxicol. 2011;34:445–453. doi: 10.3109/01480545.2011.585651. [DOI] [PubMed] [Google Scholar]
- 58.Fink BE, Mortensen DS, Stauffer SR, Aron ZD, Katzenellenbogen JA. Novel structural templates for estrogen-receptor ligands and prospects for combinatorial synthesis of estrogens. Chem. Biol. 1999;6:205–219. doi: 10.1016/S1074-5521(99)80037-4. [DOI] [PubMed] [Google Scholar]
- 59.Wright JS, Shadnia H, Anderson JM, Durst T, Asim M, El-Salfiti M, Choueiri C, Pratt MA, Ruddy SC, Lau R, Carlson KE, Katzenellenbogen JA, O'Brien PJ, Wan L. A-CD estrogens. I. Substituent effects, hormone potency, and receptor subtype selectivity in a new family of flexible estrogenic compounds. J. Med. Chem. 2011;54:433–448. doi: 10.1021/jm100513m. [DOI] [PubMed] [Google Scholar]
- 60.Asim M, El-Salfiti M, Qian Y, Choueiri C, Salari S, Cheng J, Shadnia H, Bal M, Christine Pratt MA, Carlson KE, Katzenellenbogen JA, Wright JS, Durst T. Deconstructing estradiol: removal of B-ring generates compounds which are potent and subtype-selective estrogen receptor agonists. Bioorg. Med. Chem. Lett. 2009;19:1250–1253. doi: 10.1016/j.bmcl.2008.12.080. [DOI] [PubMed] [Google Scholar]
- 61.Shanle EK, Hawse JR, Xu W. Generation of stable reporter breast cancer cell lines for the identification of ER subtype selective ligands. Biochem Pharmacol. 2011;82:1940–1949. doi: 10.1016/j.bcp.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J, Nilsson S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol. Pharmacol. 1998;54:105–112. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 63.Gehm BD, McAndrews JM, Chien PY, Jameson JL. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc. Natl. Acad. Sci. U. S. A. 1997;94:14138–14143. doi: 10.1073/pnas.94.25.14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mueller SO, Simon S, Chae K, Metzler M, Korach KS. Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor alpha (ERalpha) and ERbeta in human cells. Toxicol. Sci. 2004;80:14–25. doi: 10.1093/toxsci/kfh147. [DOI] [PubMed] [Google Scholar]
- 65.Muthusamy S, Andersson S, Kim HJ, Butler R, Waage L, Bergerheim U, Gustafsson JA. Estrogen receptor beta and 17beta-hydroxysteroid dehydrogenase type 6, a growth regulatory pathway that is lost in prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2011;108:20090–20094. doi: 10.1073/pnas.1117772108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katzenellenbogen JA, Johnson HJ, Jr, Myers HN. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 68.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 69.McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.