

Abstract
Current tumor immunotherapy approaches include the genetic modification of peripheral T cells to express tumor antigen-specific T-cell receptors (TCRs). The approach, tested in melanoma, has led to some limited success of tumor regression in patients. Yet, the introduction of exogenous TCRs into mature T cells entails an underlying risk; the generation of autoreactive clones due to potential TCR mispairing, and the lack of effective negative selection, as these peripheral cells do not undergo thymic selection following introduction of the exogenous TCR. We have successfully generated MART-1–specific CD8 T cells from genetically modified human hematopoietic stem cells (hHSC) in a humanized mouse model. The advantages of this approach include a long-term source of antigen specific T cells and proper T-cell selection due to thymopoiesis following expression of the TCR. In this report, we examine the molecular processes occurring on endogenous TCR expression and demonstrate that this approach results in exclusive cell surface expression of the newly introduced TCR, and the exclusion of endogenous TCR cell surface expression. This suggests that this stem cell based approach can provide a potentially safer approach for anticancer immunotherapy due to the involvement of thymic selection.
Introduction
The main focus of tumor immunotherapy is the development of an effective, long term, and safe therapeutic to target and clear tumors. To date, the majority of studies have concentrated on the manipulation of autologous peripheral T cells, which includes the ex vivo expansion of tumor specific CD8 T cells and the genetic modification of peripheral T cells with lentiviral vectors expressing antigen-specific T-cell receptors (TCRs)1,2,3 Although the above approaches have had some limited success, they are stymied by the nature of human T cells. Both the ex vivo expansion and genetic modification of T cells involve extensive manipulation that can lead to T-cell exhaustion.4,5 Furthermore, T-cell responses are short lived in nature.6,7 Finally, the introduction of an exogenous TCR harbors the risk of generating autoreactive clones that can cause lethal graft-versus-host disease,8,9 due to recombination between TCR chains produced from the endogenous and exogenous TCR genes, which are expressed simultaneously.
An alternative approach to the above is the genetic modification of human hematopoietic stem cells (hHSC) with vectors expressing an antigen-specific TCR, and the subsequent differentiation of these cells into mature transgenic T cells. This approach was first successfully tested in murine models.10,11 However, as these were not disease models and the lineage development of mice is quite distinct from that of humans, there was a need to determine whether this approach was feasible with hHSC.12
The development of the bone marrow/liver/thymus (BLT) humanized mouse system has enabled the testing of such approaches.13 In this chimeric model, human fetal thymus and liver are implanted under the kidney capsule to generate a thy/liv organoid. This is followed by transplantation with hHSC that results in full reconstitution of human immune cells. Using a modified version of this model, we recently introduced antigen-specific HLA-A*0201–restricted TCRs against melanoma (MART-1(26–35) epitope) or HIV (SL9(77–85) gag epitope) into hHSC and transplanted them into BLT humanized mice generated from HLA-A*0201 positive human fetal tissues.14,15 In both studies, the genetically modified stem cells developed through the human thymic organoid and gave rise to transgenic cytotoxic T lymphocytes (CTL) that were functional both in vivo and ex vivo. In the melanoma studies,14 through the use of HLA-matched target and nonmatched control tumors, we were able to demonstrate that the transgenic CTLs specifically targeted the HLA-matched tumors without significantly impacting the nonmatched tumors.
The use of TCR-transduced progenitors therefore has distinct advantage over the use of transduced peripheral T cells, because progenitors will provide a stable and renewable source of engineered cells as well as undergo proper T-cell selection and maturation to give rise to antigen-specific CTLs. Here, we demonstrate that this approach minimizes the risk of generating mature T cells expressing multiple TCRs simultaneously, which would minimize the potential of generating autoreactive clones that could result in graft-versus-host disease. Using the BLT mouse model, we generated transgenic CTLs expressing an exogenous TCR specific for the human MART-1 antigen and examined the impact of the introduced TCR on T-cell development and expression of the exogenous and endogenous TCRs. On the basis of our studies, although the exogenous MART-1–specific TCR does partially inhibit endogenous TCR rearrangement, it does not fully block this process. However, despite the expression of RNA from endogenous TCR Vβ chains, the resulting transgenic CTLs in the periphery express on their surface only a single TCR, that is encoded by the exogenous transgene. Therefore, our data suggest that the generation of transgenic CTLs from genetically modified human hematopoietic progenitors is not only a long-term solution but also a potentially more specific approach to target melanoma and other chronic diseases.
Results
T-cell selection and maturation occurs in the human Thy/Liv implant of BLT mice
A key advantage of using a stem cell-based approach to generate transgenic T cells is the proper T-cell development and selection of the genetically modified progenitors. As a consequence, this would facilitate the elimination of autoreactive clones formed by mispairing of the exogenous MART-1–specific TCR chains with endogenous TCR chains. Furthermore, the TCR transgene, in our case the MART-1–specific TCR, should only influence development past the T-cell progenitor stage, as other hematopoietic lineages would not express TCR on the cell surface, due to the lack of CD3 coexpression, which is required for the transport of TCR to the cell surface. The modified BLT mouse model used in our previous studies was used herein to test this hypothesis. To confirm that transduced human progenitors use the thy/liv implant in the BLT mouse for T-cell selection, we set up our BLT mouse model (Figure 1), in an HLA-A*0201 positive or negative background. Genes encoding a human TCR specific for the MART-1 peptide presented in the context of HLA-A*0201 were introduced into human hematopoietic progenitor cells via lentiviral vectoring before introduction into the humanized mice. In BLT mice constructed with HLA-A*0201+ human tissues, which possess a human thymic organoid which expresses HLA-A*0201, the TCR-transduced cells developed normally, as demonstrated by the large numbers of CD8 single positive thymocytes expressing the transgenic TCR within the thymic implant (Figure 2a). Furthermore, we observed expression of the transgenic TCR exclusively on CD8+ T cells in the periphery, as we have in our previous studies (14.2 ± 12.6% MART-1 tetramer positive CD8 T cells, n = 19, 95% CI = 7.2–21.7%) (Figure 2b).14 In addition, as with our previous studies which demonstrated ex vivo antitumor lytic activity as well as in vivo efficacy against HLA-A*0201+ melanomas,14 these transgenic CTL were functional as they limited the growth of major histocompatibility complex–matched melanoma tumors (Supplementary Figure S1; Supplementary Materials and Methods) in vivo. Moreover, in our previous work, the levels of MART-1 expressing CD8 T cells correlated with the levels of tumor regression. This indicates that proper lineage commitment was occurring, directed by the transgenic TCR. Conversely, in the HLA-A*0201- background, we were able to detect expression of thymocytes expressing the transgenic TCR mainly in the immature double positive thymocyte fraction (Figure 2c). The single CD4 and CD8 thymocytes expressed very low levels, if any, of this TCR. Furthermore, we were unable to detect mature human CD4 or CD8 T cells which expressed this transgenic TCR in the spleens of these animals (Figure 2d). Similarly, this TCR was not expressed on mature human T cells in the blood of these mice (data not shown). Our data indicate that the transduced progenitors go through the normal positive selection stages of T-cell development, as we observed expression of the transgenic TCR on double positive immature thymocytes regardless of the HLA background, but on mature single positive thymocytes only in the presence of the appropriate restricting HLA molecule. The resulting mature TCR-bearing CD8 T cells are able to mount effective lytic responses against melanoma targets. Our data further indicate that the human thy/liv implant and not the mouse thymus serves as the site of T-cell selection in this system.
Figure 1.
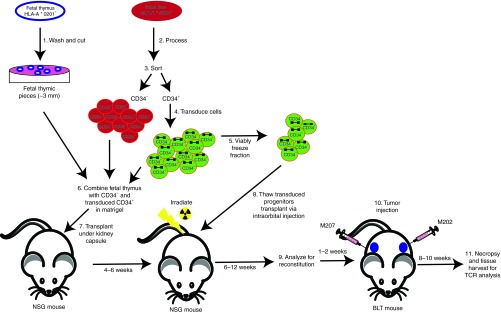
A schematic diagram of the modified BLT humanized mouse model. A schematic diagram on the modified BLT model used in these studies for the generation of chimeric mice carrying MART-1–specific T cells. HLA*0201+ fetal thymus and liver are used to generate the chimeric mice. The fetal thymus is cut into small pieces for transplantation (step 1). Concurrently the fetal liver is processed to purify CD34+ HSC (steps 2–3). The CD34 fraction is transduced with a lentiviral vector (step 4). An aliquot is frozen away (step 5) to be used for the second transplantation. The remainder of CD34 cells is combined with hepatocytes and the fetal liver and transplanted into an NSG mouse (steps 6 and 7). After 4–6 weeks, the mice are irradiated and transplanted with the CD34 from step 5 (step 8). Once reconstitution of human immune cells is established, the mice are challenged with human melanoma tumor lines. Mice are then assayed over a period of 12 weeks. M202 is an HLA*0201+MART+ cell line that could serve as a target for the transgenic CTL and M207, an HLA*0201-MART+ line that could not. M207 thus functions as a negative control for CTL activity and specificity. BLT, bone marrow/liver/thymus; CTL, cytotoxic T lymphocytes; HSC, hematopoietic stem cells.
Figure 2.

MART-1–transduced hHSC develop into T cells exclusively in the thy/liv implant. Thymocytes and splenocytes from a representative (a,b) HLA-A*0201+ or (c,d) HLA-A*0201- BLT mouse transplanted with MART-TCR engineered autologous hHSC were assayed by fluorescent activated cell sorting to detect the levels of tetramer reactive cells in the different subset populations. Panels a and c demonstrate data derived from thymocytes, whereas panels b and d from splenocytes. BLT, bone marrow/liver/thymus; hHSC, human hematopoietic stem cells; TCR, T-cell receptor.
Introduction of exogenous TCR genes suppresses endogenous TCR gene recombination
During the process of TCR development and thymic selection, the genes encoding both TCRα-chain and TCRβ-chain undergo successive rearrangements leading to the generation of a functional TCR. During TCR α gene rearrangement, the TCRδ encoding sequences that are found within the sequences encoding the TCRα-chain, are removed resulting in the formation of TCR excision circles (TREC) which can be quantitated using a PCR technique.16,17,18 Thus, the presence of TREC is an indication of proper TCRα-chain rearrangement. The introduction of an exogenous mature TCR such as the MART-1–specific TCR used here can potentially block the processes of endogenous T-cell rearrangement, as both the exogenous TCRα and β chains are already rearranged and are likely expressed before rearrangement of the endogenous TCR-encoding sequences. Such an outcome could be advantageous, as it would prevent inappropriate pairing of the exogenous TCR chains with endogenous ones and the expression of functional endogenous TCR chains would be eliminated.
To assess the impact of the transgenic TCR on endogenous TCR rearrangement, we sorted different thymocyte populations in BLT mice that were exclusively derived from HLA-A*0201+ tissues, and quantitated the levels of TREC under different conditions by real-time PCR. Thy/liv implants from these animals were excised, and thymocytes were sorted by fluorescent activated cell sorting into the following populations, based largely upon binding to a tetramer reagent that specifically detects cell surface expression of the transgenic MART-specific TCR: MART-TCR+CD4+CD8+, MART-TCR+CD8+, MART-TCR-CD4+CD8+, and MART-TCR-CD8+. Total DNA was extracted from these samples and used in a real-time PCR assay measuring TREC levels generated by rearrangement of the endogenous TCRα chain. Thymocyte populations expressing the transgenic TCR demonstrated twofold lower levels of TREC when compared with that of the populations not expressing the transgenic TCR (Figure 3). Therefore, based on these data, the introduction of the transgenic MART-specific TCR in part blocked endogenous TCRα rearrangement, thus limiting the potential for endogenous TCR expression.
Figure 3.

Overexpression of the MART-specific TCR impacts TREC levels in single and double positive thymocytes. Thymocytes from HLA-A*0201+ BLT mice transplanted with TCR engineered hHSC were sorted by fluorescent activated cell sorting into different subsets, MART-TCR+/CD8+ (MARTCD8), MART-TCR-/CD8+ (CD8), MART-TCR+/CD4+CD8+ (MARTCD4CD8), MART-TCR-/CD4+CD8+ (CD4CD8). Total cellular DNA was extracted and used in a real time PCR assay to measure the levels of TREC between the different thymocyte populations. Statistical significance was determined by a Student's t-test (**P < 0.01). BLT, bone marrow/liver/thymus; hHSC, human hematopoietic stem cells; TCR, T-cell receptor.
Analysis of endogenous Vβ chain mRNA expression in TCR-transgenic CD8 T cells
The presence of significant levels of TREC in the TCR-transgenic thymocyte populations (Figure 3) suggested that endogenous TCR chains could be expressed in some cells and potentially pair with the TCR chains encoded by the exogenous transgene. This left open the possibility that the CTL derived from genetically engineered progenitors express on their surface TCRs that either have resulted from the pairing of exogenous and endogenous TCR chains or possibly simultaneous expression of two distinct TCRs, potentially forming bispecific CTLs.
To this end, we looked at the RNA expression levels of the endogenous Vβ chains in MART-specific TCR-transgenic CD8 T cells isolated from the periphery of our humanized mice. Splenocytes from the same cohort used in the TREC assays described above were harvested and used to sort MART-TCR+CD8+ and MART-TCR-CD8+ human T cells. Subsequently, we isolated total RNA to be used in a spectratyping assay designed to detect expression of rearranged TCRβ chains (Figure 4a).19 On the basis of our results from the different mice that received transduced progenitors, transgenic CD8 T cells did express endogenous TCRβ RNA although the levels of the MART-specific Vβ13b (BV06b) chain RNA were significantly higher in all the animals. The average contribution of the transgenic Vβ chain was 50% of the total TCR RNA expression (Figure 4b). These results would suggest that several scenarios might be operating (i) at least some of the RNA corresponding to the Vβ sequences not encoding the transgene may be the equivalent of out-of-frame TCR chain expression,16 (ii) that there are other mature TCR protein chains expressed in the transgenic cells,20,21,22 or (iii) that there is posttranscriptional silencing of these chains.23,24
Figure 4.
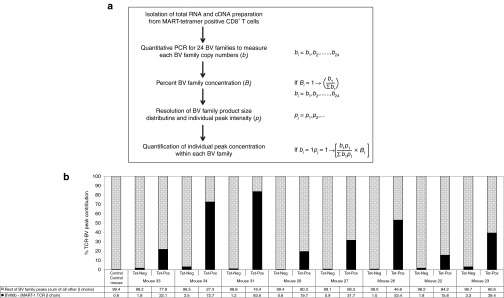
Spectratyping reveals expression of endogenous Vβ chains. (a) Scheme of Quantitative TCR-BV spectratyping and the calculation applied to assess repertoire diversity and individual peak concentration. Where, “bi” represents the level of “ith” BV family specific transcripts and “Bi” is the percent concentration of “ith” BV family among all 24 BV families. Also, “pj” represents peak intensity measured as area under the curve of “jth” peak within a BV family. (b) A graphic representation of the fraction of sum of the endogenous TCR β chains versus the fraction of the MART-1–specific TCR β chain (BV06b). Splenocytes were sorted into MART-TCR+CD8+ (Tet-Pos) and MART-TCR-CD8+ (Tet-Neg) human T cells. Total RNA was extracted and used for spectratyping analysis to determine the percent contribution of the exogenous Vβ chain (BV06b) in the transgenic MART-1–specific TCR expressing CD8 T cells. TCR, T-cell receptor.
Endogenous TCRs are not coexpressed with the transgenic TCR on the surface of transgenic CD8 T cells
The presence of endogenous TCRβ RNA prompted us to determine whether the endogenous TCRβ chains were coexpressed with the transgenic TCR at the surface of the cell. When we transduced peripheral CD8 T cells from healthy human donors with the lentiviral vector expressing the MART-specific TCR, we could easily detect the cell surface expression of both the exogenous and endogenous TCRs concurrently (Figure 5a). Using splenocytes from the same mouse cohort used in Figure 4, we analyzed by flow cytometry the protein expression of a number of Vβ chains (Vβ5.1, 5.3, 9, 13, 20) in the MART-specific TCR-transgenic BLT mice. As shown in Figure 5b, we did not see coexpression of Vβ5.1 in the populations staining positive with the tetramer reagent that specifically detects cell surface expression of the transgenic MART-specific TCR. Similar patterns were seen with the other Vβ chains (see Supplementary Figures S2–S6). The lack of endogenous TCR expression on the cell surface can be attributed to the high levels of MART-1 TCR expression siphoning off available CD3 thus preventing the assembly of endogenous TCR to the surface.16 To address this possibility, we also looked for the intracellular expression of the other Vβ chains to determine whether the endogenous TCR chains were recycled and never reached the cell surface. On the basis of our data (Figure 5c and Supplementary Figures S7–S11), we did not detect intracellular endogenous TCR proteins in the MART-specific TCR positive populations. The lack of expression of endogenous TCR proteins is consistent with the possibility that the TCRβ RNA seen in our spectratyping studies was due to incomplete or inappropriate endogenous TCR rearrangement that would not lead to expression of a mature TCR protein chain.16
Figure 5.

Lack of coexpression of transgenic TCR and other Vβ chains. (a) Peripheral CD8 T cells were transduced with the MART-1–specific TCR expressing lentiviral vector used in our studies. Two days after transduction, the cells were analyzed by flow cytometry for the expression of the transgenic TCR (using MART-1–specific tetramers) and endogenous TCRs (using Vβ chain specific antibodies). (b) Splenocytes from HLA-A*0201+ BLT mice transplanted with MART-1–specific TCR engineered hHSC were analyzed by flow cytometry for the expression of the transgenic TCR and endogenous TCRs. (c) Splenocyte samples from b were also used to determine presence of endogenous TCRs intracellularly. BLT, bone marrow/liver/thymus; hHSC, human hematopoietic stem cells; TCR, T-cell receptor.
Discussion
Engineering the immune system to increase tumor specific T cells to battle cancer has shown great promise. The introduction of melanoma-specific TCRs into peripheral T cells has led to some limited success. However, the extensive manipulation of these cells as well as inherent risks due to TCR chain miss-pairing underscore the need for the development of safer alternatives1,2,3,8.
One such approach is to genetically modify hematopoietic stem cells using lentiviral vectors expressing antigen-specific TCRs. Studies in mouse models demonstrated that such an approach was feasible. As human T-cell development is quite distinct from that of mice, the use of humanized mouse models proved very useful in testing this approach. Recent studies by our group have shown that we can introduce a tumor specific (MART-1, an antigen found in melanoma cells) or a virus specific (SL9, HIV gag) TCR in human hematopoietic progenitors and generate functional transgenic CTLs in vivo in humanized mice. One of the advantages of this model is that it allows us to study factors contributing to the development of functional T cells. On the basis of a previous work by others and us genetically modified HSC have shown to engraft well and demonstrate normal lineage development despite the introduction of exogenous vectors. In the BLT mouse model, we see the development of T cells, B cells, monocytes/macrophages, dendritic cells, and granulocytes.14,15,25 In these studies, we demonstrated that generating human transgenic T cells from genetically modified human progenitors results in the generation of transgenic CTL expressing a single functional TCR on their surface. Although endogenous TCR chains were detected at the RNA level, they were not detectable at the protein level.
Our data suggest that the overexpression of an exogenous TCR does impact endogenous TCR rearrangement. This was demonstrated by the decreased levels of α TREC in thymocyte populations expressing a transgenic exogenous TCR. However, there was not a complete shutdown of TCRα rearrangement suggesting that some endogenous α chains could be expressed. As our TREC assay focused on the TCRα chain, we are not certain on how this impacted the TCRβ rearrangement process.
Consequently, as some endogenous rearrangement has taken place, we would expect that endogenous TCR chains would appear in the mature CD8 T-cell population. To this end, we looked at the expression of Vβ chain RNA in the TCR-transgenic CD8 T-cell population. Interestingly, we did see the expression of other Vβ chains but the Vβ family that would encode the transgenic TCR was expressed at significantly higher levels. This also suggested that endogenous TCRs could be coexpressed along with the TCR transgene. Reports have shown in both humans and mice that during the T-cell selection process there is some allelic inclusion albeit at a low level.26,27,28 It is suggested that the thymic selection processes remove the majority of autoreactive clones yet allow a small fraction to be eliminated by peripheral selection mechanisms.16
To determine if any endogenous TCR chains were expressed at the protein level, we proceeded to examine if the TCR chains detected by RNA analysis were expressed at the protein level and would assemble at the cell surface along with the transgenic TCR. However, we did not detect coexpression of the transgenic TCR and endogenous TCR chains in our transgenic cells, although we could easily detect coexpression when we transduced mature peripheral CD8 T cells with the genes encoding an exogenous TCR. The lack of coexpression of endogenous TCRs in mature cells derived from TCR-transduced hematopoietic progenitors points to two possible mechanisms of action. One would suggest that following the overexpression of the transgenic TCR, there is a limited allelic inclusion of in-frame endogenous chains. Studies have suggested that the presence of bi-allelic T cells is not prohibitive as other mechanisms such as peripheral tolerance can limit the development of autoreactive clones. In addition, posttranscriptional mechanisms can block the expression of in-frame alleles.16,24 The other option is that the RNA species detected in our assays are the products of out-of-frame expression. The inclusion of out-of-frame alleles is possible during the process of T-cell rearrangement, and thus they would be expressed at the RNA but not the protein level.16
In conclusion, our data suggest that a stem cell approach to engineer T cells expressing antigen-specific TCRs has a distinct advantage over the introduction of new TCRs into peripheral T cells for the development of monospecific transgenic effector T cells. Such an approach could complement and support existing protocols to melanoma treatment. More specifically, transduced peripheral T cells with a therapeutic TCR can be given as a first line of defense while differentiation of modified progenitors will follow, so they can replenish the immune system with naïve melanoma-specific CTLs. Such an outcome will result to an increased precursor frequency and thus more efficient and lasting tumor clearance. Although some studies have suggested that peripheral T-cell genetic modification does not result in the generation of autoreactive clones, this possibility has not been eliminated. Finally, our BLT mouse model can serve as a useful platform to study TCR rearrangement and selection.
Materials and Methods
Isolation and transduction of CD34 hHSC. Fresh human fetal liver was obtained from Advanced Bioscience Resources (Alameda, CA). The tissue was processed, and CD34 cells were purified as previously described14 and were transduced (multiplicity of infection = 5) using retronectin (Takara Bio, Mountain View, CA). The lentiviral vector pF5-sr39tk expressing the F5 TCR chains and the sr39tk reporter was constructed as previously described.14 Lentiviral vector stocks were produced by transfection of 293FT cells with the pF5-sr39tk, pCMV-ΔR8.2-Δvpr (packaging construct), and pCMV-VSV-G (envelope) plasmids. Cells then were washed and used for the generation of the BLT mice. A fraction of the transduced cells was frozen to be used in CD34 cell injections.
Humanized BLT mice. Mice were generated as previously described.14 Three weeks after the generation of humanized BLT mice, the mice were irradiated at 300 rad using a cobalt-60 source to remove endogenous cells. Then, the frozen transduced CD34 cells were thawed and injected. Mice were bled 4 and 6 weeks after injection to assess reconstitution. All animal protocols were performed under the approval of the University of California, Los Angeles (UCLA) Animal Research Committee.
Quantitative human TCR β chain (TCR-BV) repertoire analysis. Human TCR-BV chain repertoire distribution was quantitatively measured in thymocyte and/or splenocyte harvested from the TCR-transgenic mice using quantitative TCR (qTCR) spectratyping as described and calculated the percent level of transgenic TCR-BV chain among the pool of all TCR-BV chain transcripts.19 Basically, the complementary-DNA of total RNA isolated from thymocyte or splenocyte was prepared using random primers. Using these cDNA, 24 different TCR-BV family specific real-time PCR was performed in the presence BV family specific forward primer and fluorescent labeled TCR β constant region specific reverse primer (FAM/VIC/NED) along with 5′-labeled probe (Cy5-TGTTCCCACCCGAGGTCGC-BHQ2). The resulted amplified products covering the V-D-J junction of each BV family were resolved through ABI-3130 Genetic analyzer (Applied Biosystem, Carlsbad, CA). The peak distributions of every BV family and area under the curve of individual peak within each BV family was analyzed by Genemapper v3.7 software (Applied Biosystem). On the basis of the concentration measured by quantitative real-time PCR for each BV family and the peak intensity measured as area under the curve, the TCR-BV repertoire distribution and the percent concentration of each BV peaks were calculated as described in Figure 4a.
Flow cytometry and cell sorting. Cells were phenotypically analyzed using fluorochrome-conjugated monoclonal antibodies specific for human CD3 (BD Biosciences, San Jose, CA), CD4, CD8, CD25, CD27, CD45, CD45RA, CD45 RO, CD56, CD57, CD62L, and CCR7 (Beckman Coulter, Brea, CA). Tetramer-expressing cells were identified using major histocompatibility complex class I tetramer containing the MART-1 (26–35) peptide conjugated to allophycocyanin or phycoerythrin (Beckman Coulter). Cells were processed by a Fortessa instrument (BD Biosciences) and results were analyzed FlowJo (TreeStar, Ashland, OR) software.
Cells (thymocytes or splenocytes) were also stained for CD3, CD8, CD45, and MART-1 tetramer for sorting. Samples were processed using a FACS Aria. Sorted populations were gated on CD45 to eliminate murine cells and then CD3 to eliminate non–T-cell populations.
Statistical analysis. Statistical analysis tests were performed as indicated in the article and figure legends. Data were analyzed with Student's t-test or one-way analysis of variance and Bonferroni multiple post hoc comparisons. All statistical analyses were performed with GraphPad Prism v5.0 (GraphPad Software, La Jolla, CA). Values with P < 0.05 were considered significant.
SUPPLEMENTARY MATERIAL Figure S1. MART-1 specific CTLs limit growth of MART-1/HLA-A*0201 (M202) melanoma tumors. Figure S2– 6. Surface expression of the MART-1 specific TCR and other endogenous Vβ chains; Vβ13.1 (S3), Vβ9 (S4), Vβ5.3 (S5), Vβ23 (S6), Vβ20 (S7). Figure S7– 11. Intracellular expression of endogenous Vβ chains in transgenic CD8 T cells; Vβ20 (S8), Vβ23 (S9), Vβ5.3 (S10), Vβ13.1 (S11), Vβ9 (S12). Materials and Methods.
Acknowledgments
We thank the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Flow Cytometry Core. We thank the University of California, Los Angeles (UCLA) AIDS Institute/UCLA Center for AIDS Research Mouse/Human Chimera Core for the development and housing of BLT mice. This work was funded in part by National Institutes of Health (NIH) Grant P50 CA086306; California Institute for Regenerative Medicine (CIRM) Grants RC1-00149-1 (to J.A.Z.); UCLA Center for AIDS Research NIH/National Institute of Allergy and Infectious Diseases Grant 5P30 AI028697; and by the UCLA AIDS Institute. The authors declared no conflict of interest.
Supplementary Material
References
- Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM.et al. (2006Cancer regression in patients after transfer of genetically engineered lymphocytes Science 314126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles JJ, Silins SL., and, Burrows SR. Engineered T cell receptors and their potential in molecular medicine. Curr Med Chem. 2006;13:2725–2736. doi: 10.2174/092986706778521959. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA., and, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers CA, Kuhns MS, Egen JG., and, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L., and, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ.et al. (2002Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes Science 298850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M.et al. (2000Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of t cell-mediated vitiligo J Exp Med 1921637–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A.et al. (2010Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy Nat Med 16565–70, 1p following 570. [DOI] [PubMed] [Google Scholar]
- Willemsen RA, Weijtens ME, Ronteltap C, Eshhar Z, Gratama JW, Chames P.et al. (2000Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR Gene Ther 71369–1377. [DOI] [PubMed] [Google Scholar]
- Yang L., and, Baltimore D. Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proc Natl Acad Sci USA. 2005;102:4518–4523. doi: 10.1073/pnas.0500600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Qin XF, Baltimore D., and, Van Parijs L. Generation of functional antigen-specific T cells in defined genetic backgrounds by retrovirus-mediated expression of TCR cDNAs in hematopoietic precursor cells. Proc Natl Acad Sci USA. 2002;99:6204–6209. doi: 10.1073/pnas.092154599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne KJ., and, Crooks GM. Immune-cell lineage commitment: translation from mice to humans. Immunity. 2007;26:674–677. doi: 10.1016/j.immuni.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA.et al. (2006Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1 Nat Med 121316–1322. [DOI] [PubMed] [Google Scholar]
- Vatakis DN, Koya RC, Nixon CC, Wei L, Kim SG, Avancena P.et al. (2011Antitumor activity from antigen-specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells Proc Natl Acad Sci USA 108E1408–E1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen SG, Levin BR, Bristol G, Rezek V, Kim S, Aguilera-Sandoval C.et al. (2012In vivo suppression of HIV by antigen specific T cells derived from engineered hematopoietic stem cells PLoS Pathog 8e1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady BL, Steinel NC., and, Bassing CH. Antigen receptor allelic exclusion: an update and reappraisal. J Immunol. 2010;185:3801–3808. doi: 10.4049/jimmunol.1001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek D. Thymic output and HIV infection: on the right TREC. Immunity. 2004;21:744–745. doi: 10.1016/j.immuni.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF.et al. (1998Changes in thymic function with age and during the treatment of HIV infection Nature 396690–695. [DOI] [PubMed] [Google Scholar]
- Balamurugan A, Ng HL., and, Yang OO. Rapid T cell receptor delineation reveals clonal expansion limitation of the magnitude of the HIV-1-specific CD8+ T cell response. J Immunol. 2010;185:5935–5942. doi: 10.4049/jimmunol.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleckman BP, Khor B, Monroe R., and, Alt FW. Assembly of productive T cell receptor delta variable region genes exhibits allelic inclusion. J Exp Med. 1998;188:1465–1471. doi: 10.1084/jem.188.8.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarukhan A, Garcia C, Lanoue A., and, von Boehmer H. Allelic inclusion of T cell receptor alpha genes poses an autoimmune hazard due to low-level expression of autospecific receptors. Immunity. 1998;8:563–570. doi: 10.1016/s1074-7613(00)80561-0. [DOI] [PubMed] [Google Scholar]
- Jia J, Kondo M., and, Zhuang Y. Germline transcription from T-cell receptor Vbeta gene is uncoupled from allelic exclusion. EMBO J. 2007;26:2387–2399. doi: 10.1038/sj.emboj.7601671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinel NC, Brady BL, Carpenter AC, Yang-Iott KS., and, Bassing CH. Posttranscriptional silencing of V beta DJ beta C beta genes contributes to TCR beta allelic exclusion in mammalian lymphocytes. J Immunol. 2010;185:1055–1062. doi: 10.4049/jimmunol.0903099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady BL, Oropallo MA, Yang-Iott KS, Serwold T, Hochedlinger K, Jaenisch R.et al. (2010Position-dependent silencing of germline V beta segments on TCR beta alleles containing preassembled V beta DJ beta C beta 1 genes J Immunol 1853564–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N.et al. (2010A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model Blood 1151534–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez MG, Kane M, Liu S, Gauld SB, Cambier JC, Torres RM.et al. (2007Ig allotypic inclusion does not prevent B cell development or response J Immunol 1791049–1057. [DOI] [PubMed] [Google Scholar]
- Casellas R, Zhang Q, Zheng N-Y, Mathias MD, Smith K., and, Wilson PC. Ig kappa allelic inclusion is a consequence of receptor editing. J Exp Med. 2007;204:153–160. doi: 10.1084/jem.20061918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucontet L, Sepulveda N, Carneiro J., and, Pereira P. Mechanisms controlling termination of V-J recombination at the TCR gamma locus: Implications for allelic and isotypic exclusion of TCR gamma chains. J Immunol. 2005;174:3912–3919. doi: 10.4049/jimmunol.174.7.3912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.