

Abstract
The industrial development of active immunotherapy based on live-attenuated bacterial vectors has matured. We developed a microsyringe for antigen delivery based on the type III secretion system (T3SS) of P. aeruginosa. We applied the “killed but metabolically active” (KBMA) attenuation strategy to make this bacterial vector suitable for human use. We demonstrate that attenuated P. aeruginosa has the potential to deliver antigens to human antigen-presenting cells in vitro via T3SS with considerable attenuated cytotoxicity as compared with the wild-type vector. In a mouse model of cancer, we demonstrate that this KBMA strain, which cannot replicate in its host, efficiently disseminates into lymphoid organs and delivers its heterologous antigen. The attenuated strain effectively induces a cellular immune response to the cancerous cells while lowering the systemic inflammatory response. Hence, a KBMA P. aeruginosa microsyringe is an efficient and safe tool for in vivo antigen delivery.
Introduction
Since William Coley's discovery in the 19th century, bacterial-based cancer treatment has been envisioned.1 First, the bacteria ability to stimulate the immune system was powerful enough particularly in the bladder cancer treatment with a live Bacillus Calmette–Guerin therapy. Then, more recently, numerous preliminary proof-of-concept experiments demonstrated the vast capacity of bacteria for treating cancer and illustrated the large number of effective tools possessed by these robot factories.2,3 More precisely, heterologous-antigen-specific bacterial-based vaccine has emerged for active immunotherapy.4,5,6,7 In the preclinical and clinical stages, three virulence-attenuated strains of pathogenic bacteria, i.e., Listeria monocytogenes, Salmonella spp. and Pseudomonas aeruginosa, have been used for active immunotherapy.4,8,9,10 These bacterial vaccines enable the in vivo delivery of heterologous antigens and the activation of antigen-presenting cells via their pathogen-associated molecular patterns.
P. aeruginosa possesses a type III secretion system (T3SS), a macromolecular, needle-like apparatus necessary for infection11 by the injection of exotoxins S, T, and U. Here, we use the OST strain, in which all of the major secreted exotoxins are absent (ExoU), or deleted (ExoS, ExoT), and in which an episomal copy of the ExsA activator of T3SS is under the control of an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible promoter.12 This system guarantees the controlled formation of a preassembled needle before bacterial injection into the host (Figure 1a). The first 54 residues of exotoxin S served as secretion tag (S54) to conduct the protein of interest through the T3SS needle and into the cellular cytoplasm, where recombinant protein is processed. Furthermore, it's an attractive strategy for the construction of multivalent vaccine.13 This T3SS-based vaccination with diverse pathogenic bacteria has been proven effective for priming antigen-specific CD8+ T cells6,14,15 against a variety of pathogens (including viruses, intracellular bacteria, such as Listeria16,17,18,19 and parasites, such as Schistosoma japonicum)20 and for antitumoral immunotherapy purposes.14,21
Figure 1.

An overview of the P. aeruginosa “killed but metabolically active” T3SS-based vaccination. (a) The P. aeruginosa strain is cultivated under T3SS-inducing conditions. The protein used for vaccination is overexpressed by bacteria before bacterial inactivation with PCT in the presence of amotosalen. Then, P. aeruginosa is injected subcutaneously (s.c.), and its active T3SS allows protein injection into the host cells. (b) Colony-forming units (CFU) of OST and OSTAB after treatment with increasing amotosalen concentration. The mean of five experiments (performed in duplicate) is represented. (c) Analysis of the membrane integrity of the P. aeruginosa OSTAB strain with the Live/Dead BacLight bacterial viability kit (Invitrogen). OSTAB (live), heat-killed (HK), or photochemically treated P. aeruginosa in presence of 0–15 µmol/l amotosalen concentrations. (d) Western blot analysis of the protein S54-OVA in the supernatant of cultivated OSTAB S54-OVA strain with or without EGTA (i.e., closed or opened syringe), after PCT and in the presence of various amotosalen concentrations. IPTG was added for 3 hours after PCT. Representative blot of three experiments. EGTA, ethylene glycol tetraacetic acid; IPTG, isopropyl β-D-1-thiogalactopyranoside; PCT, photochemical treatment.
The efficacy of an attenuated vaccine vector relies on a subtle balance between minimal toxicity and maximal stimulation of an immune response. This cornerstone, described by Blanders et al., is fundamental for vaccines.22 Although auxotrophic strains (in particular, those that are auxotrophic for aromatic amino acids) have been used with several pathogens, including Salmonella, Listeria, Shigella, and Pseudomonas spp,23 for the production of live-attenuated vaccine strains,23,24,25,26,27 single-deletion mutants, such as aroA mutants, retain sufficient virulence to make them unacceptable for human vaccines.28 Moreover, the potential risks of reversion to a virulent state, viability/propagation in environments other than the intestines and unintentional transfer to other individuals29 are unacceptable. In 2005, Brockstedt et al. developed the concept of vaccines that are “killed but metabolically active” (KBMA) using L. monocytogenes.30 The deletion of two uvr genes (A and B), coding for Exonucleotidase A and B subunits, renders bacteria sensitive to psoralen-induced crosslinking.31 In other words, the ΔuvrAB nucleotide excision repair mutants cannot replicate after photochemical treatment (PCT) with a synthetic, highly reactive psoralen known as amotosalen and upon exposure to long-wavelength ultraviolet A (UVA) light because of the presence of infrequent and randomly distributed crosslinks.32 Recently, this new class of vaccine and its broad applications has been reviewed.33 However, it is unclear whether this PCT could be applied to P. aeruginosa bacteria and whether it preserves T3SS function. To confirm this assumption, we made a ΔuvrAB deletion mutant in P. aeruginosa (named OSTAB, for description see Supplementary Table S1), optimized PCT and studied the capacity of KBMA P. aeruginosa to translocate β-lactamase (Bla) proteins into cells via the T3SS. In vitro, KBMA P. aeruginosa activated human dendritic cells (DCs) and, once activated, the DCs presented an immunogenic peptide to T lymphocytes. Because the KBMA bacteria cannot replicate and induced a lower systemic response than wild type, we sought to characterize the pharmacodynamics of KBMA P. aeruginosa in mice. Finally, the KBMA vaccine elicited functional T CD8+ cells in mice, which correlates with efficacy in mouse models of cancer. Taken together, our findings highlight the potential of using a KBMA P. aeruginosa microsyringe for in vivo antigen delivery, making a step towards a safer vaccine for human use. Furthermore, we provide a promising new therapeutic tool for antigen delivery that stimulates immunity to fight cancer and infectious diseases that are caused by intracellular pathogens.
Results
PCT blocks the replication of an OSTAB mutant without affecting the T3SS
Nucleotide excision repair mutants were obtained by removing the uvrAB genes from the P. aeruginosa strain by allelic exchange.34 We determined the relative sensitivity to photochemical inactivation by testing the new OSTAB and its parental strain over a range of amotosalen concentrations and a UVA dose of 7.2 J/cm2, using conditions established previously for gram-negative bacteria.35 As expected, the nucleotide excision repair–deficient strain, OSTAB, was much more sensitive to PCT than OST (Figure 1b). With 10 µmol/l of amotosalen, there was ~1 live replicating organism per 1.25 × 108 bacteria for OSTAB compared with 2.5 × 106 live bacteria for OST. Higher doses of amotosalen completely abolished bacterial replication, but the associated high adduct frequency in the genome may interfere with protein production. To determine bacterial viability and membrane integrity of the photochemically treated P. aeruginosa OSTAB strain, we used the Live/Dead BacLight assay (Invitrogen, Carlsbad, CA) (Figure 1c). After PCT, bacteria were unable to replicate in the nutrient medium, but they may be scored as “alive” due to their intact membranes, contrary to heat-killed (HK) bacteria. An essential feature of the previously described KBMA organisms is their capacity to transcribe and translate antigenic proteins after PCT.30 As our vaccine is based on antigen injection by the T3SS, we determined the secretion efficiency of a model protein (chicken ovalbumin fragment) fused with S54 (S54-OVA) after PCT with various concentrations of amotosalen (0–20 µmol/l). We assessed the production of secreted S54-OVA into the supernatant of the cultivated strain with or without ethylene glycol tetraacetic acid (EGTA) and with IPTG after PCT. Indeed, calcium depletion induced by EGTA triggers T3SS activation in P. aeruginosa, along with the secretion of T3SS effectors (or S54-OVA) into the culture medium.36 The level of secreted S54-OVA decreased when applying high concentrations of amotosalen from 100% at 0 µmol/l to 30% at 10 µmol/l of amotosalen (relative amount to 0 µmol/l of amotosalen) (Figure 1d). The best compromise between the absence of replication and conserved secretion was observed at 10 µmol/l of amotosalen. Therefore, we applied this condition in the following experiments to obtain a KBMA P. aeruginosa.
We then further investigated the T3SS functionality of our KBMA strain vaccine in different growth conditions. IPTG was added before PCT to induce the production of ExsA and, hence, to control the presence of a microsyringe before PCT. When adding IPTG and EGTA to induce secretion (Figure 2a), we recovered S54-OVA protein in the supernatant of the nucleotide excision repair–deficient strain. Using KBMA S0-OVA, expressing a non-secretable form of OVA, there was no S0-OVA in the supernatant, indicating the absence of bacterial lysis with PCT.
Figure 2.
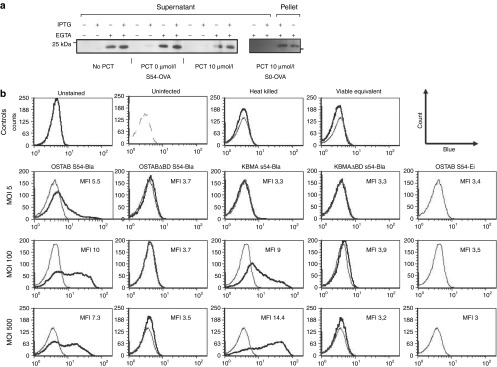
KBMA P. aeruginosa produces, secretes, and injects antigen into cells. (a) KBMA P. aeruginosa secretes S54-OVA antigen in the culture medium in vitro. Bacterial OSTAB S54-OVA were cultivated for 3 hours in the presence or absence of IPTG and EGTA in three different conditions (i.e., No PCT or after PCT with or without 10 µmol/l amotosalen treatment) (b) HL60 target cells were infected for 3 hours with OSTAB, OSTABΔpopBD, KBMA, and KBMAΔpopBD strains with pEiS54 empty (S54-Ei) or associated to the β-lactamase (S54-Bla) at MOIs of 5:1, 100:1, and 500:1. CCF2-AM cleavage by Bla was measured by flow cytometry. Control histograms corresponding to OSTAB S54-Ei for each MOI are represented with thin grey line. A representative of at least three experiments is shown. Mean fluorescence intensity (MFI) is indicated in each panel. EGTA, ethylene glycol tetraacetic acid; KBMA, “killed but metabolically active” IPTG, isopropyl β-D-1-thiogalactopyranoside; MOI, multiplicity of infection; PCT, photochemical treatment.
To better assess the export of proteins into eukaryotic cells by KBMA P. aeruginosa's T3SS, we used strains that deliver Bla fused to ExoS54 (Figure 2b). HL60 cells were incubated for 3 hours with P. aeruginosa strains as previously described.37 The HL60 cells were then loaded with CCF2-AM (Invitrogen) for 30 minutes. CCF2-AM is a fluorescent substrate of Bla; enzymatic cleavage modifies its emitted fluorescence from green to blue.37 Flow cytometry analysis revealed intracellular blue fluorescence, indicating increased CCF2-AM cleavage by the injected Bla for the KBMA S54-Bla strain at multiplicity of infections (MOIs) of 100 and 500. No blue fluorescence was observed in cells that were unstained, uninfected or infected with an equivalent dose of viable bacteria (i.e., 1 live replicating bacteria per 1.25 × 108 exposed bacteria corresponds to an MOI of approximately 500) or in strains harboring an empty plasmid (pEi), a T3SS-deficient S54-Bla plasmid (OSTABΔpopBD S54-Bla) or a KBMA S54-Bla T3SS deficiency (KBMAΔpopBD S54-Bla) (Figure 2b). Taken together, these data demonstrate that the KBMA P. aeruginosa obtained after PCT in 10 µmol/l amotosalen was unable to replicate on a nutrient agar medium but produce and inject proteins by T3SS.
The KBMA P. aeruginosa vaccine is less toxic for cells and avirulent in mice
DCs are the most potent antigen-presenting cells. They play a crucial role in the initiation and modulation of specific immune responses. Moreover, these cells are particularly sensitive to P. aeruginosa.38 Therefore, we examined the cytotoxicity of KBMA P. aeruginosa on DCs. When using human monocyte-derived dendritic cells (moDCs), we observed that live OSTAB kills moDCs in a concentration-dependent manner after 3 hours of coincubation. KBMA is less cytotoxic than a live strain, killing only 17% of cells at its highest MOI (100:1) (Figure 3a). In addition, flow cytometry analysis revealed that OSTAB induced 90% of moDC death (Annexin V+ and/or 7AAD+) 24 hours after infection at all investigated MOIs (Figure 3b). KBMA bacteria is less toxic than live bacteria at lower MOIs, but it killed cells at levels comparable with live bacteria at MOIs greater than 20:1.
Figure 3.

KBMA P. aeruginosa is poorly cytotoxic and leads to a mild inflammatory response in vivo. (a) The cytotoxicity of KBMA P. aeruginosa on human moDCs measured by LDH released from dead cells 3 hours after infection at different MOI (1:1; 5:1; 20:1; 100:1) (Mean and SD from five experiments) and (b) the viability analyzed by 7AAD/AnnexinV flow cytometry assay 24 hours after infection with increasing MOI (SD from three different donors). The “-” represents not treated cells. (c) A multiplex immunoassay was used to determine the expression levels of 23 different cytokines in mouse plasma samples that were obtained 24 hours after vaccination (n = 3) with KBMA S54-OVA (triangles) or OSTAB S54-OVA (squares) (5 × 107 bacteria s.c.) versus the phosphate-buffered saline (PBS) control (circles). Only the” cytokines that were significantly different from the control (*P < 0.05) are shown in mice vaccinated with KBMA. The bar represents the mean of the data. KBMA, “killed but metabolically active” MFI, mean fluorescence intensity; MOI, multiplicity of infection.
To evaluate the toxicity of KBMA P. aeruginosa in vivo, we examined the survival of 6-week-old female C57BL/6J mice after a single subcutaneous (s.c.) injection of 5 × 107, 5 × 108 or 5 × 109 bacteria (Supplementary Table S3). All of the mice injected with a 5 × 107 dose of OST S54-OVA died within the first 24 hours. No death was observed in the group injected with 5 × 107 KBMA S54-OVA or in groups injected with 5 × 106 bacteria. We then assessed the lethality of the 5 × 108 and 5 × 109 doses of KBMA S54-OVA. One of the three mice injected with a 5 × 108 dose died, and all of the mice died with a 5 × 109 bacterial dose.
A multiplex immunoassay screening for cytokines and chemokines was used to measure the proinflammatory effect of KBMA P. aeruginosa vaccinations. Multiplex xMAP technology was performed on mice vaccinated with phosphate-buffered saline (PBS), OSTAB S54-OVA and KBMA S54-OVA (5 × 107 bacteria). Among the molecules analyzed, levels of IL-1α, Kc, IL-13, G-CSF, MIP-1α and RANTES were significantly higher (P = 0.00495) in KBMA-vaccinated mice than in PBS-injected animals (Figure 3c). At this dose, all of the cytokines and chemokines analyzed were significantly higher in OSTAB S54-OVA vaccinations than in those with PBS (P < 0.005) and in OSTAB S54-OVA vaccinations than in those with KBMA S54-OVA (Supplementary Table S4). Interestingly, IL-17 levels were significantly lower in KBMA-vaccinated mice than in OSTAB- or PBS-vaccinated mice (P = 0.00495).
On the basis of the induced systemic responses described, these experiments demonstrate that KBMA S54-OVA bacteria are less toxic and less reactogenic in vivo than the OSTAB strain.
KBMA P. aeruginosa disseminates in lymphoid organs, similar to the OSTAB strain
As the systemic response decreased after vaccination with the KBMA strain, P. aeruginosa dissemination was evaluated following s.c. injection. When live P. aeruginosa were injected, bacteria are recovered on Pseudomonas isolation agar plates containing gentamicin (guarantying OSTAB GmR strain isolation) from various homogenized organs, such as the lymph nodes, spleen, liver, skin, lungs, and kidneys (Figure 4a). In comparison, no replicating bacteria were detected in the animals when they were injected with KBMA P. aeruginosa, indicating an irreversible attenuation process following in vivo injection. Radiolabeling was employed as an alternative technique to evaluate KBMA dissemination due to its inability to replicate. Live and KBMA bacteria were labeled with 99mTc to enable their detection with high sensitivity. Bacterial labeling did not modify their replication on agar plates and therefore did not affect the viability of the live strain. As demonstrated by nanoSPECT/CT molecular imaging, a slow dissemination was observed following s.c. injection of 5 × 107 bacteria, with ~85% and 50% of the injected dose remaining at the injection site at 3 and 24 hours after injection, respectively, in both live and KBMA strains. In comparison, free 99mTc diffused rapidly, with only 3% injected dose remaining at the injection site after 24 hours (Figure 4b). This discrepancy between 99mTc-labeled bacteria and free 99mTc diffusion strongly supports the in vivo stability of the radiolabeling. However, some free 99mTc was detected 24 hours after injection of the labeled bacteria, as demonstrated by the activity found in tissues that are known to express the Na/I symporter, e.g., salivary glands, stomach and thyroid (Figure 4c). This activity might be attributed to a loss of 99mTc from living bacteria or, more likely, to 99mTc release following bacterial degradation by the immune system. In all other evaluated tissues, comparable dissemination was observed between OSTAB and KBMA strains, with only moderately significant differences (24 and 29%) in the liver and the spleen (P < 0.05). Therefore, we conclude that KBMA P. aeruginosa dissemination is similar to OSTAB dissemination.
Figure 4.

KBMA P. aeruginosa dissemination in vivo. (a) The effect of photochemical treatment on the number of colony-forming units 24 hours after subcutaneous (s.c.) injection of 5 × 107 bacteria. (−) Absence of CFU/ml, (+) ~101 CFU/ml, (++) ~102 CFU/ml, (+++) >5.102 CFU/ml. (n = 3 per group) (b) Representative SPECT/CT images of 5 × 107 99mTc-radiolabeled bacteria 3 hours and 24 hours following s.c. injection in the flank (arrowhead). Free 99mTc is shown as control. (c) Comparison of 99mTc-labeled OSTAB and KBMA bacteria retrieved from different organs 24 hours following s.c. injection (n = 6 mice per group). ID, injected dose; KBMA, “killed but metabolically active.”
KBMA P. aeruginosa activates and injects antigens into human dendritic cells, which then stimulates antigen-specific CD8+ T cells
The DC maturation process coordinates the regulation of antigen processing and presentation, the expression of costimulatory molecules, and the secretion of cytokines that prime CD8+ and CD4+ T-cell responses. We evaluated whether DC maturation occurred when they are exposed to KBMA P. aeruginosa delivering the flu M1 antigen. Immature day 5 moDCs displayed typical morphology and an immature phenotype (moderate levels of CD80, CD86, CD40, and CD83) (data not shown). Immature moDCs were incubated for 3 hours with one of three IPTG-induced OSTAB strains: live (MOI 5:1), KBMA (MOI 100:1), or HK (MOI 100:1). Functional features were assessed 24 hours later. Phenotypic analysis revealed that stimulation with both the KBMA S54-M1 and HK S54-M1 strains led to increases in the surface expression levels of costimulatory molecules (CD80, CD86, CD40) and maturation-associated (CD83) molecules (Figure 5a). The degrees of upregulation were comparable with the activation induced by the controls (lipopolysaccharide and CD40L). We did not observe upregulation of CD80, CD86, CD40, and CD83 with live strain (OSTAB S54-M1) or equivalent live bacteria (eqCFU) but probably due to cytotoxicity effect at early stage of injection.
Figure 5.

The KBMA P. aeruginosa vaccine activates human moDCs and induces antigen-specific CD8+ T-cell responses in vitro. (a) Expression of the maturation markers (CD80, CD86, CD83, and CD40) on the surface of human moDCs infected with live or KBMA strains, at MOI 100:1. (a,b) Not treated lipopolysaccharide (LPS) and CD40L were used as controls. (b) Cytokine secretion by human moDCs following live-strain and KBMA stimulation (n = 5); (c,d) Cross-presentation of flu M1 epitope by DCs submitted to OSTAB or KBMA strains assayed by intracellular IFN-γ labeling within tetramer flu-specific CD8+ T cells from cocultures. T2 and mDC loaded with the epitope were used as controls. (c) One representative experiment, (d) mean of four experiments. (e) Cross-presentation of flu M1 epitope by DCs submitted to OSTAB or KBMA strains assayed by IFN-γ measurement in the supernatants of the cultures (n = 5–8 experiments). HK, heat-killed; KBMA, “killed but metabolically active” MOI, multiplicity of infection.
We have analyzed whether the tested strains induce cytokine secretion by moDC. KBMA P. aeruginosa and the positive controls (lipopolysaccharide and CD40L) induced the secretion of TNF-α, IL-6, IL-8, and IL-12 to comparable levels. KBMA induced higher cytokine secretion levels than the OSTAB strain (Figure 5b), provided the tests were conducted at high MOIs. KBMA induced higher levels of IL-1β than the positive control or the OSTAB strain.
As shown previously by Epaulard et al.,21 recombinant protein injected via P. aeruginosa's T3SS is efficiently processed by the major histocompatibility complex class I pathway. Here, we used OVA-specific CD8+ T-cell hybridoma cells (B3Z)39 to determine the capacity of primary C57BL/6J mouse BMDCs infected with KBMA P. aeruginosa to process and present the immunodominant major histocompatibility complex class I OVA epitope SIINFEKL to CD8+ T-cell hybridoma (Supplementary Figure S1). We incubated BMDCs for 3 hours with each strain (live or KBMA) after IPTG preinduction of the T3SS. TCR-mediated B3Z activation was obtained after incubation with DCs that had been exposed to the KBMA S54-OVA strain. In contrast, we observed no activation with BMDCs infected with HK OSTAB S54-OVA and equivalent live bacteria, i.e., 10 bacteria per ml. Hence, the signal obtained for the KBMA S54-OVA could not be due to this viable equivalent.
In complementary experiments, we evaluated priming of antigen-specific T cell by human moDCs that are currently used for cellular immunotherapy40,41 treated by KBMA strain injecting the antigen. We choose the viral (flu) M1 antigen to address this question rather than a tumor antigen because it's hard to produce tumor-specific CD8 T cells from healthy donors. DCs infected with OSTAB S54-M1 or KBMA S54-M1 were used to activate flu M158–66 HLA-A*0201-specific CD8+ T cells. Activation was assessed by measuring interferon γ (IFN-γ) production via intracellular staining within tetramer+ CD8+ T cells (Figure 5d). IFN-γ secretion was observed in the presence of flu-peptide-loaded T2 cells or moDCs, and in the presence of moDCs that processed the bacteria vaccine containing the flu M158–66 epitope (OSTAB and KBMA). As expected, IFN-γ secretion was not observed in control empty vectors (pEi). The results indicate that the flu peptide was effectively processed and presented by the moDCs infected by OSTAB and KBMA S54-M1 strains. Figure 5c is a representative row data of the intracellular IFN-γ labeling within tetramer-positive CD8 T cells whereas Figure 5d represents the pool of several experiments. Figure 5c clearly shows that the IFN-γ levels obtained with OSTAB M1—and KBMA M1—pulsed moDCs (11.1 and 8.5% respectively) are higher compared with the level obtained in control conditions (mDC loaded with control peptide, 1.7%). Figure 5d is a pool of six experiments with differential background levels. Therefore, the final mean abrogates the intraexperiment difference between the conditions. To be more consistent on this important point, we performed additional experiments by assessing the IFN-γ release in the supernatants of the cocultures (Figure 5e). This figure clearly shows a difference between the control condition (−) and OASTB M1 and KBMA M1 conditions.
In brief, KBMA P. aeruginosa is able to activate and mature DCs and can load them with antigens that will be processed in an major histocompatibility complex class I restricted presentation to CD8+ T cells.
Vaccination with KBMA P. aeruginosa induces specific cellular immunity and protects mice from tumor development
To assess the capacity of the KBMA vector to raise specific CD8+ T lymphocytes in vivo, C57BL/6J mice were injected s.c. with 5 × 106 or 5 × 107 bacteria or with multisite 2X 5 × 107 or 4X 5 × 107 bacteria on day 1 and 8. Spleens were harvested on day 14. Enzyme-linked immunosorbent spot (ELISpot) assays were performed from the splenocytes stimulated with the OVA peptide epitope SIINFEKL. The spleens of mice that were immunized with an OVA-secreting strain had a higher frequency of IFN-γ secreting cells than the control PBS-immunized mice (Figure 6a). The number of spot-forming cells per 4 × 105 splenocytes was significantly higher in the KBMA strain than in HK S54-OVA P. aeruginosa. Therefore, the KBMA strain induced a SIINFEKL peptide-specific T cell response in vivo as assessed in vaccinated mice. KBMA vaccination in a 4X multisite injection procedure with 2 × 108 bacteria reach the same rate of peptide-specific T CD8+ cells as the OSTAB strain vaccination (5 × 106 bacteria).
Figure 6.
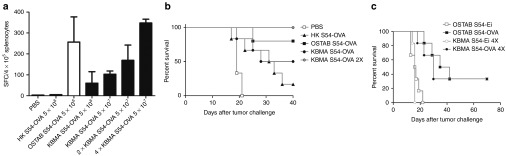
The KBMA P. aeruginosa immunotherapy stimulates the production of specific lymphocyte CD8+ T cells in vivo and prolongs survival of mice bearing B16-OVA tumor. (a) The average number of spot-forming cells (SFC) per 4 × 105 splenocytes of mice subcutaneous (s.c.) injected with phosphate-buffered saline (PBS), heat-killed (HK) S54-OVA, OSTAB S54-OVA, or KBMA S54-OVA strains at 5 × 106, 5 × 107, 2X 5 × 107, or 4X 5 × 107 bacteria. Mice were s.c. injected on day 14 and 7 in the right flank (1X vaccination), in the right and left flank (2X vaccinations), and in the right and left flanks and the right and left shoulders (4X vaccinations) with 5.107 KBMA S54-OVA bacteria/injection site. ELISpot was used as indicated by the manufacturer. The results presented are the means of three independent experiments. (b–c) C57BL/6J mice received s.c. B16-OVA tumor cells (2 × 105 cells) on day 0. (b) The survival curves of PBS-treated control mice and mice that were prophylactically vaccinated on 14 and 7 days in the right flank with OST S54-OVA (5 × 106), KBMA S54-OVA (5 × 107 in one or two sites of injection), and HK OSTAB S54-OVA (5 × 107). Log-rank test, KBMA S54-OVA 2X versus PBS, P = 0.0006. (c) The survival curves of mice challenged with B16-OVA and vaccinated sixfold (d+1 and every 4 days) with KBMA P. aeruginosa or live strains. Animals were killed when tumor diameters exceeded 10 mm in any dimension for both vaccination schemes. The population size was n = 6 per group. Log-rank test, OSTAB S54-Ei 4X versus OSTAB S54-OVA 4X group, P = 0.0012. KBMA, “killed but metabolically active.”
We then evaluated the potential of a KBMA P. aeruginosa microsyringe for cancer immunotherapy. This evaluation was performed using a mouse model of syngenic B16 melanoma cells that express OVA antigen (B16-OVA cell line). The injection of B16-OVA cells into C57BL/6J mice resulted in rapid tumor development. The capacity to stimulate immunity against B16-OVA cells with a KBMA P. aeruginosa vaccine was first evaluated in a prophylactic vaccination scheme. A conventional prime (day 14) and boost (day 7) immunization dosing regimen and a tumor challenge at day 0 was conducted. We observed significantly better survival in mice vaccinated with two injections of KBMA S54-OVA (2X 5 × 107 bacteria) as compared with HK S54-OVA or PBS (P = 0.0006) (Figure 6b). In contrast, there was no significant difference between OST S54-OVA and KBMA S54-OVA (2X 5 × 107 bacteria).
To confirm the curative efficacy of the vaccine, we further investigated the survival of mice vaccinated according to our previously established therapeutic vaccination schema.13 B16-OVA cells were implanted at day 0, and mice were then vaccinated six times with KBMA P. aeruginosa or live strains (1 day after and every 4 days). We observed that, in contrast to the non-immunized group (OST-EI), all of the OVA-expressing strains exhibited efficient tumor protection. The best tumor rejection efficacy occurred with 4X injections of KBMA and OSTAB S54-OVA (Figure 6c) (P = 0.0012).
Hence, KBMA is an efficient strain to stimulate specific immune response in vivo and to cure tumor.
Discussion
Here, we report for the first time on an attenuated, KBMA P. aeruginosa T3SS-based vaccine. We used P. aeruginosa for two reasons: (i) P. aeruginosa is a pathogen that serves as an adjuvant, and we know that the host's perceived level of danger directly influences the type and intensity of the immune response;22 and (ii) P. aeruginosa's T3SS is a microsyringe, which is useful for cellular protein delivery and it helps to stimulate effector and memory T lymphocytes. In this study, the motivation for inhibiting P. aeruginosa's replication was based on two major observations: (i) although its toxicity was attenuated with multiple virulent gene deletions,24 P. aeruginosa was not considered safe enough to be administered to immune-compromised patients with cancer; and (ii) bacterial strain vaccines should not disseminate in the environment and potentially infect other individuals.
According to the KBMA attenuation strategy, we rendered attenuated P. aeruginosa strains sensitive to PCT. We obtained an optimized KBMA P. aeruginosa strain with a combination of 10 µmol/l of amotosalen and UVA exposure. Furthermore, we show for the first time that P. aeruginosa's T3SS is still able to inject/deliver heterologous proteins into human cells after PCT in vitro.
Moreover, our results demonstrated that KBMA P. aeruginosa is less cytotoxic to murine and human DCs during early infection (3–4 hours) and that DCs viability is higher 24 hours after KBMA infection than with live strain. Because KBMA P. aeruginosa has attenuated cytotoxicity, we evaluated the “host danger response” by analyzing global cytokine production in vivo following vaccination. We found that KBMA-vaccinated mice had higher levels of proinflammatory cytokines IL-1, IL-6 and Kc (IL-8 in humans) in vivo than the control PBS-vaccinated mice, but their cytokine levels were significantly lower than those in mice injected with live bacteria (P < 0.05). Therefore, KBMA P. aeruginosa induced proinflammatory cytokine production in the vaccinated host. This response is necessary for successful treatment without systemic inflammation, which is deleterious for the patient (for instance systemic inflammatory response syndrome or compensatory anti-inflammatory response syndrome severe infection). Contrary to the in vivo results, the coincubation of KBMA with human moDCs in vitro led to higher cytokine levels than those in the live strain. This result illustrates the potent local adjuvant effect of KBMA P. aeruginosa and the benefits of lower cytotoxicity in terms of the activation/maturation of antigen-presenting cells. On the basis of our current knowledge on DC activation by microbial products, it is not surprising that KBMA induces DC maturation in vitro. However, the DC maturation state is an important parameter for determining the clinical effectiveness of DC-based immunotherapy.
Because the systemic response to the vaccine was decreased in mice, we decided to examine the dissemination of the KBMA P. aeruginosa vaccine after s.c. injection in the right flank using nanoSPECT/CT nuclear imaging. Slow bacterial dissemination was observed. We recovered bacteria in secondary lymphoid organs, such as the spleen and lymph nodes, where injection to DCs could be important for the response.
When compared with non-treated P. aeruginosa, injections of KBMA S54-OVA stimulated antigen-specific CD8+ T cells and protected mice from tumor challenge for 40 days. The benefit of using KBMA P. aeruginosa is that a controlled amount of bacteria can be injected. Indeed, with less live bacteria, there is a non-controlled proliferation of the bacterial vector within the host that depends on complex, individual host/microbe interactions. The results in mice harboring OVA-expressing tumors demonstrated tumor regression in 33% of the multisite-vaccinated mice, representing a promising approach to vaccination with live-attenuated bacterial strains.
In conclusion, our data indicate that the T3SS in KBMA P. aeruginosa is an effective in vivo antigen delivery system. PCT of P. aeruginosa allows greater amounts of antigen to be delivered to DCs than when using a live strain without an increased systemic response. No live bacteria are released into the environment, and the patient has no risk of developing an infection. The future of live bacteria-based vaccines is linked to the success of the first product line developed by three biotech companies, AduroBiotech, Advaxis, and APCure.
Materials and Methods
Bacterial strains, plasmids, cell lines, and mice. The bacterial strains, plasmids, cell lines, mice, and primers used in this work are listed in Supplementary Tables S1 and S2. The OSTAB mutants were generated from the CHA-OST21 P. aeruginosa strain by Cre/lox-based mutagenesis.34
Human peripheral blood mononuclear cells were obtained from HLA-A*0201 + healthy donors by Ficoll-Hypaque density gradient centrifugation (Eurobio, Les Ulis, France). moDCs were differentiated from blood monocytes using 500 U/ml of GM-CSF and 10 ng/ml of IL-4 (TEBU Peprotech, Le Perrayen-Yvelines, France) for 6 days. This study was conducted under a procedure approved by the French Blood Agency's Institutional Review Board. All donors signed informed consent forms.
BMDCs from C57BL/6J mice were prepared by C. Villiers at IAB (Grenoble, France) as previously described and were given on day 10.21
Amotosalen/UVA inactivation of bacteria and viability assay. For PCT, the ΔuvrAB strain was cultivated in LB medium at 37 °C and 250 rpm until an OD600 of 0.5 was reached. Various concentrations of amotosalen (a gift from Grenoble EFS) were added, and cultures were grown for 1 hour. One milliliter of culture (OD600 of 1) was then transferred in each well of a six-well culture plate for UVA irradiation at a dose of 7.2 J/cm2 in a Stratalinker 1800 device (Stratagen, La Jolla, CA). Viability of the photochemically inactivated cultures was assessed by serial dilution and plating on PIA for colony-forming units. Points represent mean values of triplicate plates counted at the most appropriate dilution (Figure 1b). The Live/Dead BacLight Bacterial Viability kit (Invitrogen) was used with manufacturer's instructions to assay the membrane integrity of the photochemically treated bacteria. With an appropriate mixture of the SYTO 9 and the propidium iodide stains, bacteria with intact cell membranes stain fluorescent green, whereas bacteria with damaged membranes stain fluorescent red.
T3SS analysis by secretion assay and Bla injection. OSTAB was transformed with the pEiS54-OVA plasmid. Resulting strains were grown overnight in LB containing 300 mg/l of carbenicillin. The next day, strains were grown in LB medium during PCT. After a washing step, the cells were resuspended in LB with or without 0.5 mmol/l of IPTG, 5 mmol/l of EGTA, and 20 mmol/l of MgCl2. The resuspension was cultivated for 3 hours at 37 ° C. The EGTA-induced calcium depletion triggers the activation of P. aeruginosa's T3SS and the secretion of T3SS effectors in the culture medium.21 Supernatants were precipitated with 15% perchloric acid as described by Derouazi et al.42 and were analyzed by SDS-PAGE (Mini-PROTEAN Tris Glycine 12% precast gel (Bio-Rad, Milan, Italy)). Immunoblotting was performed with polyclonal rabbit anti-chicken ovalbumin antibody (AbDserotec, Oxford, UK) at 1/5,000 in tris-buffered saline with 0.5 mg/ml bovine serum albumin to test for the presence of the secreted fusion protein S54-OVA. A control strain (not exposed to UVA) was conserved at 4 °C and OD600 = 1 during the PCT of the other strains. When necessary, we quantify the relative amount of OVA by densitometry analysis with ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, MD; http://imagej.nih.gov/ij/, 1997–2012). We also tested for the presence of the OVA antigen, as previously described by Derouazi et al.,42 to verify its secretion in all vaccination protocols (ELISPOT or Tumor challenge vaccination). For the Bla injection assay,37 HL60 cells were washed once in a complete RPMI medium without antibiotics and were seeded in 48-well tissue culture plates at 2.105 cells/well in a complete RPMI medium without antibiotics. After 3 hours of infection at 37 °C and 5% CO2, the P. aeruginosa strain expressed the S54-Bla fusion protein. Cells were then incubated in a freshly prepared 6xCCF2/AM solution (1 μmol/l final concentration; Invitrogen) for 30 minutes in the dark at room temperature. The percentage of cells that received S54-Bla reporter fusions was quantified by flow cytometry (FACS Moflo; Dako Cytomation, Brea, CA). The results are expressed as the percentage of cells that exhibited blue fluorescence. Uninfected cells incubated with CCF2-AM were used as a negative control.
Cytotoxicity assay. Cytotoxicity assays on murine and human DCs were assessed in vitro. Prior to infection, C57BL/6J BMDCs were seeded in 96-well plates at 5 × 104 cells/well for 4 hours at 37 °C with 5% CO2. Both PCT and non-PCT bacteria were washed two times in Iscove's modified Dulbecco's medium supplemented with 1X non-essential amino acids and 1% fetal calf serum. BMDCs were infected at an MOI of 10:1, and the infection was performed for 3 hours in 200 μl of Iscove's modified Dulbecco's medium supplemented in a candle jar at 37 °C. Cytotoxicity was assessed by the release of lactate dehydrogenase into the supernatants of the infected cells using a cytotoxicity detection kit (LDH; Roche, Mannheim, Germany).43 The percentage of cytotoxicity in each experiment was calculated with the following equation: (experimental value − control with only cells)/(control with 1% Triton X-100 − control with only cells) × 100.
Dissemination in mouse. For a count of colony-forming units, three mice were injected s.c. with 5.107 bacteria. The animals were sacrificed 24 hours after injection, and their various organs were immediately collected and placed into 100 µl of medium. The organs were then mechanically homogenized under sterile conditions. A serial tenfold dilution was performed, and the solutions were plated on PIA Gm.
Labeling with Technetium-99m. The bacterial suspension was centrifuged (13,000g, room temperature for 3 minutes). The supernatant was discarded, and the pellet was mixed with 0.2 ml of 480 MBq 99mTc and 10 µl of a SnCl2 solution (1 mg/ml). After stirring and 20 minutes of incubation at room temperature, the labeled bacteria were washed twice in 0.9% NaCl by centrifugation. To prevent reduced Tc reoxidization, the pellet was resuspended each time in 0.9% NaCl solution containing 0.25 g/l of ascorbic acid as a reducing agent. Radiolabeled bacteria were resuspended in 0.2 ml NaCl 0.9% NaCl, and 10 µl was used for enumeration of viable organisms. The viability of the radioactive inoculum was similar to that of mock-labeled bacteria that had been processed under the same conditions. The in vivo experiments were performed by s.c. inoculation of 100 µl of bacteria (5 × 107) in PBS into the rear left flanks of C57BL/6J mice. Dual modality SPECT/CT acquisitions were performed under anesthesia (1.5% isoflurane) using a dedicated preclinical camera (nanoSPECT/CT; Bioscan, Washington, DC). The animals were placed in a temperature-controlled bed, and whole body acquisitions were performed using a four-head system equipped with high-sensitivity and high-resolution multipinhole collimators (9 × 1.4 mm pinholes per head) using the following parameters. CT acquisitions were performed at 45 keVp, with 240 projections and 1 second per projection. SPECT acquisitions were performed using a 140 ± 20% KeV window, with 24 projections and a 10- or a 30-minute scan. CT and SPECT acquisitions were reconstructed fused and analyzed using dedicated software (InVivoScope, Bioscan).
moDC analysis. Human moDCs were incubated for 3 hours in RPMI 1,640–10% fetal bovine serum with P. aeruginosa strains at MOIs of 5:1 or 100:1. The DCs were then incubated with 250 µg/ml of gentamicin (Sigma, St Louis, MO) to kill live bacteria for an additional 24 hours in RPMI 1,640–10% fetal bovine serum. The moDCs were then washed three times with PBS. For the positive controls, moDCs were matured with 1 µg/ml of soluble recombinant human CD40L and 1 µg/ml of enhancer following the manufacturer's instructions (Alexis Biochemicals, San Diego, CA) or 1 µg/ml of lipopolysaccharide (Sigma).
Antihuman mouse monoclonal fluorescent-conjugated antibodies were used to control moDC differentiation and maturation (i.e., anti-CD14, CD40, CD80, and CD83 (Beckman Coulter, Brea, CA) and CD209, CD11c, and isotype controls—BD Biosciences, San Jose, CA). Viability was assessed by flow cytometry via double-negative staining with FITC-conjugated Annexin V and 7-amino-actinomycin D (7AAD) (Beckman Coulter). Cytokine levels were assayed by Cytometric Bead Array (BD Biosciences) following the manufacturer's instructions.
For in vitro functional assays, antiflu M1 cytotoxic T lymphocytes were generated as previously described.44 Briefly, GEN DCs that had been loaded with 1 µmol/l flu M158–66 (GILGFVFTL) peptide (NeoMPS) and irradiated (30 Gy) were cocultured with peripheral blood mononuclear cells from an HLA-A*0201+ donor at a 1:10 ratio in complete medium. After 1 week, cells were recovered and enriched in CD8+ T cells by magnetic beads following the manufacturer's recommendation (StemCell, Grenoble, France). Specificity was controlled by iTag HLAA*0201 tetramers (Beckman Coutler, Fullerton, CA).
IFN-γ secretion by tetramer+ CD8+ T cells was analyzed as follows. T cells were first labeled with iTAg HLA-A*0201 tetramer-PE for 30 minutes at room temperature, then washed and restimulated with peptide-pulsed T2 cells (10:1 ratio) or mDC preincubated with KBMA for 5 hours 30 minutes. Brefeldin A (1 µl/ml, BD Biosciences) was added for the last 3 hours. Cells were then surface-labeled with anti-CD3-PC7 and anti-CD8-APC antibodies and were subjected to IFN-γ intracellular staining (BD Biosciences). IFN-γ staining was analyzed on the tetramer+ CD8+ T cells.
B3Z T cell hybridoma activation. C57BL/6J BMDCs (1 × 105 cells) were incubated with P. aeruginosa at MOI 5:1 for 3 hours at 37 °C and 5% CO2. Then, gentamicin was added at 250 µg/ml to eliminate bacteria. Supernatants were discarded after 30 minutes. B3Z T cells and lacZ-inducible CD8+ T-cell hybridomas specific to the OVA257–264 (SIINFEKL) epitope presented on the murine H-2Kb class I molecule were then added at a DC/T cell ratio of 1:3 for 16 hours in RPMI with 10% fetal bovine serum, 50 µmol/l of β-mercaptoethanol, 500 µg/ml of geneticin, and 250 µg/ml of gentamicin. After centrifugation, β-galactosidase activity was determined as previously described.45
Multiplex bead-based immunoassay. Retro-orbital blood sampling (200 µl) from PBS-, KBMA-, and OSTAB S54-OVA-infected mice (n = 3) were collected in citrated tubes at 24 hours p.i. and were immediately centrifuged at 13,000g for 10 minutes at 4 °C. Plasma samples were stored at −80 °C. Samples were frozen and thawed only once. A mouse cytokine 23-Bio-Plex assay kit was used to perform measurements (Bio-Rad). IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, Kc, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-17, Eotaxin, G-CSF, GM-CSF, IFN-γ, MCP-1, MIP-1α, MIP-1β, RANTES, and TNF-α were measured using Luminex technology according to the manufacturer's instructions (BioRad Laboratories, Hercules, CA). The plasma samples were diluted 1:4 and were tested in duplicate. Data collection and analysis were performed on a Bio-Plex 200 instrument equipped with Bio-Plex Manager software (version 4.1) (BioRad Laboratories) using a five-parameter non-linear regression formula to compute sample concentrations from the standard curves. The cytokine detection limit ranged between 0.2 and 45,000 pg/ml. Concentrations outside the detection limit were assigned the lowest or highest value from the corresponding standard curve.
Animal experiments. Female C57BL/6J mice were purchased at an age of 5–6 weeks from the Elevage Janvier (Le Genestet St. Isle, France) and were kept under specific pathogen-free conditions in the PHTA animal facility at the University Joseph Fourier (Grenoble, France). All animal experiments were approved by the Animal Experiment Committee of the Region and were performed in accordance with institutional and national guidelines. Mice were anesthetized with sevoflurane prior to s.c. injection of bacteria or tumor cells. If mice had tumors larger than 10 mm in the greatest dimension or if skin ulceration occurred, they were euthanized.
IFN-γ ELISpot. Mice were immunized with bacteria in 100 µl PBS twice in a 1-week interval (day 14 and day 7). On day 0, splenocytes were prepared, harvested, and resuspended in RPMI with 10% fetal bovine serum at 4 × 106 cells/ml. ELISpot analysis for detecting OVA-specific CD8+ T cells was performed according to the manufacturer's protocol (Diaclone, Besançon, France). Briefly, cells were stimulated with 5 µg/ml of OVA (SIINFEKL) peptide in an IFN-γ capture ELISpot plate (MultiScreen HTS filter plates; Millipore, Danvers, MA) for 18–20 hours. After that, the number of spot-forming cells was counted using an automated ELISpot reader (Bioreader 4,000 Pro; Biosys, Karben, Germany).
Protection against tumor cell challenge. In the prophylactic assay, six mice per group were immunized according to the schedule mentioned in section IFN-γ ELISpot of the methods to analyze the capacity of the KBMA strain to stimulate immune response. Mice were s.c. injected with different strains in the right flank 14 and 7 days before the s.c. injection of 2.105 B16-OVA cells in the left flank. For the curative assay, six mice were s.c. injected with 2.105 B16-OVA cells in 100 µl of PBS on day 1, then vaccinated with different strains on the next day and every 4 days, up to six times.
Statistical analysis. Results are expressed as the means ± SD unless stated otherwise. Statistical comparisons between two groups were evaluated by the Student's t-test and corrected by ANOVA for multiple comparisons. A P value of <0.05 was considered to indicate statistical significance. Unless otherwise indicated, all experiments were conducted at least twice. Log-rank tests (Mantel–Cox) were used to compare the Kaplan–Meier tumor survival curves and were performed using GraphPad Prism (version 5.00 for Windows, GraphPad Software, San Diego, CA; www.graphpad.com). For multiplex cytokines analysis, Mann–Whitney test statistical analysis was performed.
SUPPLEMENTARY MATERIAL Figure S1. KBMA P. aeruginosa deliver OVA to BMDCs, where the antigen is loaded into the major histocompatibility complex class I pathway. Table S1. Bacterial strains, plasmids, cell lines, and mice used in this study. Table S2. Primers used in this study. Table S3. The in vivo toxicity of KBMA P. aeruginosa. Mortality of C57BL/6J mice after s.c. injection of 5 × 106, 5 × 107, 5 × 108, or 5 × 109 bacteria. Table S4. Analysis of systemic response by xMAP technology.
Acknowledgments
The authors thank O Epaulard and AL Fiser for their assistance in developing this manuscript, and G Baptist and J Geiselmann for their assistance with the live/dead BacLight experiment. This work was supported by GRAVIT grant number 081202 and Ligue contre le cancer (comité ardèche). The authors thank the “Etablissement Français du sang” for the gift of amotosalen S-59, the access to ELISPOT reader. Finally, authors thank Ina Attree at CEA (Grenoble) for the access to the moFlo FACS. ALG, BP, and BT declare competing financial interest. They are the cofounders of APCure, a start-up that produces immunotherapy treatments for patients with cancer. The work was done in Grenoble, France.
Supplementary Material
References
- Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Am J Med Sci. 1893;105:488–511. [PubMed] [Google Scholar]
- Bernardes N, Seruca R, Chakrabarty AM, Fialho AM. Microbial-based therapy of cancer: current progress and future prospects. Bioeng Bugs. 2010;1:178–190. doi: 10.4161/bbug.1.3.10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes NS. Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer. 2010;10:785–794. doi: 10.1038/nrc2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton HA. Pathogenic bacteria as vaccine vectors: teaching old bugs new tricks. Yale J Biol Med. 2010;83:217–222. [PMC free article] [PubMed] [Google Scholar]
- Guirnalda P, Wood L, Paterson Y. Listeria monocytogenes and its products as agents for cancer immunotherapy. Adv Immunol. 2012;113:81–118. doi: 10.1016/B978-0-12-394590-7.00004-X. [DOI] [PubMed] [Google Scholar]
- Hegazy WA, Xu X, Metelitsa L, Hensel M. Evaluation of Salmonella enterica type III secretion system effector proteins as carriers for heterologous vaccine antigens. Infect Immun. 2012;80:1193–1202. doi: 10.1128/IAI.06056-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevil Domènech VE, Panthel K, Winter SE, Rüssmann H. Heterologous prime-boost immunizations with different Salmonella serovars for enhanced antigen-specific CD8 T-cell induction. Vaccine. 2008;26:1879–1886. doi: 10.1016/j.vaccine.2008.01.044. [DOI] [PubMed] [Google Scholar]
- Le Gouellec A, Chauchet X, Polack B, Buffat L, Toussaint B. Bacterial vectors for active immunotherapy reach clinical and industrial stages. Hum Vaccin Immunother. 2012;8:1454–1458. doi: 10.4161/hv.21429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahabi V, Maciag PC, Rivera S, Wallecha A. Live, attenuated strains of Listeria and Salmonella as vaccine vectors in cancer treatment. Bioeng Bugs. 2010;1:235–243. doi: 10.4161/bbug.1.4.11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Wallecha A. Cancer immunotherapy using recombinant Listeria monocytogenes: transition from bench to clinic. Hum Vaccin. 2011;7:497–505. doi: 10.4161/hv.7.5.15132. [DOI] [PubMed] [Google Scholar]
- Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol. 2009;7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouazi M, Toussaint B, Quénée L, Epaulard O, Guillaume M, Marlu R, et al. High-yield production of secreted active proteins by the Pseudomonas aeruginosa type III secretion system. Appl Environ Microbiol. 2008;74:3601–3604. doi: 10.1128/AEM.02576-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gouëllec AL, Chaker H, Asrih H, Polack B, Toussaint B. Optimization of antitumor immunotherapy mediated by type III secretion system-based live attenuated bacterial vectors. J Immunother. 2012;35:223–234. doi: 10.1097/CJI.0b013e31824747e5. [DOI] [PubMed] [Google Scholar]
- Panthel K, Meinel KM, Sevil Domènech VE, Trülzsch K, Rüssmann H. Salmonella type III-mediated heterologous antigen delivery: a versatile oral vaccination strategy to induce cellular immunity against infectious agents and tumors. Int J Med Microbiol. 2008;298:99–103. doi: 10.1016/j.ijmm.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Xiong G, Husseiny MI, Song L, Erdreich-Epstein A, Shackleford GM, Seeger RC, et al. Novel cancer vaccine based on genes of Salmonella pathogenicity island 2. Int J Cancer. 2010;126:2622–2634. doi: 10.1002/ijc.24957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DT, Chen LM, Gillis J, Lin KC, Harty B, Mazzara GP, et al. Mucosal priming of simian immunodeficiency virus-specific cytotoxic T-lymphocyte responses in rhesus macaques by the Salmonella type III secretion antigen delivery system. J Virol. 2003;77:2400–2409. doi: 10.1128/JVI.77.4.2400-2409.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüssmann H, Shams H, Poblete F, Fu Y, Galán JE, Donis RO. Delivery of epitopes by the Salmonella type III secretion system for vaccine development. Science. 1998;281:565–568. doi: 10.1126/science.281.5376.565. [DOI] [PubMed] [Google Scholar]
- Shams H, Poblete F, Rüssmann H, Galán JE, Donis RO. Induction of specific CD8+ memory T cells and long lasting protection following immunization with Salmonella typhimurium expressing a lymphocytic choriomeningitis MHC class I-restricted epitope. Vaccine. 2001;20:577–585. doi: 10.1016/s0264-410x(01)00363-2. [DOI] [PubMed] [Google Scholar]
- Rüssmann H, Igwe EI, Sauer J, Hardt WD, Bubert A, Geginat G. Protection against murine listeriosis by oral vaccination with recombinant Salmonella expressing hybrid Yersinia type III proteins. J Immunol. 2001;167:357–365. doi: 10.4049/jimmunol.167.1.357. [DOI] [PubMed] [Google Scholar]
- Chen G, Dai Y, Chen J, Wang X, Tang B, Zhu Y, et al. Oral delivery of the Sj23LHD-GST antigen by Salmonella typhimurium type III secretion system protects against Schistosoma japonicum infection in mice. PLoS Negl Trop Dis. 2011;5:e1313. doi: 10.1371/journal.pntd.0001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epaulard O, Toussaint B, Quenee L, Derouazi M, Bosco N, Villiers C, et al. Anti-tumor immunotherapy via antigen delivery from a live attenuated genetically engineered Pseudomonas aeruginosa type III secretion system-based vector. Mol Ther. 2006;14:656–661. doi: 10.1016/j.ymthe.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol. 2012;12:215–225. doi: 10.1038/nri3167. [DOI] [PubMed] [Google Scholar]
- Stocker BA. Auxotrophic Salmonella typhi as live vaccine. Vaccine. 1988;6:141–145. doi: 10.1016/s0264-410x(88)80017-3. [DOI] [PubMed] [Google Scholar]
- Epaulard O, Derouazi M, Margerit C, Marlu R, Filopon D, Polack B, et al. Optimization of a type III secretion system-based Pseudomonas aeruginosa live vector for antigen delivery. Clin Vaccine Immunol. 2008;15:308–313. doi: 10.1128/CVI.00278-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterle J, Bergmann B, Yone CL, Hotz C, Meyer SR, Spreng S, et al. Cancer immunotherapy based on recombinant Salmonella enterica serovar Typhimurium aroA strains secreting prostate-specific antigen and cholera toxin subunit B. Cancer Gene Ther. 2008;15:85–93. doi: 10.1038/sj.cgt.7701109. [DOI] [PubMed] [Google Scholar]
- Kotton CN, Lankowski AJ, Scott N, Sisul D, Chen LM, Raschke K, et al. Safety and immunogenicity of attenuated Salmonella enterica serovar Typhimurium delivering an HIV-1 Gag antigen via the Salmonella Type III secretion system. Vaccine. 2006;24:6216–6224. doi: 10.1016/j.vaccine.2006.05.094. [DOI] [PubMed] [Google Scholar]
- Priebe GP, Brinig MM, Hatano K, Grout M, Coleman FT, Pier GB, et al. Construction and characterization of a live, attenuated aroA deletion mutant of Pseudomonas aeruginosa as a candidate intranasal vaccine. Infect Immun. 2002;70:1507–1517. doi: 10.1128/IAI.70.3.1507-1517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring G, Pier GB. Vaccines and immunotherapy against Pseudomonas aeruginosa. Vaccine. 2008;26:1011–1024. doi: 10.1016/j.vaccine.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Detmer A, Glenting J. Live bacterial vaccines–a review and identification of potential hazards. Microb Cell Fact. 2006;5:23. doi: 10.1186/1475-2859-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockstedt DG, Bahjat KS, Giedlin MA, Liu W, Leong M, Luckett W, et al. Killed but metabolically active microbes: a new vaccine paradigm for eliciting effector T-cell responses and protective immunity. Nat Med. 2005;11:853–860. doi: 10.1038/nm1276. [DOI] [PubMed] [Google Scholar]
- Sancar A, Sancar GB. DNA repair enzymes. Annu Rev Biochem. 1988;57:29–67. doi: 10.1146/annurev.bi.57.070188.000333. [DOI] [PubMed] [Google Scholar]
- Wollowitz S. Fundamentals of the psoralen-based Helinx technology for inactivation of infectious pathogens and leukocytes in platelets and plasma. Semin Hematol. 2001;38 4 Suppl 11:4–11. doi: 10.1016/s0037-1963(01)90118-0. [DOI] [PubMed] [Google Scholar]
- Dubensky TW, Jr, Skoble J, Lauer P, Brockstedt DG. Killed but metabolically active vaccines. Curr Opin Biotechnol. 2012;23:917–923. doi: 10.1016/j.copbio.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Quénée L, Lamotte D, Polack B. Combined sacB-based negative selection and cre-lox antibiotic marker recycling for efficient gene deletion in pseudomonas aeruginosa. BioTechniques. 2005;38:63–67. doi: 10.2144/05381ST01. [DOI] [PubMed] [Google Scholar]
- Lankowski AJ, Hohmann EL. Killed but metabolically active Salmonella typhimurium: application of a new technology to an old vector. J Infect Dis. 2007;195:1203–1211. doi: 10.1086/512618. [DOI] [PubMed] [Google Scholar]
- Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verove J, Bernarde C, Bohn YS, Boulay F, Rabiet MJ, Attree I, et al. Injection of Pseudomonas aeruginosa Exo toxins into host cells can be modulated by host factors at the level of translocon assembly and/or activity. PLoS ONE. 2012;7:e30488. doi: 10.1371/journal.pone.0030488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worgall S, Martushova K, Busch A, Lande L, Crystal RG. Apoptosis induced by Pseudomonas aeruginosa in antigen presenting cells is diminished by genetic modification with CD40 ligand. Pediatr Res. 2002;52:636–644. doi: 10.1203/00006450-200211000-00006. [DOI] [PubMed] [Google Scholar]
- Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci USA. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouazi M, Wang Y, Marlu R, Epaulard O, Mayol JF, Pasqual N, et al. Optimal epitope composition after antigen screening using a live bacterial delivery vector: application to TRP-2. Bioeng Bugs. 2010;1:51–60. doi: 10.4161/bbug.1.1.9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacheux D, Attree I, Schneider C, Toussaint B. Cell death of human polymorphonuclear neutrophils induced by a Pseudomonas aeruginosa cystic fibrosis isolate requires a functional type III secretion system. Infect Immun. 1999;67:6164–6167. doi: 10.1128/iai.67.11.6164-6167.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspord C, Charles J, Leccia MT, Laurin D, Richard MJ, Chaperot L, et al. A novel cancer vaccine strategy based on HLA-A*0201 matched allogeneic plasmacytoid dendritic cells. PLoS ONE. 2010;5:e10458. doi: 10.1371/journal.pone.0010458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford KJ, Higgins DE, Pasquini S, Cheadle EJ, Carta L, Jackson AM, et al. A recombinant E. coli vaccine to promote MHC class I-dependent antigen presentation: application to cancer immunotherapy. Gene Ther. 2002;9:1455–1463. doi: 10.1038/sj.gt.3301812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.