

Abstract
Recently, cell-penetrating peptides have been proposed to translocate antibodies, proteins, and other molecules in targeted drug delivery. The proposed study presents the synthesis and characterization of a peptide-based chitosan nanoparticle for small interfering RNA (siRNA) delivery, in-vitro. Specifically, the synthesis included polyethylene glycol (PEG), a hydrophilic polymer, and trans-activated transcription (TAT) peptide, which were chemically conjugated on the chitosan polymer. The conjugation was achieved using N-Hydroxysuccinimide-PEG-maleimide (heterobifunctional PEG) as a cross-linker, with the bifunctional PEG facilitating the amidation reaction through its N-Hydroxysuccinimide group and reacting with the amines on chitosan. At the other end of PEG, the maleimide group was chemically conjugated with the cysteine-modified TAT peptide. The degree of substitution on chitosan with PEG and on PEG with TAT was confirmed using colorimetric assays. The resultant polymer was used to form nanoparticles complexing siRNA, which were then characterized for particle size, morphology, cellular uptake, and cytotoxicity. The nanoparticles were tested in-vitro on mouse neuroblastoma cells (Neuro2a). Particle size and surface charge were characterized and an optimal pH condition and PEG molecular weight were determined to form sterically stable nanoparticles. Results indicate 7.5% of the amines in chitosan polymer were conjugated to the PEG and complete conjugation of TAT peptide was observed on the synthesized PEGylated chitosan polymer. Compared with unmodified chitosan nanoparticles, the nanoparticles formed at pH 6 were monodispersed and of <100 nm in size, exhibiting maximum cell transfection ability and very low cytotoxicity. Thus, this research may be of significance in translocating biotherapeutic molecules for intracellular delivery applications.
Keywords: chemical conjugation, peptide, transfection, polymer, synthesis, in-vitro
Introduction
Cell-penetrating peptides (CPPs) have gained much attention due to their ability to drugs, genes, and large therapeutic molecules intracellularly. One commonly used CPP is the transcription activating factor of the human immunodeficiency virus (HIV), termed “trans-activated transcription” (TAT) peptide. Its efficient cell-penetrating characteristics are linked to a domain rich in arginine and lysine amino acid residues that interacts with the negatively charged proteoglycans on the cell surface.1,2 It has been reported that TAT peptide may enhance cellular uptake due to its strong cell adherence, with no dependence on receptors, temperature, nor an energy-dependent pathway.3–5 Moreover, the basic domain of TAT peptide is also involved in RNA binding6 and the nuclear localization of proteins.7 TAT peptide can be conjugated covalently or non-covalently to cationic polymers like liposomes, poly-l-lysine, poly(ethyleneimine) (PEI).8–10 It may also be used as a carrier of large therapeutic molecules, proteins, small interfering RNA (siRNA), quantum dots etc.1,11–13 Because of these properties, it is emerging as a new strategy to achieve efficient cellular nuclear targeting.
Lipid nanoparticles have also been used for drug delivery targeting the brain.14 However, such formulations lack active targeting to the tissues of interest. Boado and Pardridge have explained the use of a monoclonal antibody as a targeting moiety on nonviral liposome nanoparticles targeting the brain.15 Various other blood–brain barrier (BBB)-targeting antibodies have been detailed by Jones and Shusta.16 However, the high molecular weight (HMW) of antibodies makes the overall size of the nanoparticles large, which hinders optimal distribution at the targeted site. BBB-specific peptides are also being utilized for specific targeting.17,18 Further, other researchers have used cyclodextrins to deliver therapeutics across the BBB, but the penetration enhancer effect of cyclodextrins has been found to disrupt the integrity of the BBB, leading to toxicity.19 A detailed review of supramolecular devices for the treatment of brain diseases has been published elsewhere.20
Cellular toxicity of transfection agents made of synthetic cationic polymers is an important concern, because high cationic charge density may lead to cell membrane disruption. The advantage of cationic polymers is their endosomal buffering capacity by protonated amino groups in the endosomal environment (pH 5.5). This buffering protects the endocytosed gene from lysosomal degradation.21–25 To reduce the cytotoxicity, hydrophilic molecules like polyethylene glycol (PEG) have been employed. PEG not only reduces toxicity but also prevents any unintended interaction of the encapsulated therapeutic molecule with cellular or serum proteins, thus enhancing stability of the polyplex and avoiding macrophage uptake.26 Although PEG increases the stability of the polyplex, it has also been observed to considerably reduce the transfection ability.27,28 PEG conjugation with cationic polymers is typically achieved through functionally reactive amino groups on the polymer, such as chitosan (CS).29 These amino groups contribute cationic properties, rendering the polymer an efficient transfection agent. However, PEG conjugation via amino groups masks the overall positive charge of the polyplex, hindering the polymer’s transfection ability. In this situation, PEGylating CS polymer can also be synthesized via the hydroxyl groups present at the C6 position of the CS polymer, where the amine groups are protected before PEGylation.30 Thus, the transfection ability of the PEGylated-CS nanoparticles is not compromised since the amine groups are deprotected following PEGylation, and the polymer shows efficient transfection with minimal cytotoxicity.30 However, in the study reported here, a CPP was incorporated to enhance the transfection.
Our study aimed to develop a CS-PEG-TAT nanoparticle formulation for the delivery of siRNA in-vitro. Heterobifunctional PEG was employed as a linker between the two cationic moieties – that is, the polymer (CS) and the peptide (TAT). CS is a nontoxic,31 biodegradable,32,33 and biocompatible34,35 polysaccharide.36 CS has a number of biomedical applications, including as a drug, gene, peptide, vaccine delivery vehicle and as a scaffold for targeted delivery and tissue engineering.37–40 CS has similar characteristics to PEI in terms of cell transfection efficiency and the “proton sponge” effect but is comparatively less cytotoxic. Various cell types, such as THP-1,41 HEK 293,42,43 HeLa,42 Cos-7,43 CHO K1,44 Caco-2,45 HepG2,46 and NIH 3T3,47 have shown effective transfection by derivatized form of CS/DNA complexes. In the presented work, PEG functions as a linker, reduces the steric hindrance between the two cationic moieties and stabilizes the polycomplex. It creates a protective shield that prevents degradation of the therapeutic molecule. Linking a cationic peptide, TAT, to a polymer, enhances the amount of DNA complexation as well as the stability of the polycomplex.48
In this study, CS conjugation was achieved through the interaction of its amino groups with N-Hydroxysuccinimide (NHS) on a bifunctional PEG. This conjugate was further modified with the cysteine-terminated TAT peptide using the maleimide group present on the other end of the bifunctional PEG. The functionalized polymer was allowed to self-assemble into nanoparticles with siGLO Green Transfection Indicator (Thermo Fisher Scientific, Waltham, MA, USA) (scrambled siRNA). These nanoparticles were then used to transfect mouse neuroblastoma (Neuro2a) cells, as neuronal cells are typically challenging to transfect successfully. This study not only developed a novel nanoparticle formulation but also investigated the effect of the molecular weight (MW) of PEG and the pH of the solution on the obtained particle size and surface charge, an important factor in the cellular uptake of polyplexes. The optimization and characterization of the novel nanoparticle formulation was achieved using a particle sizer, by determining zeta potential, and using transmission electron microscopy (TEM).
Materials and methods
Materials
Low-molecular-weight (LMW) CS with a degree of deacetylation of 80% was obtained from Wako (Richmond, VA, USA). Sodium triphosphate pentabasic (TPP) and glacial acetic acid of analytical grade were obtained from Sigma-Aldrich (St Louis, MO, USA). siGLO Green Transfection Indicator, a scrambled siRNA (20 nmole); SM(PEG)4 (NHS-PEG4-maleimide) of LMW 513.50; Zeba Desalting Spin Columns; 2,4,6-Trinitrobenzenesulfonic acid solution (TNBS) reagent; and Ellman’s reagent were obtained from Thermo Fisher Scientific. NHS-PEG-Malhex of HMW 5000 was obtained from Rapp Polymere (Tuebingen, Germany). Cysteine-terminated TAT peptide (GRKKRRQRRRGC-SH) with a MW of 1556.88 was synthesized by Sheldon Biotech, McGill University (Montreal, Canada). CellTiter 96® AQueous One Solution Reagent was purchased from Promega (Fitchburg, WI, USA) to perform cell viability assays. Eagle’s minimum essential medium (EMEM) was obtained from the American Type Culture Collection (Manassas, VA, USA) and supplemented with 10% fetal bovine serum. For dilution purposes, ultrapure double-distilled water (ddH2O) was used from a laboratory-installed Barnstead Nanopure Diamond™ (Thermo Fisher Scientific) water supply unit.
Synthesis of CS-PEG-TAT polymer
LMW CS was dissolved in 1% (v/v) acetic acid to yield a concentration of 0.5% (w/v) CS and the pH was adjusted to 5 using 1 M NaOH. Either NHS-PEG-maleimide of PEG with a MW of 513.50 (LMW) or 5000 (HMW) was added to CS solutions in 10 M excess. The reaction proceeded for 3 hours under inert conditions at room temperature. Following the reaction, the pH was adjusted to 7 using 1 M NaOH. The reaction was allowed to stir overnight under similar conditions. Cysteine-terminated TAT was added in a one to one or one to four molar ratio of PEG:TAT and the reaction stirred continuously for another 4 hours under similar conditions. Unreacted PEG and TAT were removed from the reaction mix using a Zeba Desalting Spin gel filtration column with an exclusion limit of a MW of 7000. The filtrate obtained consisted of CS-PEG-TAT polymer, used to make nanoparticles under varying pH values.
Degree of substitution (DS)
Quantifying free amine groups: TNBS assay
The CS-PEG polymer was analyzed by TNBS assay to quantify the unmodified free amino groups present on the CS, which were representative of the DS of PEG on the CS. Following the overnight reaction of CS with NHS-PEG-maleimide, the mixture was passed through a gel filtration Zeba Desalting Spin Column and the filtrate was freeze-dried. CS-PEG conjugate (1 mg) was rehydrated in 250 μL of ddH2O and incubated with 250 μL of 4% (w/v) NaHCO3 and 0.1% (v/v) TNBS reagent at 37°C for 2 hours. Then, 250 μL of 2 M HCl was added and the absorbance was recorded at 344 nm using a Cary® 100 Bio UV-Vis Spectrophotometer (Varian, Palo Alto, CA, USA). The DS of PEG on CS was calculated using a standard curve (R2 = 0.9959) using 4.0, 2.0, 1.0, 0.5, 0.25, 0.125, 0.0625, and 0.03125 mg/mL concentrations of unmodified CS. The free amino group content was quantified according to the following equation:
| (1) |
where “C” and “CP” stand for amino group content in CS and in CS-PEG polymer, respectively.
Quantifying free sulfhydryl groups: Ellman’s assay
Ellman’s assay was used to determine the amount of free cysteine/sulfhydryl groups present in the reaction mix of CS-PEG-TAT polymer. This assay was performed prior to the removal of unreacted TAT peptide from the reaction mix, using Zeba Desalting Spin Columns. The unreacted TAT peptide bearing a cysteine group at its C-terminal end was quantified using this assay. In brief, sodium phosphate 0.1 M and 1 mM ethylenediaminetetraacetic acid (EDTA) reaction buffer at pH 8.0 was prepared. The stock solution of Ellman’s reagent was prepared by dissolving 4 mg of Ellman’s reagent in 1 mL reaction buffer. According to the manufacturer’s protocol, 250 μL of the sample was added to a test tube containing 50 μL Ellman’s reagent solution and 2.5 mL reaction buffer. The mix was incubated for 15 minutes at 37°C and the absorbance was measured at 412 nm. A serial dilution of cysteine hydrochloride monohydrate was used to generate a standard curve (R2 = 0.9743) using 1.5, 1.25, 1.0, 0.75, 0.5, 0.25, 0.125, 0.0625, and 0.03125 mM concentrations. The free sulfhydryl group content was quantified according to the following equation:
| (2) |
where “T” and “CPT” stand for sulfhydryl group content in TAT peptide and in CS-PEG-TAT polymer, respectively.
Preparation of siGLO Green-complexed CS-PEG-TAT nanoparticles
siGLO Green was complexed with CS-PEG-TAT polymer to form nanoparticles. Briefy, the pH of the CS-PEG-TAT, obtained during the previously outlined step, was adjusted to various pH values of 5.5, 6.0, 6.5, and 7.0, using 1 M HCl or 1 M NaOH, as required. This was done to determine the effect of pH on particle size, cellular uptake, and cytotoxicity. Finally, siGLO Green (20 μM stock) was mixed with 200 μL of TPP (anionic cross-linker) pH 3 (0.07% [w/v] stock in ddH2O) before adding it dropwise to 800 μL of the pH-adjusted CS-PEG-TAT (0.05% [w/v] stock in ddH2O) polymer solution. The final concentration of siGLO Green used was 2 μg per mL of polymer solution, with a CS to siGLO Green ratio of 200:1 (w/w), as optimized in our previous study.49 Specific N:P ratio (molar ratio of chitosan amino groups/siRNA phosphate groups) was calculated using, 325 Da as mass-per-phosphate for siRNA and 161.16 (molecular weight of repeating units of chitosan) as a mass-per-charge for chitosan. The N:P ratio calculated was 103. The solution was stirred for 1 hour at room temperature. The electrostatic interaction between the cationic polymer and anionic siGLO Green led to the formation of polyelectrolyte complexes that were observed by TEM on a JEOL JEM-2000fx transmission electron microscope (Tokyo, Japan).
Characterization of siGLO Green-complexed CS-PEG-TAT polyelectrolyte complexes
The nanoparticles were characterized in terms of their size (mean particle diameter, based on the mean count of nanoparticles) and size distribution using a Brookhaven BI-90 Particle Nanosizer (Holtsville, NY, USA). Nanoparticle surface charge (zeta potential) determination was performed using a Malvern Instruments Zetasizer zetaPALS 31362 (Malvern, UK) at 25°C and morphological observation was performed under high-resolution TEM. A drop of each sample was placed on a carbon film-based copper mesh grid and allowed to air dry before TEM observation.
Transfection efficiency
Transfection studies were performed on mouse neuroblastoma cells (Neuro2a). The cells were sub-cultured after reaching a confluency of approximately 85% and seeded in a 96-well plate at a density of 20,000 cells per well. The seeding was undertaken 24 hours prior to transfection, with 200 μL of complete growth media (EMEM with 10% fetal bovine serum) without antibiotics in each well. After 24 hours, the transfection media, containing 100 μL of serum-free medium, along with 50 μL of a siGLO Green-complexed nanoparticle sample (each sample with a different pH and/or MW of PEG) were added onto the cells. The cells were then incubated at 37°C with 5% CO2 for 24 hours, after which they were observed under a fluorescence microscope (Nikon Eclipse TE2000-U, Tokyo, Japan) at a wavelength of 490 nm. The quantitative measurement of transfection was done using a microtiter plate reader (PerkinElmer Victor3 V 1420 Multilabel Plate Counter, Waltham, MA, USA) at 490 nm.
Cytotoxicity assay
The cytotoxicity of the nanoparticles was determined with relation to cell viability. Neuro2a cells were seeded at a density of 20,000 cells per well in a 96-well microplate with EMEM complete growth media. After 4 hours of incubation at 37°C and 5% CO2, the media was replaced with 100 μL of serum-free media along with 50 μL of the sample treatment (0.133 mg/mL of polymer, carrying 2 μg/mL of siGLO Green) per well. The cells were then incubated and the cytotoxicity assay was performed after 24 hours. The cytotoxicity assay involved adding 20 μL of CellTiter 96® AQueous One Solution Reagent per well followed by 4 hours’ incubation. We used this assay rather than 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay because it does not require a further step for solubilizing formazan crystals. After 4 hours of incubation, the absorbance was recorded at 490 nm using the microtiter plate reader.
Statistical analysis
Experimental results are expressed herein as means ± standard deviation. Statistical analysis was carried out using SPSS software (v 17.0; IBM New York, NY, USA). Statistical comparisons were carried out using the general linear model, and Tukey’s post-hoc analysis. Statistical significance was set at P < 0.05 and P values < 0.01 were considered highly significant.
Results and discussion
Synthesis of CS-PEG-TAT polymer
Synthesis of the CS-PEG-TAT polymer, described in Figure 1, was achieved using a heterobifunctional NHS-PEG-maleimide as a linker. The NHS group reacts with the amines (–NH2) on CS and the maleimide reacts with the sulfhydryl (–SH) containing cysteine-modified TAT peptide. Since the pKa value of CS is ~6.5,50 above which it precipitates, the use of a hydrophilic molecule like PEG improves the water solubility of the polymer and reduces conjugant aggregation. According to the manufacturer’s specifications, the reaction specificity for both the NHS and maleimide group of PEG are in the range of 7.0–9.0 and 6.5–7.5, respectively. In addition, it is noted that NHS esters are more unstable than the maleimides and can easily undergo hydrolytic degradation at a higher pH. Thus, both reactions were performed at a pH of 7.0. The extra PEG and the reaction byproducts were removed using a Zeba Desalting Spin Column to maintain the pH conditions of the reaction, with repeated buffer change.
Figure 1.

Synthesis of chitosan-g-polyethylene glycol (PEG)-trans-activated transcription (TAT) polymer.
Abbreviation: NHS, N-Hydroxysuccinimide.
Another similar characterization study using CS, PEG, and TAT has been published by our group.51 However, the nanoparticles formed in that study utilized a different synthetic scheme that followed eight intermediate reaction steps. The current study presented here reports nanoparticles prepared using a much simpler, three-step reaction method. The synthesis scheme used in this study did not employ any organic solvents and the reaction conditions were very mild. However, since the reaction conditions modulated the ionic interactions, the evaluation of pH and the effect of MW on particle size and surface charge became very important considerations prior to in-vivo studies.
Quantification of the DS: TNBS and Ellman’s assay
The conjugation reactions of the PEG on CS and the TAT peptide on PEG were confirmed using TNBS and Ellman’s assay, respectively. The TNBS assay was used to indirectly quantify the amount of free amine groups present on the CS following conjugation with PEG. When compared with the unmodified CS, it was observed that 7.5% of the amines in chitosan were successfully modified with PEG. Similarly, Ellman’s assay was performed to quantify the number of free sulfhydryl groups present in the solution. At the same concentration of TAT peptide and CS-PEG-TAT polymer, formed in the ratio of one to one (PEG:TAT), Ellman’s assay was found to be negative, confirming complete binding and consumption of the free thiols of TAT peptide by the heterobifunctional PEG. This was in agreement with a recently published study for the determination of free thiols using Ellman’s assay.52
Optimization and characterization of CS-PEG and CS-PEG-TAT nanoparticles
PEGylated-CS nanoparticles with and without TAT peptide were allowed to form self-assembled polyelectrolyte complexes using TPP as a cross-linker. The formed nanoparticles were characterized with respect to particle size and zeta potential due to changes in pH conditions (Figure 2), conjugation of TAT peptide (Figure 3), and the MW of PEG (Figure 4). A similar characterization with different molecular weights of CS has been reported previously by our group.49 In this study, the developed CS nanoparticles, PEGylated and conjugated with or without TAT peptide were evaluated for their transfection ability and cytotoxicity on Neuro2a cells, as represented in Figures 5 and 6, respectively.
Figure 2.
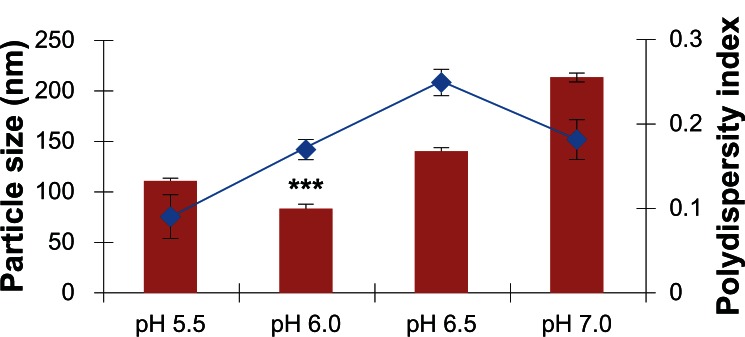
Effect of different pH conditions on the size of chitosan-polyethylene glycol (PEG) nanoparticles, formed using a low molecular weight PEG.
Notes: Data presented are mean ± standard deviation, n = 3 (***P < 0.001). The blue line indicates polydispersity index.
Figure 3.

The nanoparticle size and polydispersity index of different nanoparticle formulations, formed at pH 6.0 using high-molecular-weight polyethylene glycol (PEG) and with different ratios of trans-activated transcription (TAT) peptide.
Notes: Data shown are mean ± standard deviation, n = 3 (*P < 0.05, ***P < 0.001). The blue line indicates, polydispersity index.
Figure 4.

Effect of polyethylene glycol (PEG) molecular weight on the size of chitosan-PEG-trans-activated transcription (TAT) nanoparticles, formed at different pH conditions.
Notes: Data presented are mean ± standard deviation, n = 3 (**P < 0.01).
Abbreviations: LMW, low molecular weight; HMW, high molecular weight.
Figure 5.

Transfection efficiency of chitosan-polyethylene glycol (PEG)-trans-activated transcription nanoparticles formed with low-molecular-weight (LMW) and high-molecular-weight (HMW) PEG, for the delivery of siGLO Green Transfection Indicator (Thermo Fisher Scientific, Waltham, MA, USA) to mouse neuroblastoma (Neuro2a) cells. Results were analyzed after 4 hours of incubation with various nanoparticle formulations developed at various conditions: (A) LMW PEG at pH 5.5, particle size 1010 nm ± 4.58; (B) HMW PEG at pH 5.5, particle size 143 nm ± 7.51; (C) LMW PEG at pH 6.0, particle size 234 nm ± 4.05; (D) HMW PEG at pH 6.0, particle size 185 nm ± 4.85; (E) LMW PEG at pH 6.5, particle size 691 nm ± 214; (F) HMW PEG at pH 6.5, particle size 1730 nm ± 719.
Note: Scale: 20 μm; magnification 400×.
Figure 6.

(A) Transfection efficiency performed on mouse neuroblastoma (Neuro2a) cells with chitosan (CS)-polyethylene glycol (PEG)-trans-activated transcription (TAT) nanoparticles, formed at various pH conditions, with high- and low-molecular-weight PEG. (B) Cytotoxicity of unmodified and modified CS nanoparticle formulations evaluated on Neuro2a cells using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay at 490 nm.
Notes: Data shown are mean ± standard deviation, n = 3 (*P < 0.05, **P < 0.01, ***P < 0.001).
Abbreviations: HMW, high molecular weight; LMW, low molecular weight.
Effect of pH on PEGylated-CS nanoparticles
CS, being cationic in nature, dissolves under acidic conditions due to the protonation of its amine groups. However, an acidic pH in transfection is not ideal as it promotes cytotoxicity. Following modification, the PEGylated nanoparticles were assumed to show enhanced water solubility and, as such, be soluble at different pH conditions. In this study, prior to nanoparticle formation, the pH of some of the solution was adjusted variously to 5.5, 6.0, 6.5, and 7.0. The lowest particle size with PEGylated-CS nanoparticles was obtained using pH 6 (P < 0.0001). This identified pH 6.0 as ideal for nanoparticle formation. The particle size gradually increased above a pH of 6.0, with nanoparticles of approximately 142 ± 1.61 nm at pH 6.5 and 215 ± 3.71 nm at pH 7.0. The increase in nanoparticle size was attributed to the unreacted amines, which were not in protonated form for interaction with the negatively charged TPP. This result was in accordance with the study performed by another group, who demonstrated an increase in particle size at the 1 point of CS (pKa = 6.5), decreasing the electrostatic repulsion, leading to particle aggregation.53,54 In this study, there was an increase in particle size at a pH > 6.0 (Figure 2) but the nanoparticles did not precipitate completely. The zeta potential, as observed in Table 1A, shows a gradual decrease in the total positive surface charge of the nanoparticles, with an increase in pH conditions due to the de-protonated amines on the CS. Although the CS nanoparticles were PEGylated, the zeta potential did not fall below zero. This effect can be attributed to the use of LMW PEG, which is not dense enough to entirely shield the surface of the CS nanoparticles. Nevertheless, taking into account the particle size and zeta potential results, it was concluded that pH 6.0 was the most ideal condition for the formation of CS-PEG nanoparticles.
Table 1.
Zeta potential of various nanoparticle formulations
| A. PEGylated-CS nanoparticles formed with LMW PEG | ||||
|---|---|---|---|---|
| CS-PEG | pH 5.5 | pH 6.0 | pH 6.5 | pH 7.0 |
| With LMW PEG | 15.05 ± 2.48 | 13.19 ± 3.8 | 8.83 ± 5.44 | 5.36 ± 3.72 |
|
| ||||
| B. Nanoparticle formulation incorporating TAT peptide, formed with HMW PEG | ||||
|
| ||||
| Nanoparticles | CS | CS-PEG | CS-PEG-TAT (1:1) | CS-PEG-TAT (1:4) |
| With HMW PEG | 17.94 ± 0.84 | 0 ± 10 | 3.73 ± 4.18 | 10.73 ± 2.63 |
|
| ||||
| C. TAT peptide-tagged CS nanoparticle formulations formed with LMW and HMW PEG | ||||
|
| ||||
| CS-PEG-TAT | pH 5.5 | pH 6.0 | pH 6.5 | |
| With LMW PEG | 16.01 ± 1.13 | 21.56 ± 1.23 | 21.96 ± 1.24 | |
| With HMW PEG | 8.78 ± 1.88 | 14.76 ± 0.98 | 15.14 ± 2.17 | |
Abbreviations: CS, chitosan; HMW, high molecular weight; LMW, low molecular weight; PEG, polyethylene glycol; TAT, trans-activated transcription.
Conjugation of TAT peptide: size and zeta potential of PEGylated-CS nanoparticles
As mentioned, the PEGylated-CS nanoparticles formed at pH 6.0 yielded the smallest particle size. Thus, this pH was used for conjugating TAT peptide on the PEGylated-CS nanoparticles. This conjugation was optimized using different concentrations of HMW PEG, as the LMW PEG did not completely mask the surface charge, as shown in Table 1A. Two different molar ratios of PEG to TAT (1:1 and 1:4) were used for optimization. The synthesized nanoparticles were characterized for size and zeta potential. As observed in Figure 3 and Table 1B, an increase in particle size and zeta potential was observed in the nanoparticles following TAT conjugation. The CS nanoparticles prepared at pH 6.0 were 114.6 ± 4.5 nm in size but had a polydispersity index of 0.61 ± 0.04, representing the formation of a mixture with non-homogenous particle sizes. The grafting of HMW PEG on the CS nanoparticles significantly (P = 0.036) reduced the size to 85.00 ± 0.85 nm with a polydispersity index of 0.20 ± 0.01, representing highly monodispersed nanoparticles. This monodispersity was attributed to the steric stability provided by PEG.
The complete conjugation of PEG on the nanoparticles was also confirmed by determining the zeta potential (Table 1B), where the surface charge of the PEGylated-CS nanoparticles was greatly reduced, confirming the masking of the positively charged amine groups of CS by HMW PEG, in contrast to the results obtained with LMW PEG. The conjugation of TAT peptide at a PEG to TAT ratio of 1:1 led to a significant increase (P < 0.001) in particle size as compared with the PEGylated-CS nanoparticles, with a polydispersity index of 0.28 ± 0.02 (Figure 3). A further increase in particle size and zeta potential was observed with TAT peptide conjugation at a molar ratio of one to four (PEG:TAT), with a polydispersity index of 0.20 ± 0.01. This increase was significantly higher (P < 0.001) than TAT-tagged PEGylated-CS nanoparticles formed at one to one molar ratio. This indicates the availability of more maleimide groups on bifunctional PEG conjugated to the cysteine group of TAT at a higher molar ratio, which results in an increase in particle size and zeta potential (Table 1B).
Effect of different PEG MW and pH on size and zeta potential of CS-PEG-TAT nanoparticles
Both LMW and HMW PEG were used as linkers to investigate their effect on particle size, dispersity, and total surface charge. As can be observed in Figure 4 and Table 1C, LMW PEG yielded larger particles with a higher surface charge when compared with HMW PEG, with an exception at pH 6.5. PEG masks the surface of CS through chemical conjugation. As LMW PEG has a smaller hydrophilic chain length compared with that of HMW PEG, it does not entirely shield the positive character of cationic CS nanoparticles, making them sterically instable, which leads to aggregation of nanoparticles and larger particles. HMW PEG can better shield the positive character of CS nanoparticles, making them more sterically stable. This phenomenon has been previously described in other research using PEGylated silica or PEI nanoparticles.55,56 As observed in Figure 4, the CS nanoparticle size with LMW PEG at pH 5.5 was significantly larger (P < 0.001) than that of those nanoparticles obtained at pH 6.0. This can be attributed to the increased cationic nature of CS nanoparticles at acidic pH (pH 5.5) due to reactive amine groups. The increase in particle size for both LMW and HMW PEGylated-CS nanoparticles at pH 6.5 was attributed to particle aggregation, as the isoelectric point of CS is at approximately pH 6.5. Due to the possible aggregation of nanoparticles at pH 6.5, the LMW PEGylated-CS nanoparticles were significantly larger (P = 0.009) than the nanoparticles prepared at pH 6.0. The steric stability of nanoparticles with LMW and HMW PEG is comparable to the zeta potential data. The CS nanoparticles with LMW PEG at all pH conditions showed higher surface charge as compared with that of those with HMW PEG. The grafting density of LMW and HMW PEG has been previously described57 and has been categorized to have either mushroom- or brush-like conformation.57 With low PEG density, the polymer chains arrange in mushroom-like configurations and with high PEG density they stretch out to produce brush-like configurations.57 It has been noted that the mushroom-like conformation can be transitioned to brush-like depending on the MW of the PEG and the type of nanoparticle used.
Although PEG is known to be a non-immunogenic and non-antigenic compound and increases the residence circulating time of the nanoparticles, researchers have shown in an animal model that repeated administration of PEGylated liposomes can lead to the accelerated blood clearance (ABC) phenomenon.58 It has been proposed that the mechanism responsible for ABC is the increased production of anti-immunoglobulin PEG antibodies in the spleen that selectively bind to the PEGylated liposomes on administration of a second/repeated dose.58 The production of antibodies leads to the activation of the complement system, leading to the opsonization of and uptake by hepatic Kupffer cells.58 In addition, the same group of researchers utilized other hydrophilic polymers such as poly(N-vinyl-2-pyrrolidone), poly(4-acryloylmorpholine), poly(N,N-dimethylacrylamide), and poly(acrylamide), poly(vinyl alcohol) as PEG replacements.59 However, nanoparticles formed with these polymers resulted in no ABC/circulation times than the PEGylated nanoparticles.59 In contrast to these findings, another recent study found that the methoxy group of PEG leads to immune responses; however, if replaced with a hydroxyl group, the immune response was not pronounced.60 Moreover, nanoparticle size has a role in ABC regardless of PEG density and chain length. Nanoparticle sizes > 30 nm have been reported to induce ABC.58 Thus, size is also a determinant factor in the clearance rate of nanoparticles from the system. An excellent review on PEG grafting and its various characteristics has been published elsewhere.61
In the study presented here, results obtained on the change of particle size and zeta potential with change in pH is due to a phenomenon similar to ABC. The electrostatic interaction between the nanoparticles and PEG and TAT peptide play an important role in obtaining sterically stable nanoparticle conformations with optimal positive zeta potential for transfecting cells with a size ≤ 100 nm.
TEM study
The synthesized nanoparticles developed with HMW PEG at pH 6.0 from CS-PEG and CS-PEG-TAT polymer were observed using TEM to analyze their size and morphology. Nanoparticles formed with unmodified CS polymer (Figure 7A) were irregular in shape. After PEGylation, the nanoparticles appeared spherical (Figure 7B), which was attributed to the hydrophilic nature of the PEG, which confers steric stability to the nanoparticles. After conjugating with TAT peptide, the nanoparticles appeared oval to spherical in shape (Figure 7C).
Figure 7.

Transmission electron microscope images of (A) unmodified chitosan nanoparticles (magnification 122,000×), (B) polyethylene glycol (PEG)ylated chitosan nanoparticles (magnification 302,000×), and (C) trans-activated transcription-tagged PEGylated chitosan nanoparticles (magnification 420,000×) formed at pH 6.0, with low-molecular-weight PEG.
Note: Scale bars = 100 nm.
Cell study with CS-PEG-TAT nanoparticles
Particle size and surface charge play a key role in the intracellular delivery of siRNA. This phenomenon is more pronounced when employing nanoparticles for in-vivo delivery of therapeutics. For example, in most cancer studies, leaky vasculature leads to uptake of nanoparticles based on size due to the enhanced permeability and retention effect.62 The nanoparticles carrying siRNA as a payload are taken up by the cells via an adsorptive mechanism, in which the positive charge of the nanoparticles interacts with the negatively charged cell membranes. Due to the presence of amine groups, a “proton sponge” effect occurs in the endosome of each nanoparticle, swelling the nanoparticle and breaking open the endosome, which releases the siRNA into the cytosol. As observed in Figures 5 and 6A, maximum transfection efficiency was achieved with the nanoparticles formed with LMW and HMW PEG at pH 6 (Figure 5C and D), with significantly higher fluorescence intensity (P = 0.003 and P = 0.013, respectively) as compared with the control. Large particle sizes did not yield higher transfection. As observed in Figure 6A, nanoparticles formed at pH 5.5 with LMW (P = 0.999) and HMW (P = 0.612) PEG were not significantly different from the control. Similarly, nanoparticles formed at pH 6.5 with LMW (P = 0.557) and HMW (P = 1.000) PEG were not significantly different to the control. However, the nanoparticles formed at pH 6.5 with LMW PEG showed stronger fluorescence intensity (Figure 5) than those formed at the same pH with HMW PEG. Nonetheless, a variation in fluorescence intensity was observed when the experiment was performed in triplicate, leading to a high standard deviation and the representative P value of 0.557, which indicated no significant difference (Figure 6A). In addition, the size of the nanoparticles (Figure 4) and the surface charge (Table 1C) collectively play a key role in determining the transfection efficiency. The nanoparticles prepared at pH 6.5 had comparatively small particle size and high zeta potential compared with those of the HMW PEG nanoparticles. This difference in size and surface charge was due to the difference in the chain length of HMW and LMW PEG. The cytotoxicity of the developed nanoparticles at pH 6 was observed on Neuro2a cells 24 hours post-transfection. The results (Figure 6B) indicate that the unmodified CS nanoparticles were significantly toxic (P < 0.0001), in comparison with the control. However, the conjugation of PEG and TAT tremendously reduced the toxicity and no significant difference was observed with CS-PEG nanoparticles (P = 0.095) and CS-PEG-TAT nanoparticles (P = 0.562) when compared to the control. Thus, this study has confirmed that the conjugation of PEG and TAT on CS nanoparticles enhances the intracellular delivery of siRNA without compromising cell viability.
In future, studies will be undertaken using the developed nanoparticle formulation to deliver siRNA in-vivo. The in-vitro characterization of the nanoparticles was performed on mouse neuroblastoma cells, with a view to eventual use of the nanoparticles in an animal model of brain cancer. The toxicity, safety, immune response, and functionality of siRNA, when delivered via nanoparticles, are important features to be evaluated in-vivo. In the current study, TAT peptide was chosen as a model ligand to characterize the nanoparticles for their size, surface charge, and stability in in-vitro studies only. It may not be the best choice for in-vivo studies because of its nonselective targeting. However, it would be interesting to compare the stability and circulation of CS-PEG nanoparticles with those of CS-PEG-TAT nanoparticles. In the presented in-vitro study, nanoparticle uptake was facilitated by the interaction of positively charged amines on the nanoparticles with the negatively charged cell membrane. Thus, nanoparticle uptake resulted purely from an adsorptive mechanism.63 For our future in-vivo studies, we intend to use a specific nanoparticle moiety targeted toward specific cell-surface receptors of neuroblastoma cells. This would promote selective receptor-mediated endocytosis of nanoparticles into the cells.
Conclusion
The study reported here demonstrated the successful synthesis and characterization of TAT peptide-tagged PEGylated-CS nanoparticles formed by complexing siRNA. The synthesis scheme utilized a simple three-step reaction method and the nanoparticles produced following this scheme were sterically stable, monodispersed, and ≤100 nm in size, with a surface charge of approximately 20 mV. The study optimized the pH, the MW of PEG, and molar ratio of TAT to form nanoparticles that yielded the best transfection efficiency in mouse neuroblastoma cells. At pH 6.0, the CS nanoparticles formed the smallest particles and proved efficient at transfection with minimal toxicity in neuronal cells. Future studies should investigate serum adsorption of the developed PEG-grafted nanoparticles prior to in-vivo studies to ensure an appropriate circulation time. The ABC phenomenon should also be characterized in relation to the methoxy/hydroxyl-terminated PEG to prevent any undesired immune responses in-vivo. The prepared nanoparticles demonstrated an enhanced ability to deliver siRNA intracellularly, which may be useful in other delivery applications.
Acknowledgments
We gratefully acknowledge the assistance of the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant to Dr S Prakash and the support of a Fonds de recherché Santé Québec (FRSQ) doctoral scholarship to M Malhotra, an NSERC Alexander Graham Bell Canada Graduate Doctoral Scholarship to C Tomaro-Duchesneau, and a McGill University internal scholarship to I Kahouli. We also acknowledge the support of the Centre of Biorecognition and Biosensors at McGill University.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Fawell S, Seery J, Daikh Y, et al. Tat mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91(2):664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272(25):16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 3.Richard J, Melikov K, Vives E, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 4.Futaki S. Oligoarginine vectors for intracellular delivery: design and cellular-uptake mechanisms. Biopolymers. 2006;84(3):241–249. doi: 10.1002/bip.20421. [DOI] [PubMed] [Google Scholar]
- 5.Torchilin VP, Mohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci USA. 2001;98(15):8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weeks KM, Ampe C, Schultz SC, Steitz TA, Crothers DM. Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science. 1990;249(4974):1281–1285. doi: 10.1126/science.2205002. [DOI] [PubMed] [Google Scholar]
- 7.Ruben S, Perkins A, Purcell R, et al. Structural and functional characterization of human immunodeficiency virus tat protein. J Virol. 1989;63(1):1–8. doi: 10.1128/jvi.63.1.1-8.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudolph C, Plank C, Lousier J, Schillinger U, Muller RH, Rosenecker J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J Biol Chem. 2003;278(13):11411–11418. doi: 10.1074/jbc.M211891200. [DOI] [PubMed] [Google Scholar]
- 9.Hashida H, Miyamoto M, Cho Y, et al. Fusion of HIV-1 Tat protein transduction domain to poly-lysine as a new DNA delivery tool. Br J Cancer. 2004;90(6):1252–1258. doi: 10.1038/sj.bjc.6601680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyndman L, Lemoine JL, Huang L, Porteous DJ, Boyd AC, Nan X. HIV-1 Tat protein transduction domain peptide facilitates gene transfer in combination with cationic liposomes. J Control Release. 2004;99(3):435–444. doi: 10.1016/j.jconrel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Turner JJ, Jones S, Fabani MM, Ivanova G, Arzumanov AA, Gait MJ. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol Dis. 2007;38(1):1–7. doi: 10.1016/j.bcmd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In-vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285(5433):1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 13.Santra S, Yang H, Stanley JT, et al. Rapid and effective labeling of brain tissue using TAT-conjugated CdS:Mn/ZnS quantum dots. Chem Commun (Camb) 2005;7(25):3144–3146. doi: 10.1039/b503234b. [DOI] [PubMed] [Google Scholar]
- 14.Bondì ML, Di Gesù R, Craparo EF. Lipid nanoparticles for drug targeting to the brain. Methods Enzymol. 2012;508:229–251. doi: 10.1016/B978-0-12-391860-4.00012-4. [DOI] [PubMed] [Google Scholar]
- 15.Boado RJ, Pardridge WM. The Trojan horse liposome technology for nonviral gene transfer across the blood-brain barrier. J Drug Deliv. 2011;2011:296151. doi: 10.1155/2011/296151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24(9):1759–1771. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma HS, Castellani RJ, Smith MA, Sharma A. The blood-brain barrier in Alzheimer’s disease: novel therapeutic targets and nanodrug delivery. Int Rev Neurobiol. 2012;102:47–90. doi: 10.1016/B978-0-12-386986-9.00003-X. [DOI] [PubMed] [Google Scholar]
- 18.Sharma G, Modgil A, Sun C, Singh J. Grafting of cell-penetrating peptide to receptor-targeted liposomes improves their transfection efficiency and transport across blood-brain barrier model. J Pharm Sci. 2012;101(7):2468–2478. doi: 10.1002/jps.23152. [DOI] [PubMed] [Google Scholar]
- 19.Dehouck MP, Jolliet-Riant P, Brée F, Fruchart JC, Cecchelli R, Tillement JP. Drug transfer across the blood–brain barrier: correlation between in-vitro and in-vivo models. J Neurochem. 1992;58(5):1790–1797. doi: 10.1111/j.1471-4159.1992.tb10055.x. [DOI] [PubMed] [Google Scholar]
- 20.Paolino D, Cosco D, Molinaro R, Celia C, Fresta M. Supramolecular devices to improve the treatment of brain diseases. Drug Discov Today. 2011;16(7–8):311–324. doi: 10.1016/j.drudis.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Alexis F, Lo SL, Wang S. Covalent attachment of low molecular weight Poly(ethylene imine) improves Tat peptide mediated gene delivery. Adv Mater. 2006;18(16):2174–2178. [Google Scholar]
- 22.Boussif O, Lezoualc’h F, Zanta MA, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in-vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92(16):7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. J Control Rel. 1999;60(2–3):149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 24.Putnam D, Gentry CA, Pack DW, Langer R. Polymer-based gene delivery with low cytotoxicity by a unique balance of side-chain termini. Proc Natl Acad Sci USA. 2001;98(3):1200–1205. doi: 10.1073/pnas.031577698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang GP, Guo HY, Alexis F, et al. Low molecular weight polyethylenimines linked by β-cyclodextrin for gene transfer into the nervous system. J Gene Med. 2006;8(6):736–744. doi: 10.1002/jgm.874. [DOI] [PubMed] [Google Scholar]
- 26.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134(4):2139–2147. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 27.Sung SJ, Min SH, Cho KY, et al. Effect of polyethylene glycol on gene delivery of polyethylenimine. J Biol Pharm Bull. 2003;26(4):492–500. doi: 10.1248/bpb.26.492. [DOI] [PubMed] [Google Scholar]
- 28.Suh J, Choy K, Lai SK, et al. PEGylation of nanoparticles improves their cytoplasmic transport. Int J Nanomedicine. 2007;2(4):735–741. [PMC free article] [PubMed] [Google Scholar]
- 29.Chan P, Kurisawa M, Chung JE, Yang YY. Synthesis and characterization of chitosan-g-poly(ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery. Biomaterials. 2007;28(3):540–549. doi: 10.1016/j.biomaterials.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 30.Malhotra M, Lane C, Tomaro-Duchesneau C, Saha S, Prakash S. A novel method for synthesizing PEGylated chitosan nanoparticles: strategy, preparation and in-vitro analysis. Int J Nanomedicine. 2011;6:485–494. doi: 10.2147/IJN.S17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kean T, Thanou M. Biodegradation, biodistribution and toxicity of chitosan. Adv Drug Deliv Rev. 2010;62(1):3–11. doi: 10.1016/j.addr.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto H, Amaike M. Biodegradation of cross-linked chitosan gels by a microorganism. Macromolecules. 1997;30(13):3936–3937. [Google Scholar]
- 33.Tomihata K, Ikada Y. In-vitro and in-vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials. 1997;18(7):567–575. doi: 10.1016/s0142-9612(96)00167-6. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Bai Y, Zhu D, et al. Evaluation of the impact of arginine-chitosan/DNA nanoparticles on human naive CD4+ T cells. J Biomed Mater Res A. 2011;96(1):170–176. doi: 10.1002/jbm.a.32966. [DOI] [PubMed] [Google Scholar]
- 35.Risbud MV, Bhonde RR. Polyacrylamide-chitosan hydrogels: in-vitro biocompatibility and sustained antibiotic release studies. Drug Deliv. 2000;7(2):69–75. doi: 10.1080/107175400266623. [DOI] [PubMed] [Google Scholar]
- 36.Shahidi F, Synowiecki J. Isolation and characterization of nutrients and value-added products from snow crab (Chionoecetes opilio) and shrimp (Pandalus borealis) processing discards. J Agric Food Chem. 1991;39(8):1527–1532. [Google Scholar]
- 37.Bernkop-Schnurch A. Chitosan and its derivatives: potential excipients for peroral peptide delivery systems. Int J Pharm. 2000;194(1):1–13. doi: 10.1016/s0378-5173(99)00365-8. [DOI] [PubMed] [Google Scholar]
- 38.Illum L, Jabbal-Gill I, Hinchcliffe M, Fisher AN, Davis SS. Chitosan as a novel nasal delivery system for vaccines. Adv Drug Deliv Rev. 2001;51(1–3):81–96. doi: 10.1016/s0169-409x(01)00171-5. [DOI] [PubMed] [Google Scholar]
- 39.Guang Liu W, De Yao K. Chitosan and its derivatives – a promising non-viral vector for gene transfection. J Control Release. 2002;83(1):1–11. doi: 10.1016/s0168-3659(02)00144-x. [DOI] [PubMed] [Google Scholar]
- 40.Madihally SV, Matthew HW. Porous chitosan scaffolds for tissue engineering. Biomaterials. 1999;20(12):1133–1142. doi: 10.1016/s0142-9612(99)00011-3. [DOI] [PubMed] [Google Scholar]
- 41.Chellat F, Grandjean-Laquerriere A, Le Naour R, et al. Metalloproteinase and cytokine production by THP-1 macrophages following exposure to chitosan-DNA nanoparticles. Biomaterials. 2005;26(9):961–970. doi: 10.1016/j.biomaterials.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Turan K, Nagata K. Chitosan-DNA nanoparticles: the effect of cell type and hydrolysis of chitosan on in-vitro DNA transfection. Pharm Dev Technol. 2006;11(4):503–512. doi: 10.1080/10837450600940873. [DOI] [PubMed] [Google Scholar]
- 43.Gao Y, Xu Z, Chen S, Gu W, Chen L, Li Y. Arginine-chitosan/DNA self-assemble nanoparticles for gene delivery: In-vitro characteristics and transfection efficiency. Int J Pharm. 2008;359(1–2):241–246. doi: 10.1016/j.ijpharm.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 44.Kadiyala I, Loo Y, Roy K, Rice J, Leong KW. Transport of chitosan-DNA nanoparticles in human intestinal M-cell model versus normal intestinal enterocytes. Eur J Pharm Sci. 2010;39(1–3):103–109. doi: 10.1016/j.ejps.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katas H, Alpar HO. Development and characterization of chitosan nanoparticles for siRNA delivery. J Control Release. 2006;115(2):216–225. doi: 10.1016/j.jconrel.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 46.Lee MK, Chun SK, Choi WJ, et al. The use of chitosan as a condensing agent to enhance emulsion mediated gene transfer. Biomaterials. 2005;26(14):2147–2156. doi: 10.1016/j.biomaterials.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 47.Howard KA, Rahbek UL, Liu X, et al. RNA interference in-vitro and in-vivo using a novel chitosan/siRNA nanoparticle system. Mol Ther. 2006;14(4):476–484. doi: 10.1016/j.ymthe.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Xie W, Liu J, Qiu M, Yuan J, Xu A. Design, synthesis and biological activity of cell-penetrating peptide-modified octreotide analogs. J Pept Sci. 2010;16(2):105–109. doi: 10.1002/psc.1205. [DOI] [PubMed] [Google Scholar]
- 49.Malhotra M, Kulamarva A, Sebak S, et al. Ultrafine chitosan nanoparticles as an efficient nucleic acid delivery system targeting neuronal cells. Drug Dev Ind Pharm. 2009;35(6):719–726. doi: 10.1080/03639040802526789. [DOI] [PubMed] [Google Scholar]
- 50.Anthonsen MW, Smidsrød O. Hydrogen ion titration of chitosans with varying degrees of N-acetylation by monitoring induced 1H-NMR chemical shifts. Carbohydr Polym. 1995;26(4):303–305. [Google Scholar]
- 51.Malhotra M, Tomaro-Duchesneau C, Prakash S. Synthesis of TAT peptide-tagged PEGylated chitosan nanoparticles for siRNA delivery targeting neurodegenerative diseases. Biomaterials. 2013;34(4):1270–1280. doi: 10.1016/j.biomaterials.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 52.Iqbal J, Shahnaz G, Dünnhaupt S, Müller C, Hintzen F, Bernkop-Schnürch A. Preactivated thiomers as mucoadhesive polymers for drug delivery. Biomaterials. 2012;33(5):1528–1535. doi: 10.1016/j.biomaterials.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luangtana-Anan M, Limmatvapirat S, Nunthanid J, Chalongsuk R, Yamamoto K. Polyethylene glycol on stability of chitosan microparticulate carrier for protein. AAPS Pharm Sci Tech. 2010;11(3):1376–1382. doi: 10.1208/s12249-010-9512-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gan Q, Wang T, Cochrane C, McCarron P. Modulation of surface charge, particle size and morphological properties of chitosan-TPP nanoparticles intended for gene delivery. Colloids Surf B Biointerfaces. 2005;44(2–3):65–73. doi: 10.1016/j.colsurfb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Xu H, Yan F, Monson EE, Kopelman R. Room-temperature preparation and characterization of poly (ethylene glycol)-coated silica nanoparticles for biomedical applications. J Biomed Mater Res. 2003;66A(4):870–879. doi: 10.1002/jbm.a.10057. [DOI] [PubMed] [Google Scholar]
- 56.Mao S, Neu M, Germershaus O, et al. Influence of polyethylene glycol chain length on the physicochemical and biological properties of poly(ethylene imine)-graft-poly(ethylene glycol) block copolymer/SiRNA polyplexes. Bioconjug Chem. 2006;17(5):1209–1218. doi: 10.1021/bc060129j. [DOI] [PubMed] [Google Scholar]
- 57.Hak S, Helgesen E, Hektoen HH, et al. The effect of nanoparticle polyethylene glycol surface density on ligand-directed tumor targeting studied in-vivo by dual modality imaging. ACS Nano. 2012;6(6):5648–5658. doi: 10.1021/nn301630n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishida T, Kiwada H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm. 2008;354(1–2):56–62. doi: 10.1016/j.ijpharm.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 59.Ishihara T, Maeda T, Sakamoto H, et al. Evasion of the accelerated blood clearance phenomenon by coating of nanoparticles with various hydrophilic polymers. Biomacromolecules. 2010;11(10):2700–2706. doi: 10.1021/bm100754e. [DOI] [PubMed] [Google Scholar]
- 60.Sherman MR, Williams LD, Sobczyk MA, Michaels SJ, Saifer MG. Role of the methoxy group in immune responses to mPEG-protein conjugates. Bioconjug Chem. 2012;23(3):485–499. doi: 10.1021/bc200551b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. PEGylated nanoparticles for imaging and therapy. Nanomedicine (Lond) 2011;6(4):715–728. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:186–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 63.Malhotra M, Prakash S. Targeted drug delivery across blood-brain-barrier using cell penetrating peptides tagged nanoparticles. Curr Nanosci. 2011;7(1):81–93. [Google Scholar]