

Abstract
Sulfate is the second most abundant anion (behind chloride) in modern seawater, and its cycling is intimately coupled to the cycling of organic matter and oxygen at the Earth’s surface. For example, the reduction of sulfide by microbes oxidizes vast amounts of organic carbon and the subsequent reaction of sulfide with iron produces pyrite whose burial in sediments is an important oxygen source to the atmosphere. The concentrations of seawater sulfate and the operation of sulfur cycle have experienced dynamic changes through Earth’s history, and our understanding of this history is based mainly on interpretations of the isotope record of seawater sulfates and sedimentary pyrites. The isotope record, however, does not give a complete picture of the ancient sulfur cycle. This is because, in standard isotope mass balance models, there are more variables than constraints. Typically, in interpretations of the isotope record and in the absence of better information, one assumes that the isotopic composition of the input sulfate to the oceans has remained constant through time. It is argued here that this assumption has a constraint over the last 390 Ma from the isotopic composition of sulfur in coal. Indeed, these compositions do not deviate substantially from the modern surface-water input to the oceans. When applied to mass balance models, these results support previous interpretations of sulfur cycle operation and counter recent suggestions that sulfate has been a minor player in sulfur cycling through the Phanerozoic Eon.
The sulfur cycle is intimately coupled to the cycles of carbon, oxygen, and many other elements, forming one of the great biogeochemical cycles of the surface Earth (e.g., refs. (1, 2). It interacts with the carbon cycle mainly through the process of sulfate reduction, an anaerobic metabolism forming sulfide from sulfate during organic matter oxidation (3). Sulfide produced by sulfate reduction reacts with iron for ultimate removal as pyrite (FeS2) in sediments. The burial of pyrite (FeS2) represents a net source of oxygen to the atmosphere, whereas the oxidation of pyrite on land represents an oxygen sink, with both processes providing an important regulation of atmospheric oxygen concentrations (1, 4, 5). Sulfur is also removed from the oceans as gypsum, a sulfate mineral precipitating in evaporites. Sulfate evaporites are mainly found during the Phanerozoic Eon (although conspicuous deposits are also found during some periods of the Proterozoic Eon, ref. 6), and it has been suggested that their formation on a grand scale was enabled as sulfate concentrations rose above Precambrian levels (7, 8). This increase has been linked to sediment mixing and the resultant pyrite oxidation in sediments by bioturbating animals as they came to prominence through the early stages of the Paleozoic Era (8).
The increase in sulfate concentration enabled more active sulfate removal as gypsum in evaporites, and modeling suggests that the pathways of sulfur removal from the oceans have shifted from pyrite burial early in the Paleozoic Era to sulfate evaporite burial in the late Paleozoic Era and thereafter (8–10). In this view, the influence of the sulfur cycle on oxygen regulation has also decreased through the Phanerozoic Eon as pyrite burial has decreased in magnitude (11). These models, however, are based on isotope mass balance considerations and they require assumptions about the history of the isotopic composition of sulfate input to the oceans.
In a clever new approach, sulfate burial rates have been quantified independently from a macrostratigraphic database available for the North American Continent and the Caribbean (12). These data have been scaled to global rates of sulfate burial through various assumptions about the evolution of the environments available for sulfate deposition and sulfate loss by weathering through time. This analysis suggests that sulfate burial rates as obtained from the traditional analysis of the isotope record have been far overestimated, and that pyrite cycling has dominated the sulfur cycle through the whole of the Phanerozoic Eon. There emerge, therefore, two opposing views on the evolution of sulfur cycle dynamics, and these views can be tested with knowledge of the isotopic composition of sulfur input to the oceans through time. It was suggested many years ago that the isotopic composition of sulfur in coal might provide a measure of the isotopic composition of the ancient terrestrial sulfate reservoir (13). In the present contribution, this idea is tested against a modern understanding of how the isotopic composition of surface water sulfate is transferred from plants to peat and eventually to coal. Following this, available data of the isotopic composition of sulfur in coal are compiled. It is argued that the coal sulfur isotope record provides a history of the evolution of the surface water sulfate reservoir through time, thus providing a critical constraint of the evolution of the sulfur cycle.
Modeling Sulfur Cycle Evolution
Our interpretations of sulfur cycle evolution are based mainly on the isotope records of sulfide and sulfate as preserved in marine sediments and evaporites through time. The relationship between pyrite and sulfate dynamics sediments is normally assessed through mass balance expressions, where
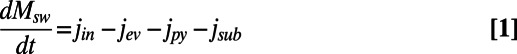 |
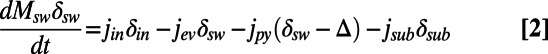 |
In these equations, Msw is the mass of seawater sulfate at any time in the past, and j represents the fluxes either into the ocean (jin) or the output fluxes including pyrite burial (jpy), sulfate burial as evaporites (jev) and any sulfur (pyrite or sulfate) subducted back into the mantle (jsub) (9). The input flux jin represents a variety of sources including volcanic input (both terrestrial and submarine), ocean crust weathering, and riverine input. Of these, the river flux has probably dominated sulfur input during the Phanerozoic Eon (8, 9). Rivers obtain their sulfur, transported as sulfate, mainly from a combination of sulfide oxidation and sulfate evaporite dissolution. These two sulfur sources contribute about equally to the natural (nonanthropogenic) sulfate burden in modern rivers (14). The δ-terms represent the isotopic composition of the inputs or outputs at any time (the isotopic composition of evaporite sulfate is assumed the same as seawater sulfate at the time of evaporite formation), whereas Δ is the difference between the isotopic compositions of seawater sulfate and the pyrite removed from the oceans at any time.
In typical discussions of the Phanerozoic sulfur cycle, nonriverine inputs are ignored as is the output flux by subduction (however, see ref. 9). In the traditional analysis of sulfur cycle dynamics, jin and δin are held constant, and a series of pseudosteady states are usually assumed (in other words, 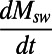 = 0). With these assumptions, Eqs. 1 and 2 can be solved for the fraction of pyrite burial fpy through time:
= 0). With these assumptions, Eqs. 1 and 2 can be solved for the fraction of pyrite burial fpy through time:
Modern δin has been estimated at between 3 and 8% (15, 16), and ref. 12 has used a value of 8% to calculate fpy from the isotope histories of pyrite and seawater sulfate through time. These results are reproduced in Fig. 1 (δin-constrained). The range in results accommodates variability imposed by assuming a dynamic mass balance which allows changes in Msw through time (in other words, 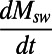 ≠ 0) as constrained from evaporite fluid inclusion data. Overall, this makes little difference to the model results. This history of fpy through time as presented in Fig. 1 is very similar to previous histories (8–10). The model trend shows a pronounced evolution in fpy through the Phanerozoic Eon, where pyrite burial dominated the Paleozoic Era, giving way to dominant sulfate burial after about 400 Ma, as described above.
≠ 0) as constrained from evaporite fluid inclusion data. Overall, this makes little difference to the model results. This history of fpy through time as presented in Fig. 1 is very similar to previous histories (8–10). The model trend shows a pronounced evolution in fpy through the Phanerozoic Eon, where pyrite burial dominated the Paleozoic Era, giving way to dominant sulfate burial after about 400 Ma, as described above.
Fig. 1.

Different model predictions for the burial proportion of pyrite sulfur fpy into marine sediments through most of Phanerozoic time. All model results are derived from the isotopic compositions of marine sulfate and pyrite sulfur through time as shown in Fig. 2, but with different assumptions. The δin-constrained mass balance model is determined by assuming that the sulfur input flux jin and its isotopic composition δin have remained constant through time. The “je-constrained mass balance” model results have been computed with constraints on the burial rates of evaporite sulfur from a macrostratigraphic database, and with various assumptions about the burial rates of pyrite sulfur jpy through time. In this model, the isotopic composition of input sulfur δin is adjusted to achieve mass balance (Fig. 2). The results for both model outputs are redrawn from ref. 12 with permission from AAAS. Also shown are calculations assuming δin-values of between 11 and 12 per mil for the time window of 160–220 Ma as indicated by the coal sulfur isotope record in coal as shown in Fig. 2.
Modeling from Metastratigraphic Data
Modeling based on the analysis of the macrostratigraphic database (12) yields a quite different result. Thus, by constraining sulfate burial with the macrostratigraphic data, and by assuming values for jpy, values for fpy are calculated (12). In this case, isotope mass balance in the sulfur cycle is achieved by solving for the isotopic composition of the sulfate input to the oceans, δin (12). These calculations yield a range in fpy values (plotted as je-constrained in Fig. 1), with the overall conclusion that pyrite burial has dominated the sulfur cycle through all of the Phanerozoic Eon, with evaporitic sulfate removal playing only a minor role. This is quite in contrast with the results constrained by assumptions of constant δin as discussed above (Fig. 1). These results suggest a sulfur cycle dominated by pyrite weathering and burial, and furthermore, with high values of fpy, these results demonstrate a much more active role for pyrite cycling in atmospheric oxygen regulation than previously imagined (12). However, these values for fpy come with a testable prediction. That is, that the isotopic composition of the sulfur input to the oceans δin was much more depleted in 34S than previously assumed, as shown in Fig. 2. As mentioned above, these values for δin are required in the modeling to achieve mass balance.
Fig. 2.

Various aspects of the evolution of the sulfur cycle through most of the Phanerozoic Eon. The evolution of the δ34S of seawater sulfate and average values for the δ34S of pyrite sulfur are shown. The trends are redrawn from ref. 12, with permission from AAAS. Model results for the evolution of the isotopic composition of sulfur input to the oceans δin are shown assuming sulfate burial constraints from the macrostratigraphic database. The variability in model results comes from different assumptions about the burial fluxes of pyrite sulfur, jpy. These results are redrafted from ref. 12. Overlain is the isotopic compositions of sulfur in coal through time. Data from China are presented as box plots showing 5th, 10th, 25th, 50th, 75th, 90th, and 95th percentile uncertainties. For these data, the isotopic composition of total sulfur is plotted. The other data, plotted as circles, represent the isotopic composition of organic sulfur, with low-sulfur coals (<1 wt % total S) plotted as black circles. Coal data come from refs. 13, 30, 32, 33, 36, and 37.
Isotopic Composition of Sulfur in Coal Precursors
It is suggested here that the isotopic composition of sulfur in coal provides a measure of the isotopic composition of terrestrial sulfur pools, and thus a constraint on δin. This, then, provides a test of the various models predicting values of fpy through time and hence the evolution of the sulfur cycle through the Phanerozoic Eon. The logic is as follows: Coal is composed of degraded and thermally matured aquatic plant material. If sulfate is limiting during plant growth, as is typical in terrestrial environments, then the sulfur in the growing plants is assimilated with an isotopic composition of only about 1.5 per mil depleted in 34S on average compared with the isotopic composition of the surface waters providing the source sulfate (17). This sulfur is delivered further into the peats forming from the plants, and ultimately into the coal itself (18).
This cascade of events can be illuminated in various examples from nature. For example, peats from the Okefenokee Swamp, southeastern Georgia, form in a freshwater environment under sulfate limitation (19). Due to this limitation, the peats have low organic sulfur contents (0.32 ± 0.09 wt % S) with δ34S values similar to the plants forming the peats and the surface waters in the swamp (18, 19). Furthermore, in a study of moss and underlying peats from the Karkonosze Mountains in Southwestern Poland (20), the isotope signal from the moss was transferred to the underlying peat (within about 2 per mil), and in the peat bog “Nad Jagniecym Potokiem” in the Izerska Mountains in Southwest Poland, the δ34S of Sphagnum and Polytrichum mosses were consistently within 2–3 per mil of the surface water sulfate source (21). In a comprehensive study of Sphagnum moss from a variety of spruce forest floors in Europe, the δ34S of the plant sulfur resembled that of the source sulfate, but with a consistent 2 per mil shift to 34S-depleted values during assimilation (22).
In some cases, and particularly when sulfate is abundant, early diagenesis can alter the isotope signal of the accumulating organic biomass. For example, many peats in the Florida Everglades form in a high-sulfate environment, with sulfate supplied from the sea. These peats are enriched in organic sulfur (1.96 ± 0.93 wt % S) (19) and their isotopic compositions deviate substantially from the sulfate source. When sulfate is particularly abundant, the organic sulfur is up to 40% depleted in 34S compared with the sulfate, and this depletion is reduced as the sulfate becomes more limiting. These peats clearly show the influence of microbial sulfate reduction in supplying 34S-depleted sulfide to the peat, and they also demonstrate that organic sulfur is enriched into the peat during early diagenesis (18, 19, 23). Similarly, for high-sulfate bogs from the western British Isles with a clear marine influence, Sphagnum moss is some 5–10 per mil depleted in 34S compared with the isotopic composition of the rain supplying the sulfate to these sites (24). In what appears to be a rather unusual case, peats from the New Jersey pinelands record the addition of significant amounts of 34S-depleted sulfur during diagenesis, with organic sulfur reaching values as light as −15 per mil compared with values ranging from 2 to 11 per mil for surface water and rainwater in the area (25). Presumably the light sulfur is incorporated from sulfide produced from microbial sulfate reduction in the peats, but strangely, the sulfate concentrations in the peat pore waters are only in the range of 10–30 μM, too low to produce significant fractionations during sulfate reduction as we currently understand the process (26). Perhaps there is some non-steady-state aspect to the development of the isotopic signal in these peats.
Nevertheless, from these studies we can conclude that despite diagenesis, the isotopic composition of the mosses and peats in low-sulfate areas to a first approximation (generally within 4 or 5 per mil or less) reflects the isotopic composition of the sulfate available for plant growth. When sulfate is abundant, the isotopic composition of the accumulating peats can deviate substantially from the input sulfate. In this case, microbial sulfate reduction can supply 34S-depleted sulfide to the peat. Enrichments in 34S in peat sulfur are also possible. This is believed to occur during the late stages of sulfate reduction when sulfate is nearly depleted and the 34S of the sulfide produced may also be 34S-enriched (e.g., ref. 27). This has been argued for the peats accumulating in the area of Forsinard, Scotland, which have a strong marine sulfate source (28). Therefore, low-sulfur peat, and by analogy, low-sulfur coals, should provide the best targets for obtaining the isotopic composition of the source sulfate at the time of peat formation. This point has also been previously made (13, 18, 29, 30), and it is generally assumed that coals with low sulfur contents (total sulfur < 1 wt % and/or organic sulfur < 0.8 wt %) are attributed to a freshwater origin, whereas high-sulfur coals are viewed as having experienced a marine influence (13, 29, 30).
Still, we must also consider the source of sulfate to the peats forming coal. This will be variable, with sources ranging from rainwater, to the weathering of rock minerals, to the recycling of sulfur compounds within the water shed. The mix of these variable sources will depend on the proximity of the peats to groundwater flow, to riverine input, and on the amounts of sulfate delivered by rain, all of which will change from peat to peat (e.g., refs. 20, 25, 31). In many modern peats, rainwater is often cited as a major source of sulfate to the plants forming the peat (e.g., refs. 21, 25, 31). This view, however, is heavily biased by the large fluxes of anthropogenic sulfur to the atmosphere from multiple vectors including biomass burning and fossil fuel combustion. These sources contribute an estimated 84% of the current terrestrial sources of sulfur to the atmosphere (14). Therefore, rain would have been a much less significant source of sulfur to peats on the preanthropogenic Earth. Of the preanthropogenic nonmarine sulfur sources, volcanoes dominate, with biomass burning and biogenic sulfur sources contributing perhaps one-third as much sulfur (14). Whereas biomass burning and biogenic gas production largely recycle plant sulfur, volcanoes provide a unique sulfur source with a distinct isotope signal. Still, in the absence of anthropogenic input, volcanoes only contribute about 7% of the sulfate flux through the terrestrial system and out to the oceans, and it seems unlikely that they would contribute the dominant source of sulfate to peats through time. Therefore, it is concluded here that low-sulfate coals provide a reasonably good indicator of the isotopic composition of surface water sulfate in the environment where the organic material ultimately forming coal was produced.
Isotopic Composition of Coal Through Time
A compilation of available data on the isotopic composition of coal sulfur through time is shown in Fig. 2. For most of the data, organic sulfur isotopic compositions are plotted except for the data from China (32), represented by the box plots, which show total-sulfur δ34S values. For the Chinese coals, about 15% of the isotope data represent coals with total sulfur contents > 1 wt % S, and the remaining should be considered “low-sulfur” coals. Also for the Chinese coals, the spread in isotope values increases as total sulfur content increases beyond about 0.5 wt % total S and as pyrite sulfur becomes more abundant than organic sulfur. The average isotopic values, however, are independent of both the total sulfur content and the ratio of organic to pyrite sulfur. Therefore, it seems likely that whereas the high-sulfur part of this data set contributes to scattering in the data, it does not obviously influence the average isotope values. For the other data, samples with total sulfur < 1.0 wt % are indicated separately by the black circles.
Taken together, the isotopic composition of sulfur in coals through the last 390 Ma is, with some exceptions, within the range of estimates for the modern riverine input to the ocean. The low-sulfur coals show a tighter clustering of values than the high-sulfur coals, reinforcing observations, as discussed above, that peats with an abundant sulfate supply show the greatest range in isotope variability. The most 34S-depleted low-sulfur coals are found in the end-Tertiary Powder River Basin, Wyoming. Here, in most cases where δ34S for the organic sulfur is < 0%, the pyrites are also similarly or even more depleted in 34S (33). These trends could represent real variability in the isotopic composition of input sulfur to the peats forming the coals, or to the addition of sulfide from sulfate reduction, despite the low sulfur contents of the coal.
There are also some indications for more 34S-enriched coals, and thus more 34S-enriched surface waters in the range of 11–12 per mil, during the Jurassic and Triassic periods (145–250 Ma). From a modeling perspective, this would generate lower values for fpy (Fig. 1) and therefore, even a higher proportion of sulfate burial than the traditional assumption of constant δin in the range of 3–8%. Such high rates of sulfate burial during this time window are consistent with previous estimates of the worldwide abundance of sulfate evaporites through Phanerozoic time (34).
Nearly all of the low-sulfur coal sulfur isotope data are substantially enriched in 34S compared with the predictions of riverine input isotope compositions as computed from the je-constrained mass balance (Figs. 1 and 2), which is ultimately constrained by the macrostratigraphic database from North America and the Caribbean. The coal sulfur isotope data would seem to invalidate this model. It appears that the metastratigraphic database has underestimated rates of marine sulfate evaporite formation through the last 400 Ma. The reasons for this are unclear, but could relate to the poor preservability of evaporite minerals, the poor preservability of coastal environments precipitating evaporites, or perhaps other factors.
Overall, the coal isotope data suggests that previous modeling is most correct when based on assumptions that riverine 34S has been close to the modern value. Therefore, the δin-constrained history of fpy as shown in Fig. 1 is most likely correct. This means that through the Phanerozoic Eon, the sulfur cycle has shifted from pyrite-dominated before about 400 Ma to sulfate-dominated afterward. From an oxygen regulation perspective, the sulfur cycle has likely become less significant through time.
Final Thoughts
Quantitative models of the evolution of the sulfur cycle through time rely on assumptions of key variables including the isotopic composition of sulfur entering the oceans, δin. The isotopic composition of sulfur in coals through time provides an important constraint of this variable and thus, these data provide a key insight into the workings of the sulfur cycle through Phanerozoic time. The coal sulfur isotope data also point to the possibility of further constraining δin, and our understanding of sulfur cycle evolution, through additional studies of coal or other preserved terrestrial plant material.
Acknowledgments
The author acknowledges stimulating discussions with W. Fisher and I. Halevy and valuable comments and suggestions from J. Farquhar and L. Kump. Financial support from the Danish National Research Foundation (Grant DNRF53) and the European Research Council (through the grant “Oxygen”) is greatly appreciated.
Footnotes
The author declares no conflict of interest.
References
- 1.Garrels RM, Perry EA. Cycling of carbon, sulfur, and oxygen through geologic time. In: Goldberg ED, editor. The Sea. Vol 5. New York: Wiley; 1974. pp. 303–336. [Google Scholar]
- 2.Berner RA, Raiswell R. Burial of organic carbon and pyrite sulfur in sediment over Phanerozoic time: A new theory. Geochim Cosmochim Acta. 1983;47:855–862. [Google Scholar]
- 3.Jørgensen BB. The sulfur cycle of a coastal marine sediment (Limfjorden, Denmark) Limnol Oceanogr. 1977;22:814–832. [Google Scholar]
- 4.Holland HD. Systematics of the isotopic composition of sulfur in the oceans during the Phanerozoic and its implications for atmospheric oxygen. Geochim Cosmochim Acta. 1973;37(12):2605–2616. [Google Scholar]
- 5.Berner RA, Canfield DE. A new model for atmospheric oxygen over Phanerozoic time. Am J Sci. 1989;289(4):333–361. doi: 10.2475/ajs.289.4.333. [DOI] [PubMed] [Google Scholar]
- 6.Schroeder S, Bekker A, Beukes NJ, Strauss H, van Niekerk HS. Rise in seawater sulphate concentration associated with the Paleoproterozoic positive carbon isotope excursion: Evidence from sulphate evaporites in the similar to 2.2-2.1 Gyr shallow-marine Lucknow Formation, South Africa. Terra Nova. 2008;20(2):108–117. [Google Scholar]
- 7.Grotzinger JP, Kasting JF. New constraints on Precambrian ocean composition. J Geol. 1993;101(2):235–243. doi: 10.1086/648218. [DOI] [PubMed] [Google Scholar]
- 8.Canfield DE, Farquhar J. Animal evolution, bioturbation, and the sulfate concentration of the oceans. Proc Natl Acad Sci USA. 2009;106(20):8123–8127. doi: 10.1073/pnas.0902037106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canfield DE. The evolution of the Earth surface sulfur reservoir. Am J Sci. 2004;304:839–861. [Google Scholar]
- 10.Kampschulte A, Strauss H. The sulfur isotopic evolution of Phanerozoic seawater based on the analysis of structurally substituted sulfates in carbonates. Chem Geol. 2004;204:255–286. [Google Scholar]
- 11.Canfield DE. The early history of atmospheric oxygen: Homage to Robert M. Garrels. Annu Rev Earth Planet Sci. 2005;33:1–36. [Google Scholar]
- 12.Halevy I, Peters SE, Fischer WW. Sulfate burial constraints on the Phanerozoic sulfur cycle. Science. 2012;337(6092):331–334. doi: 10.1126/science.1220224. . Available at http://www.sciencemag.org/content/337/6092/331.abstract?sid=44afde86-f17a-4314-85bc-071b0e769c71. [DOI] [PubMed] [Google Scholar]
- 13.Smith JW, Batts BD. The distribution and isotopic composition of sulfur in coal. Geochim Cosmochim Acta. 1974;38:121–133. [Google Scholar]
- 14. Berner EK, Berner RA (2012) Global Environemnt: Water, Air and Geochemical Cycles (Princeton Univ Press, Princeton), 2nd Ed.
- 15.Holser WT, Schidlowski M, Mackenzie FT, Maynard JB. Geochemical cycles of carbon and sulfur. In: Gregor CB, Garrels RM, Mackenzie FT, Maynard JB, editors. Chemical Cycles in the Evolution of the Earth. New York: Wiley; 1988. pp. 105–173. [Google Scholar]
- 16.Ivanov MV. The sulphur cycle in continental reservoirs. In: Ivanov MV, Freney JR, editors. The Global Biogeochemical Sulphur Cycle. Chichester, UK: Wiley; 1983. pp. 297–356. [Google Scholar]
- 17.Trust BA, Fry B. Stable sulphur isotopes in plants: A review. Plant Cell Environ. 1992;15:1105–1110. [Google Scholar]
- 18. Casagrande DJ (1987) Sulphur in peat and coal. Coal and Coal-Bearing Strata: Recent Advances, ed Scott AC, Geological Society Special Publication, pp 87–105.
- 19.Price FT, Casagrande DJ. Sulfur distribution and isotopic composition in peats from the Okefenokee Swamp, Georgia and the Everglades, Florida. Int J Coal Geol. 1991;17:1–20. [Google Scholar]
- 20.Skrzypek G, Jezierski P, Szynkiewicz A. Preservation of primary stable isotope signatures of peat-forming plants during early decomposition - observation along an altitudinal transect. Chem Geol. 2010;273(3–4):238–249. [Google Scholar]
- 21.Skrzypek G, Akagi T, Drzewicki W, Jedrysek MO. Stable isotope studies of moss sulfur and sulfate from bog surface waters. Geochem J. 2008;42(6):481–492. [Google Scholar]
- 22.Novak M, Bottrell SH, Prechova E. Sulfur isotope inventories of atmospheric deposition, spruce forest floor and living Sphagnum along a NW-SE transect across Europe. Biogeochemistry. 2001;53(1):23–50. [Google Scholar]
- 23. Chou C-L (1997) Geologic factors affecting the abundance, distribution, and speciation of sulfur in coals. Geology of Fossil Fuels, Proc 30th Int Geol Congress, ed Yang Q (Utrecht, The Netherlands), Vol. 18, Part B, pp 47–57. [Google Scholar]
- 24.Bottrell S, Novak M. Sulphur isotopic study of two pristine Sphagnum bogs in the western British Isles. J Ecol. 1997;85(2):125–132. [Google Scholar]
- 25.Mandernack KW, Lynch L, Krouse HR, Morgan MD. Sulfur cycling in wetland peat of the New Jersey Pinelands and its effect on stream water chemistry. Geochim Cosmochim Acta. 2000;64(23):3949–3964. [Google Scholar]
- 26.Habicht KS, Gade M, Thamdrup B, Berg P, Canfield DE. Calibration of sulfate levels in the archean ocean. Science. 2002;298(5602):2372–2374. doi: 10.1126/science.1078265. [DOI] [PubMed] [Google Scholar]
- 27.Canfield DE, Raiswell R, Bottrell S. The reactivity of sedimentary iron minerals toward sulfide. Am J Sci. 1992;292:659–683. [Google Scholar]
- 28.Coulson JP, Bottrell SH, Lee JA. Recreating atmospheric sulphur deposition histories from peat stratigraphy: Diagenetic conditions required for signal preservation and reconstruction of past sulphur deposition in the Derbyshire Peak District, UK. Chem Geol. 2005;218(3-4):223–248. [Google Scholar]
- 29.Price FT, Shieh YN. Fractionation of sulfur isotopes during laboratory synthesis of pyrite at low temperatures. Chem Geol. 1979;27:245–253. [Google Scholar]
- 30.Smith JW, Gould KW, Rigby D. The stable isotope geochemistry of Australian coals. Org Geochem. 1982;3:111–131. [Google Scholar]
- 31.Novák M, Wieder RK, Schell WR. Sulfur during early diagenesis in Sphagnum peat: Insights from δ34S ratio profiles in 210Pb-dated peat cores. Limnol Oceanogr. 1994;39(5):1172–1185. [Google Scholar]
- 32.Xiao HY, Liu CQ. The elemental and isotopic composition of sulfur and nitrogen in Chinese coals. Org Geochem. 2011;42(1):84–93. [Google Scholar]
- 33.Hackley KC, Anderson TF. Sulfur isotopic variations in low-sulfur coals from the Rocky Mountain region. Geochim Cosmochim Acta. 1986;50:1703–1713. [Google Scholar]
- 34.Hay WW, et al. Evaporites and the salinity of the ocean during the Phanerozoic: Implications for climate, ocean circulation and life. Palaeogeogr Palaeocl. 2006;240(1-2):3–46. [Google Scholar]
- 35.Wu NP, Farquhar J, Strauss H, Kim ST, Canfield DE. Evaluating the S-isotope fractionation associated with Phanerozoic pyrite burial. Geochim Cosmochim Acta. 2010;74(7):2053–2071. [Google Scholar]
- 36. Holmes CW, Brownfield ME (1992) The Distribution of Carbon and Sulfur Isotopes in Upper Cretaceous Coal of Northwestern Colorado, Geological Society of America Special Papers, Vol 267, pp 57–68.
- 37.Price FT, Shieh YN. Distribution and isotopic composition of sulfur in coals from the Illinois Basin. Econ Geol. 1979;74(6):1445–1461. [Google Scholar]