

Abstract
Heme/porphyrin-based electrocatalysts (both synthetic and natural) have been known to catalyze electrochemical O2, H+, and CO2 reduction for more than five decades. So far, no direct spectroscopic investigations of intermediates formed on the electrodes during these processes have been reported; and this has limited detailed understanding of the mechanism of these catalysts, which is key to their development. Rotating disk electrochemistry coupled to resonance Raman spectroscopy is reported for iron porphyrin electrocatalysts that reduce O2 in buffered aqueous solutions. Unlike conventional single-turnover intermediate trapping experiments, these experiments probe the system while it is under steady state. A combination of oxidation and spin-state marker bands and metal ligand vibrations (identified using isotopically enriched substrates) allow in situ identification of O2-derived intermediates formed on the electrode surface. This approach, combining dynamic electrochemistry with resonance Raman spectroscopy, may be routinely used to investigate a plethora of metalloporphyrin complexes and heme enzymes used as electrocatalysts for small-molecule activation.
Keywords: electrocatalysis, ferryl, SERRS
Electrocatalysis has taken center stage in contemporary research because of its indisputable relevance in the thriving area of renewable energy. Electrocatalytic O2 reduction, O2 evolution, H2 evolution, and H2 oxidation thus are areas of great interest and have attracted focused effort across several disciplines of science (1–11). Both natural systems [metalloenzymes such as cytochrome c oxidase (CcO), hydrogenase, and glucose oxidase] and biomimetic systems (i.e., synthetic/biochemical mimics of the natural enzyme) are known to function as electrocatalysts (12–15). In particular, it has been known for five decades that heme/porphyrin-based electrocatalysts (both natural and synthetic) catalyze electrochemical O2, H+, and CO2 reduction (14, 16–19). Several excellent metalloporphyrin-based O2 reduction electrocatalysts have been reported, some mimicking natural active sites and some inspired by their design (14, 20). Of these catalysts, the Fe-based catalysts generally have provided valuable insights into the O2 reduction mechanism and a deeper understanding of the structure–function correlations of key enzymes such as CcO, the terminal enzyme in the mitochondrial electron transport chain (1, 21). Similarly, several naturally occurring enzymes have been used as electrocatalysts for O2 reduction (e.g., CcO, microperoxidase) and substrate oxidation using O2 (e.g., cytochrome P450) (22–24). Apart from porphyrins, transition metal complexes of corroles, a related macrocycle that lacks one of the four mesocarbons of porphyrins, have been used extensively as electrocatalysts for O2 reduction and H2O oxidation (5, 6). These catalytic systems are efficient in catalyzing selective O2 activation/reduction reactions when the electrons (i.e., the reducing component) are provided from the electrode and O2 (the oxidizing component) is obtained from the bulk solution. An inherent advantage of this construct is its heterogeneity, which allows the use of water-insoluble catalysts to catalyze the reaction in an environmentally friendly solvent, such as water.
Elaborate mechanistic investigations have been conducted on these electrocatalysts; however, these investigations were limited to chemical and electrochemical perturbations (4). So far, there are no direct spectroscopic data on the intermediates formed during electrocatalysis. This is in sharp contrast to the investigation of O2 activation by iron porphyrin catalysts and heme enzymes in homogeneous solutions. In these cases, a broad spectrum of reactive intermediate species, formed during single-turnover reactions, is reported (25–29). The problem lies mainly in investigating a thin layer of catalyst (a monolayer in many cases) on an electrode and getting a good signal-to-noise ratio.
Surface enhanced resonance Raman spectroscopy (SERRS) is particularly suited for investigating metalloporphyrins and metalloenzymes attached to surfaces. The surface enhancement in addition to conventional resonance Raman (rR) effect (by exciting the intense absorption bands of these complexes) provides excellent enhancement of signals (30, 31). Additionally, metalloporphyrin systems are particularly well suited for Raman investigations. Several symmetric ligand vibrations (intense in Raman spectroscopy) have been identified that reflect the oxidation and spin states of the metals chelated by these macrocycles with remarkable fidelity (32, 33). In particular, the combination of the ν4 (∼1,330–1,375 cm−1) and the ν2 (∼1,540–1,575 cm−1) ligand vibrations are used routinely to diagnose the ground-state electronic structure of iron porphyrin complexes and enzyme active sites. SERRS has been used extensively to investigate various physical aspects of cytochrome c functionalized electrodes, some of which have been under different applied potentials on the electrode (34).
Studying the reaction mechanism of a monolayer of catalyst/protein requires accumulation of a good signal-to-noise ratio on a small time scale. For example, the catalytic cycle of CcO, which goes through several intermediates, takes less than 1 ms at room temperature (35, 36). Collecting a good signal in such short time scales, without cryogenically trapping the intermediates (cannot be achieved during electrocatalysis), presents a formidable challenge. Alternatively, reactions may be monitored under steady-state conditions. In such a case, only the species that are rate determining will accumulate under steady-state conditions. In principle, any spectroscopic data obtained will contain contributions from all the species present and may be hard to resolve. However, in the case of metalloporphyrins, the individual intermediates may be identified by using a combination of the oxidation and spin-state marker bands ν4 and ν2 in the resonance Raman spectrum (which are unique and resolvable, Table 1) and metal ligand vibrations (with appropriate isotopic substitutions). In a related approach, the intermediates formed during the electrocatalytic H2O2/O2 reduction by Bi/Pd-modified Au surfaces on a static electrode have been identified (37, 38).
Table 1.
Oxidation- and spin-state marker bands of iron porphyrins during steady-state conditions at room temperature (29, 32)
| Samples | Species | Ligation | Spin | ν4, cm−1 | ν2, cm−1 |
| Oxidized | |||||
| FeEs4 | Fe(III) (32) | 5 | 5/2 | 1,364 | 1,555 |
| FeTPP | Fe(III) (32) | 5 | 5/2 | 1,360 | 1,552 |
| Reduced | |||||
| FeEs4 | Fe(II) (32) | 5 | 2 | 1,349 | 1,547 |
| FeTPP | Fe(II) (32) | 5 | 2 | 1,344 | 1,539 |
| During steady state | |||||
| FeEs4 | Fe(III) | 6 | 1/2 | 1,369 | 1,565 |
| Fe(II) | 5 | 2 | 1,352 | 1,540 | |
| Fe(IV) = O (29) | 6 | — | 1,371 | 1,571 | |
| FeTPP | Fe(III) | 6 | 1/2 | 1,366 | 1,565 |
| Fe(II) | 5 | 2 | 1,347 | 1,535 | |
| Fe(IV) = O | 6 | — | 1,371 | 1,570 |
In this study, we report an SERRS setup modified to accommodate a conventional rotating disk electrochemistry (RDE) or rotating ring disk electrochemistry (RRDE) setup (39) bearing iron porphyrin catalysts and engaged in steady-state electrocatalytic O2 reduction. The data provide direct first-hand information about the nature of intermediates involved in steady-state O2 reduction and helps define the mechanism of these catalysts.
Results and Discussion
The experimental station is described in Fig. 1. The working disk electrode is mounted at the tip of a conventional RDE shaft b. An air-tight, water-jacketed, three-electrode electrochemical cell c with a Pyrex optical window g is built such that both the incident laser and the Raman scattering are collected through this optical window. The scattered light is collected with a lens h and reflected on mirror j mounted on holder k toward another lens, which focuses it on the entrance slit of the spectrograph. Two iron porphyrin catalysts are investigated: iron tetraphenylporphyrin (FeTPP) (40) and iron α4-tetra-2-(4-carboxymethyl-1,2,3-triazolyl)-phenylporphyrin (FeEs4) (3) (Fig. 2). Both are known to electrocatalytically reduce O2, and the latter recently was shown to have a hydrogen-bonding distal superstructure that can stabilize an Fe-O2 adduct (28).
Fig. 1.
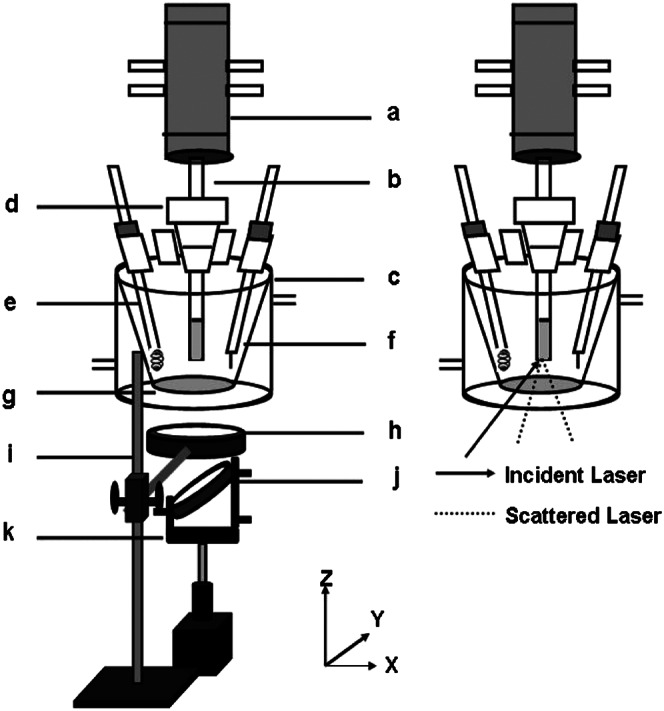
Experimental setup for SERRS-RDE.
Fig. 2.

Iron porphyrin complexes α4-FeEs4 (Left) and FeTPP (Right).
A cyclic voltammetry (CV) scan of the FeEs4 catalysts (Fig. 3A, black line) immobilized on the octanethiol (C8SH) self-assembled monolayer (SAM)-covered Au disk electrode shows the half-wave potential (E1/2) of the FeIII/II process at −100 mV vs. Ag/AgCl in a deaerated buffer solution. The integrated charge under this wave indicates a surface coverage of 8 × 1011 molecules per square centimeter. Such low surface density is consistent with the formation of a monolayer and not a multilayer (typically 1014 molecules per square centimeter) of catalyst on the SAM (41, 42). This also is suggested by the atomic force microscopy (AFM) image of the electrode, which shows a smooth surface (with a height of 1.70–1.76 nm) without any multilayer (Fig. 3B) (43). SERRS-RDE data obtained by applying positive (0 V) and negative (−0.5 V) potentials (i.e., at potentials above and below the E1/2) at 200 rpm (Pine Research Instrumentation; AFE6M rotor) rotation rate show the ν4 and ν2 vibrations (Table 1) indicative of oxidized (Fig. 4, bold black line) and reduced (Fig. 4, dashed black line) iron porphyrin complexes, respectively. Alternatively, a linear sweep voltammetry (LSV) scan performed in aerated buffer solutions using an RDE shows that as the potential of the working electrode is lowered gradually below the E1/2, a catalytic current gradually rises as the result of electrocatalytic reduction of O2 (Fig. 3A, gray line) by the FeII porphyrin produced on the electrodes at these potentials (i.e., below E1/2). This catalytic current becomes potential independent at lower potentials and becomes mass transfer limited. The current is determined by the flow of the substrate, O2, to the electrode, i.e., on the angular rotation rate of the electrode (38). This also is indicated by a plot of 1/icat (at −0.5 V) vs. 1/ω1/2, which is linear (Fig. 3C).
Fig. 3.

(A) CV and LSV data of FeEs4 physisorbed on C8SH SAM in deoxygenated and air-saturated pH 7 buffers, respectively, at a scan rate of 50 mV/s using Ag/AgCl as a reference and Pt wire as counter electrodes. The dotted lines represent the variable potentials applied during the variable-potential SERRS-RDE experiments. (B) AFM image of the C8SH SAM-covered electrode-bearing catalyst. (C) Plot of 1/icat vs. 1/ω1/2 obtained at −0.5 V.
Fig. 4.

SERRS-RDE data of FeEs4 (A) and FeTPP (B) in the low-frequency (Left) and high-frequency (Right) regions, respectively, where the oxidized spectra are indicated by bold black lines, the reduced spectra under anaerobic conditions by dashed black lines, and the reduced spectra under aerobic conditions by gray lines.
SERRS-RDE data collected on the electrode held at −0.5 V (i.e., during a bulk electrolysis experiment using RDE) show a spectrum (Fig. 4, gray line) different from the one obtained in the absence of O2 (Fig. 4, dashed black line) (38). Several ν4 and ν2 vibrations are observed in which a particular set of ν4 and ν2 vibrations uniquely represents an iron porphyrin complex having a particular oxidation, spin, and ligation state (Table 1). For the α4-FeEs4 complex, there is a clear increase in intensity at 1,369 cm−1 and 1,565 cm−1, suggesting the formation of a low-spin FeIII species (Fig. 4A, gray line) during turnover (32). Additionally, peaks are observed at 1,352 cm−1 and 1,540 cm−1 (Fig. 4A, gray line), which are characteristic of the reduced FeII species. There also is an increase in intensity at 1,371 cm−1 and 1,571 cm−1, which clearly is observed in the difference spectrum (Fig. 5B) and suggests formation of high-valent FeIV = O species (29). Thus, in addition to the resting high-spin FeIII species, a high-spin FeII species, a low-spin FeIII species, and an FeIV = O species are observed during steady-state O2 reduction by the FeEs4 catalyst. In the low-energy region of the Raman spectrum, there are increases in intensities at 779 cm−1, 631 cm−1, and 570 cm−1 (Fig. 4A, gray line).
Fig. 5.

Difference spectra of the SERRS-RDE data at different potentials −0.15 V (red), −0.25 V (yellow), −0.35 V (blue), and −0.5 V (green) from 0 V of FeEs4 (A) and FeTPP (C) in the low-frequency (Left) and high-frequency (Right) regions, respectively. The ν4 and ν2 regions of the spectrum obtained from the difference of the spectrum at −0.5 V from the spectrum at 0 V of FeEs4 and FeTPP, along with their fits, are shown in B and D, respectively. (E) The difference spectra obtained at different rotation rates of FeEs4.
For the FeTPP complex, very similar increases in intensities are observed in the high-frequency region of the spectrum, suggesting the formation of high-spin FeII, low-spin FeIII, and FeIV = O species during turnover (Fig. 4B, gray line). In the low-frequency region, peaks at 634 cm−1, 677 cm−1, 741 cm−1, 778 cm−1, and 830 cm−1 gain intensity (Fig.4B, gray line).
The O2 reduction current is potential dependent; hence, the SERRS-RDE data were collected by applying different potentials at the working electrode (rotation rate, 200 rpm). In the case of FeEs4, the difference spectrum (data at a particular potential – resting oxidized) in the low-energy region shows that as the potential is lowered, peaks at 830 cm−1, 782 cm−1, 631 cm−1, and 570 cm−1 gain intensity (Fig. 5A), whereas the peak at the 580-cm−1 region loses intensity as the electrocatalytic O2 reduction current increases and approaches a potential-independent mass transfer-controlled limit. In the higher-energy region, the ν4 and ν2 vibrations at 1,352 cm−1 and 1,540 cm−1 (high-spin FeII), 1,369 cm−1 and 1,565 cm−1 (low-spin FeIII), and 1,371 cm−1 and 1,571 cm−1 (FeIV = O) gain intensity (Fig. 5 A and C). Alternatively, the ν4 and ν2 vibrations at 1,364 cm−1 and 1,555 cm−1 (high-spin FeIII) lose intensity (Fig. 5 A and C). Thus, as the catalytic current transitions from the potential-dependent region to the mass transfer-limited region, the high-spin FeII, the low-spin FeIII, and an FeIV species gradually build up at the electrode at the expense of the high-spin FeIII species (Fig. 5B). In the case of FeTPP, the analysis is complicated slightly by decay of the catalyst during the variable potential SERRS-RDE experiments (14). However, a clear increase in the intensities of the ν4 and ν2 vibrations at 1,347 cm−1 and 1,535 cm−1, 1,366 cm−1 and 1,565 cm−1, and 1,371 cm−1 and 1,570 cm−1 indicates a buildup of high-spin FeII, low-spin FeIII, and FeIV = O species, respectively, on the electrode as the potential is lowered and the catalytic O2 reduction current increases to attain a mass transfer-limited value (Fig. 5D). The data obtained at multiple rotation rates (Fig. 5E) show that at rotation rates >100 rpm, the distribution of the species observed remains the same, indicating that the reaction is in a steady state and there is a buildup of different intermediate species involved in this multistep catalytic reaction. In the low-energy region, peaks at 596 cm−1, 638 cm−1, and 779 cm−1 gain intensity as the potential is lowered gradually to negative values, i.e., where the FeIII is reduced to FeII (Fig. 5C). [Note that the peaks at 677 cm−1, 742 cm−1, and 832 cm−1 gain intensity only at a very negative potential (Fig. 5C, green).]
Experiments were performed in 18O2-saturated pH 7 buffers with FeEs4 to gain insight into the nature of these intermediates. The variable potential SERRS-RDE data (Fig. 6 A and B), as well as the difference data, show significant differences between 16O2 and 18O2 data (Fig. 6 C and D). In particular, the 830-cm−1 band, which gains intensity in 16O2 buffer (Fig. 6 A and C), loses intensity in the 18O2-saturated buffer (Fig. 6 B and D). Similarly, the 631-cm−1 band loses intensity in the 18O2 buffer while it gains intensity in 16O2-saturated buffer, whereas the 580-cm−1 band loses intensity in 16O2 buffer but not in 18O2 buffer (Fig. 6). There is a larger increase in intensity in the 770–780-cm−1 region in the case of 18O2 buffer relative to the increase observed in 16O2 buffer. In particular, this peak, centered at 780.5 cm−1 in 18O2 buffer, is asymmetric because of the presence of a shoulder at higher energy that is not observed in 16O2 buffer. This indicates the growth of a new peak in 18O2 buffer. Furthermore, the intensity of the band at 746 cm−1 increases more in 18O2 buffer than it does in 16O2 buffer. The differences in the intensities of these vibrations in the low-energy region of the spectrum between 16O2- and 18O2-containing buffers imply the presence of Fe-Ox species derived from O2 during reduction. The 16O2/18O2 difference spectrum (obtained at different potentials) reveals a clear shift from 830 cm−1 to 783 cm−1 (Fig. 6 E and F). There is an additional shift from 631 cm−1 to 594 cm−1. Note that these differences arise only at potentials in which electrocatalytic O2 reduction is observed, and their intensities increase as the potential is lowered and hence arise as the result of incorporation of 18O2 into intermediates formed during electrocatalytic O2 reduction.
Fig. 6.

SERRS-RDE data of FeEs4 obtained at various potentials under 16O2-containing (A) and 18O2-containing (B) pH 7 buffers. The spectra obtained from the difference of various potentials from 0 V for 16O2- and 18O2-containing buffers are shown in C and D, respectively. (E) Difference spectra of 18O2 from 16O2 obtained at respective potentials. (F) The shifts obtained, along with their fits.
The peak at 782 cm−1 that gradually gained intensity in the variable-potential SERRS-RDE experiment in both 16O2 and 18O2 (Fig. 6) possibly arises from an intermediate formed during O2 reduction as well. However, a clear isotopic shift was not observed for these species. Note that a weak peak that grows in at 752 cm−1 in the 18O2 difference spectrum might be the result of an isotope shift of this species, i.e., 782 cm−1 in 16O2 to 752 cm−1 in 18O2; possibly, this arises from an FeIV = O species. Ferryl species are known to undergo fast exchange of the ferryl oxygen with water (44, 45). Thus, any FeIV = 18O formed will quickly exchange in H216O in the medium to form FeIV = 16O, leading to limited population of FeIV = 18O species. (An experiment in H2O18 might help this assignment, but a minimum of 30 mL of this buffer would be required, which makes this experiment commercially unviable.) The 830-cm−1 and 631-cm−1 bands represent the O-O and Fe-O vibrations of a low-spin FeIII-OOH species, respectively, and the 782-cm−1 band represents the Fe-O stretch of an FeIV = O species. These assignments are consistent with the observation of ν4 and ν2 bands characteristic of low-spin FeIII species and FeIV species in the variable-potential SERRS-RDE data (32). The 16/18O2 isotope analysis of FeTPP is complicated by its decay during electrocatalytic O2 reduction.
In addition to the SERRS-RDE data, the formation of the high-valent FeIV = O species also was evaluated using this species (E0 ∼ 1 V) produced during O2 reduction on the electrode to chemically oxidize [Fe(CN)6]4− in situ. The redox potential (Eo) for [Fe(CN)6]3−/4− is 250 mV vs. Ag/AgCl at pH 7. The FeIII porphyrin species used here, which have an Eo of –100 mV, are incapable of oxidizing [Fe(CN)6]4− to [Fe(CN)6]3−. In an RRDE setup, any [Fe(CN)6]3− produced on the working electrode is diffused radially to the Pt ring (as a result of the hydrodynamic current produced by the rotation of the electrode-bearing cylindrical shaft) and may be detected by reducing it back to [Fe(CN)6]4− by applying a potential of 0 V on the Pt ring (the Pt ring is independently addressable in an RRDE experiment using a bipotentiostat). In the presence of 10 mM K4[Fe(CN)6] in the buffer solution, a clear reduction current is observed in the ring and the onset of this current coincides with the onset of the O2 reduction current. This indicates that the [Fe(CN)6]4− present in the solution is getting oxidized to [Fe(CN)6]3− by the FeIV = O species produced on the electrode during O2 reduction (42), further supporting the formation of high-valent FeIV = O species on the electrode during O2 reduction. Note that FeIII-OOH species, also produced on the electrode, is proposed to oxidize C-H bonds (46). However, its oxidation proceeds by C-H bond activation and not via a classical outer/inner sphere mechanism generally operative in [Fe(CN)6]4− (47, 48). These processes are driven by free energy of electron transfer. The Eo of an FeIII-OOH species likely is close to that of the resting FeIII-OH species (Eo = −100 mV), which is much lower than the Eo of [Fe(CN)6]4− (Eo = 250 mV). Thus, electron transfer from [Fe(CN)6]4− to an FeIII-OOH species is a significantly uphill process, i.e., oxidation of [Fe(CN)6]4− by an FeIII-OOH species is not likely. Note that no current is observed in the Pt ring if there is no K4[Fe(CN)6] present in the solution or no catalyst is present on the electrode. The oxidation of [Fe(CN)6]4− by small amounts of H2O2 produced in the electrode as the result of incomplete reduction of O2 may be precluded, as it is a very slow process under these conditions (49). It is important to note that the observation of an FeIV = O species on the electrode implies that its reduction to FeIII-OH is slow despite a large driving force for electron transfer (ET) from the electrode (held at −0.5 V) to the FeIV = O species (Eo = ∼1 V). This is the case in the active site of CcO, where the ET from heme a to heme a3 (with marginal driving force) is >105 s−1 (50), whereas the ET from heme a to PR (the FeIV = O species produced during O2 reduction) is ∼103 s−1 despite having a much larger driving force (51).
Based on the intermediates identified, a mechanistic proposal of O2 reduction by these Fe porphyrins is shown in Fig. 7. The SERRS-RDE data indicate that FeII species, FeIII high-spin species, low-spin FeIII-OOH species, and FeIV = O species are present during steady-state electrocatalytic O2 reduction. Furthermore, the variable-potential SERRS-RDE data indicate that as the potential is lowered, there is an increase in the population of the high-spin FeII species, low-spin FeIII-OOH species, and FeIV = O species and a decrease in population of the resting high-spin FeIII species. Thus, at potentials in which the current is potential dependent, the population of the O2-derived species is less, as the reduction of the high-spin FeIII to the catalytically active FeII state (i → ii in Fig. 7) is the rate-determining step leading to a higher population of the resting high-spin FeIII species on the electrode. However, as steady-state O2 reduction current is obtained at low potentials, the low-spin FeIII-OOH species is observed along with a high-spin FeII species and an FeIV = O species. This implies that the decay of these species—i.e., reductive cleavage of the O-O bond of the FeIII-OOH species (iv → v in Fig. 7), O2 binding to FeII (ii → iii in Fig. 7), and reduction of FeIV = O (v → i in Fig. 7)—comprises rate-determining steps during O2 reduction. At this stage, we could not find evidence of an Fe-O2 adduct in these data. Because resonance Raman is not quantitative, the relative population of these species, i.e., the relative rates of these steps, cannot be determined using these data. However, it establishes, with confidence, the species accumulated under steady state and provides a qualitative description of the rate-determining steps in O2 reduction by these electrocatalysts.
Fig. 7.

The oxygen-reducing mechanism by an iron porphyrin, showing the various possible intermediates. The marker bands of the individual species are given in boxes, with the ν(Fe-O) values (16O2/18O2) corresponding to those observed for FeEs4.
Conclusion
A SERRS-RDE experimental setup is developed and the mechanism of O2 reduction by a set of iron porphyrin complexes is investigated using in situ resonance Raman spectroscopy on the system under steady state. The oxidation and spin-state marker bands help identify species having the Fe in different oxidation states. Experiments using isotopically labeled O2 enable identification of O2-derived intermediates accumulating under steady-state conditions. This approach, which combines dynamic electrochemistry with in situ Raman spectroscopy, allows direct identification of intermediates involved in O2 reduction by an iron porphyrin-based electrocatalyst. This technique may be used to probe electrochemical systems using synthetic as well as biological catalysts routinely used for small-molecule activation and to gain first-hand information on the reaction mechanism of these electrocatalysts. The ability to control the applied potential on the working electrode enables investigation of reaction mechanisms when the catalysis is electron transport controlled, as well as when it is mass transport controlled.
Materials and Methods
All reagents were of the highest grade commercially available and were used without further purification. Octanethiol (C8SH), potassium hexafluorophosphate (KPF6), and all buffers were purchased from Sigma–Aldrich. Disodium hydrogen phosphate dihydrate (Na2HPO4⋅2H2O) and potassium chloride (KCl) were purchased from Merck. All solvents used were of spectroscopic grade and were purchased from Merck. Au disks for the RDE and RRDE experiments and Ag disks for SERRS experiments were purchased from Pine Instruments. 18O2 (99%) was purchased from ICON Isotopes. All electrochemical experiments were performed using a CH Instruments model CHI710D electrochemical analyzer. Bipotentiostat reference electrodes were purchased from CH Instruments. The RRDE setup from Pine Research Instrumentation (E6 Series ChangeDisk tips with AFE6M rotor) was used to obtain the RRDE data. SERRS-RDE data were collected using a Trivista 555 spectrograph (Princeton Instruments) and 413.1-nm excitation from a Kr+ laser (Coherent Innova Sabre SBRC-DBW-K), rotating the electrode at 200 rpm unless noted otherwise.
Freshly cleaned Au and roughened Ag disks were immersed in an ethanolic solution of C8SH (0.4 mM) for 24 h. A dilute solution of the catalyst (1 mM) in chloroform was dropped on these electrodes and kept for 30 min. The unabsorbed or loosely absorbed catalyst was washed off using CHCl3. The electrochemical experiments were done in Au electrodes, and the SERRS-RDE experiments were performed on roughened Ag disks, because SERRS cannot be performed on Au surfaces at <600 nm and the rR experiments on porphyrin complexes need excitation near 400 nm. All these surfaces show similar coverage and O2 reduction currents. It is assumed that the spectroscopic data obtained on SAM-covered Ag surfaces are valid for SAM-covered Au surfaces as well.
Acknowledgments
We thank Dr. Somdatta Ghosh Dey for helpful discussions and acknowledge the reviewers for their constructive criticism. This work was funded by Department of Science and Technology, India, Grant DST/SR/IC-35-2009. K.S. and S.C. acknowledge support from Council of Scientific and Industrial Research (CSIR) senior research fellowship and CSIR junior research fellowship, respectively. S.S. acknowledges the integrated doctoral program of Indian Association for the Cultivation of Science.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
References
- 1.Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem Rev. 2004;104(2):561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 2.Kanan MW, Nocera DG. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+ Science. 2008;321(5892):1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- 3.Samanta S, Sengupta K, Mittra K, Bandyopadhyay S, Dey A. Selective four electron reduction of O2 by an iron porphyrin electrocatalyst under fast and slow electron fluxes. Chem Commun (Camb) 2012;48(61):7631–7633. doi: 10.1039/c2cc32832a. [DOI] [PubMed] [Google Scholar]
- 4.Carver CT, Matson BD, Mayer JM. Electrocatalytic oxygen reduction by iron tetra-arylporphyrins bearing pendant proton relays. J Am Chem Soc. 2012;134(12):5444–5447. doi: 10.1021/ja211987f. [DOI] [PubMed] [Google Scholar]
- 5.Schechter A, Stanevsky M, Mahammed A, Gross Z. Four-electron oxygen reduction by brominated cobalt corrole. Inorg Chem. 2012;51(1):22–24. doi: 10.1021/ic202139f. [DOI] [PubMed] [Google Scholar]
- 6.Dogutan DK, McGuire R, Jr, Nocera DG. Electocatalytic water oxidation by cobalt(III) hangman β-octafluoro corroles. J Am Chem Soc. 2011;133(24):9178–9180. doi: 10.1021/ja202138m. [DOI] [PubMed] [Google Scholar]
- 7.Karunadasa HI, et al. A molecular MoS₂ edge site mimic for catalytic hydrogen generation. Science. 2012;335(6069):698–702. doi: 10.1126/science.1215868. [DOI] [PubMed] [Google Scholar]
- 8.Valdez CN, Dempsey JL, Brunschwig BS, Winkler JR, Gray HB. Catalytic hydrogen evolution from a covalently linked dicobaloxime. Proc Natl Acad Sci USA. 2012;109(39):15589–15593. doi: 10.1073/pnas.1118329109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith SE, Yang JY, DuBois DL, Bullock RM. Reversible electrocatalytic production and oxidation of hydrogen at low overpotentials by a functional hydrogenase mimic. Angew Chem Int Ed Engl. 2012;51(13):3152–3155. doi: 10.1002/anie.201108461. [DOI] [PubMed] [Google Scholar]
- 10.Jacques P-A, Artero V, Pécaut J, Fontecave M. Cobalt and nickel diimine-dioxime complexes as molecular electrocatalysts for hydrogen evolution with low overvoltages. Proc Natl Acad Sci USA. 2009;106(49):20627–20632. doi: 10.1073/pnas.0907775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pijpers JJH, Winkler MT, Surendranath Y, Buonassisi T, Nocera DG. Light-induced water oxidation at silicon electrodes functionalized with a cobalt oxygen-evolving catalyst. Proc Natl Acad Sci USA. 2011;108(25):10056–10061. doi: 10.1073/pnas.1106545108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincent KA, Parkin A, Armstrong FA. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem Rev. 2007;107(10):4366–4413. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- 13.Foster CE, et al. Inhibition of [FeFe]-hydrogenases by formaldehyde and wider mechanistic implications for biohydrogen activation. J Am Chem Soc. 2012;134(17):7553–7557. doi: 10.1021/ja302096r. [DOI] [PubMed] [Google Scholar]
- 14.Collman JP, et al. A cytochrome c oxidase model catalyzes oxygen to water reduction under rate-limiting electron flux. Science. 2007;315(5818):1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collman JP, et al. Role of a distal pocket in the catalytic O2 reduction by cytochrome c oxidase models immobilized on interdigitated array electrodes. Proc Natl Acad Sci USA. 2009;106(18):7320–7323. doi: 10.1073/pnas.0902285106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bettelheim A, Kuwana T. Rotating-ring-disk analysis of iron tetra(N-methylpyridyl)porphyrin in electrocatalysis of oxygen. Anal Chem. 1979;51(13):2257–2260. [Google Scholar]
- 17.Anson FC, Ni CL, Saveant JM. Electrocatalysis at redox polymer electrodes with separation of the catalytic and charge propagation roles. Reduction of dioxygen to hydrogen peroxide as catalyzed by cobalt(II) tetrakis(4-N-methylpyridyl)porphyrin. J Am Chem Soc. 1985;107(12):3442–3450. [Google Scholar]
- 18.Durand RR, Jr, Bencosme CS, Collman JP, Anson FC. Mechanistic aspects of the catalytic reduction of dioxygen by cofacial metalloporphyrins. J Am Chem Soc. 1983;105(9):2710–2718. [Google Scholar]
- 19.Chang CJ, Loh Z-H, Shi C, Anson FC, Nocera DG. Targeted proton delivery in the catalyzed reduction of oxygen to water by bimetallic pacman porphyrins. J Am Chem Soc. 2004;126(32):10013–10020. doi: 10.1021/ja049115j. [DOI] [PubMed] [Google Scholar]
- 20.Collman JP, Fu L, Herrmann PC, Zhang X. A functional model related to cytochrome c oxidase and its electrocatalytic four-electron reduction of O2. Science. 1997;275(5302):949–951. doi: 10.1126/science.275.5302.949. [DOI] [PubMed] [Google Scholar]
- 21.Kim E, Chufán EE, Kamaraj K, Karlin KD. Synthetic models for heme-copper oxidases. Chem Rev. 2004;104(2):1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 22.Haas AS, et al. Cytochrome c and cytochrome c oxidase: monolayer assemblies and catalysis. J Phys Chem B. 2001;105(45):11351–11362. [Google Scholar]
- 23.Mayhew MP, Reipa V, Holden MJ, Vilker VL. Improving the cytochrome P450 enzyme system for electrode-driven biocatalysis of styrene epoxidation. Biotechnol Prog. 2000;16(4):610–616. doi: 10.1021/bp000067q. [DOI] [PubMed] [Google Scholar]
- 24.Yang M, et al. Electrocatalytic drug metabolism by CYP2C9 bonded to a self-assembled monolayer-modified electrode. Drug Metab Dispos. 2009;37(4):892–899. doi: 10.1124/dmd.108.025452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collman JP, Decréau RA, Yan Y, Yoon J, Solomon EI. Intramolecular single-turnover reaction in a cytochrome C oxidase model bearing a Tyr244 mimic. J Am Chem Soc. 2007;129(18):5794–5795. doi: 10.1021/ja0690969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J-G, Shimizu Y, Ohta T, Naruta Y. Formation of an end-on ferric peroxo intermediate upon one-electron reduction of a ferric superoxo heme. J Am Chem Soc. 2010;132(11):3672–3673. doi: 10.1021/ja1001955. [DOI] [PubMed] [Google Scholar]
- 27.Halime Z, Kotani H, Li Y, Fukuzumi S, Karlin KD. Homogeneous catalytic O2 reduction to water by a cytochrome c oxidase model with trapping of intermediates and mechanistic insights. Proc Natl Acad Sci USA. 2011;108(34):13990–13994. doi: 10.1073/pnas.1104698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mittra K, et al. A hydrogen bond scaffold supported synthetic heme FeIII-O2- adduct. Chem Commun. 2012;48(85):10535–10537. doi: 10.1039/c2cc35162e. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto S, Mizutani Y, Tatsuno Y, Kitagawa T. Resonance Raman characterization of ferric and ferryl porphyrin. pi. cation radicals and the FeIV:0 stretching frequency. J Am Chem Soc. 1991;113(17):6542–6549. [Google Scholar]
- 30.Sharma B, Frontiera RR, Henry A-I, Ringe E, Van DRP. SERS: Materials, applications, and the future. Mater Today (Oxford, UK) 2012;15:16–25. [Google Scholar]
- 31.Murgida DH, Hildebrandt P. Electron-transfer processes of cytochrome C at interfaces. New insights by surface-enhanced resonance Raman spectroscopy. Acc Chem Res. 2004;37(11):854–861. doi: 10.1021/ar0400443. [DOI] [PubMed] [Google Scholar]
- 32.Burke JM, et al. Structure-sensitive resonance Raman bands of tetraphenyl and “picket fence” porphyrin-iron complexes, including an oxyhemoglobin analog. J Am Chem Soc. 1978;100(19):6083–6088. [Google Scholar]
- 33.Abe M, Kitagawa T, Kyogoku Y. Vibrational assignments of resonance Raman lines of ni(octaethylporphyrin) on the basis of a normal coordinate treatment. Chem Lett. 1976;5(3):249–252. [Google Scholar]
- 34.Grochol J, Dronov R, Lisdat F, Hildebrandt P, Murgida DH. Electron transfer in SAM/cytochrome/polyelectrolyte hybrid systems on electrodes: A time-resolved surface-enhanced resonance Raman study. Langmuir. 2007;23(22):11289–11294. doi: 10.1021/la701470d. [DOI] [PubMed] [Google Scholar]
- 35.Ferguson-Miller S, Babcock GT. Heme/copper terminal oxidases. Chem Rev. 1996;96(7):2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 36.Kaila VRI, Verkhovsky MI, Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110(12):7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Gewirth AA. Peroxide electroreduction on bi-modified Au surfaces: Vibrational spectroscopy and density functional calculations. J Am Chem Soc. 2003;125(23):7086–7099. doi: 10.1021/ja034125q. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Gewirth AA. SERS study of hydrogen peroxide electroreduction on a Pb-modified Au electrode. J Raman Spectrosc. 2005;36(6–7):715–724. [Google Scholar]
- 39.Bard AJ, Faulkner LR. Electrochemical Methods. New York: Wiley; 1980. p. 300. [Google Scholar]
- 40.Collman JP, et al. “Picket fence porphyrins.” Synthetic models for oxygen binding hemoproteins. J Am Chem Soc. 1975;97(6):1427–1439. doi: 10.1021/ja00839a026. [DOI] [PubMed] [Google Scholar]
- 41.Bettelheim A, Chan RJH, Kuwana T. Electroanalysis of oxygen reduction: Part III. Selective reduction to hydrogen peroxide or water using polymeric attachment of metalloporphyrins. J Electroanal Chem Interfacial Electrochem. 1980;110:93–102. [Google Scholar]
- 42.Sengupta K, Chatterjee S, Samanta S, Bandyopadhyay S, Dey A. Resonance Raman and electrocatalytic behavior of thiolate and imidazole bound iron porphyrin complexes on self assembled monolayers: Functional modeling of cytochrome P450. Inorg Chem. 2013;52(4):2000–2014. doi: 10.1021/ic302369v. [DOI] [PubMed] [Google Scholar]
- 43.Porter MD, Bright TB, Allara DL, Chidsey CED. Spontaneously organized molecular assemblies. 4. Structural characterization of n-alkyl thiol monolayers on gold by optical ellipsometry, infrared spectroscopy, and electrochemistry. J Am Chem Soc. 1987;109(12):3559–3568. [Google Scholar]
- 44.Hashimoto S, Tatsuno Y, Kitagawa T. Resonance Raman evidence for oxygen exchange between the FeIV = O heme and bulk water during enzymic catalysis of horseradish peroxidase and its relation with the heme-linked ionization. Proc Natl Acad Sci USA. 1986;83(8):2417–2421. doi: 10.1073/pnas.83.8.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee KA, Nam W. Determination of reactive intermediates in iron porphyrin complex-catalyzed oxygenations of hydrocarbons using isotopically labeled water: Mechanistic insights. J Am Chem Soc. 1997;119(8):1916–1922. [Google Scholar]
- 46.Derat E, Kumar D, Hirao H, Shaik S. Gauging the relative oxidative powers of compound I, ferric-hydroperoxide, and the ferric-hydrogen peroxide species of cytochrome P450 toward C-H hydroxylation of a radical clock substrate. J Am Chem Soc. 2005;128(2):473–484. doi: 10.1021/ja056328f. [DOI] [PubMed] [Google Scholar]
- 47.Taube H, Gould ES. Organic molecules as bridging groups in electron-transfer reactions. Acc Chem Res. 1969;2(11):321–329. [Google Scholar]
- 48.Taube H. Recent progress in the study of inner-sphere electron transfer reactions. Pure Appl Chem. 1970;24(2):289–305. [Google Scholar]
- 49.Kistiakowsky W. Experiments on the sensitiveness to light of hydrogen peroxide in aqueous solutions on addition of potassium ferro- and ferricyanide. Z Phys Chem. 1900;35:431–439. [Google Scholar]
- 50.Adelroth P, Brzezinski P, Malmström BG. Internal electron transfer in cytochrome c oxidase from Rhodobacter sphaeroides. Biochemistry. 1995;34(9):2844–2849. doi: 10.1021/bi00009a014. [DOI] [PubMed] [Google Scholar]
- 51.Hallén S, Nilsson T. Proton transfer during the reaction between fully reduced cytochrome c oxidase and dioxygen: pH and deuterium isotope effects. Biochemistry. 1992;31(47):11853–11859. doi: 10.1021/bi00162a025. [DOI] [PubMed] [Google Scholar]