

Abstract
Deregulated Toll-like receptor (TLR)-triggered inflammatory responses that depend on NF-κB are detrimental to the host via excessive production of proinflammatory cytokines, including TNF-α. Stat2 is a critical component of type I IFN signaling, but it is not thought to participate in TLR signaling. Our study shows that LPS-induced lethality in Stat2−/− mice is accelerated as a result of increased cellular transmigration. Blocking intercellular adhesion molecule-1 prevents cellular egress and confers survival of Stat2−/− mice. The main determinant of cellular egress in Stat2−/− mice is the genotype of the host and not the circulating leukocyte. Surprisingly, lethality and cellular egress observed on Stat2−/− mice are not associated with excessive increases in classical sepsis cytokines or chemokines. Indeed, in the absence of Stat2, cytokine production in response to multiple TLR agonists is reduced. We find that Stat2 loss leads to reduced expression of NF-κB target genes by affecting nuclear translocation of NF-κB. Thus, our data reveal the existence of a different mechanism of LPS-induced lethality that is independent of NF-κB triggered cytokine storm but dependent on cellular egress.
Sepsis is a complex clinical disorder that may culminate in multiorgan failure and death (1, 2). Many infectious agents induce a severe inflammatory response by triggering Toll-like receptor (TLR) signaling pathways. In Gram-negative sepsis, bacterial LPS binds TLR4 and relays signals through adapter molecules and transcription factors, including the NF-κB family, to mount an inflammatory response (3). This includes production of cytokines (e.g., TNF-α, IL-6, and IFN-γ) and chemokines [e.g., regulated on activation, normal T cell expressed and secreted (RANTES) and monocyte chemoattractant protein-1 (MCP-1)] that recruit innate and adaptive components of inflammation to sites of injury.
Infusion of LPS into rodent models recapitulates cytokine surges that correlate with the pathophysiology of sepsis (4, 5). Mice deficient in genes encoding components of the TLR signaling pathway [Tlr4 (6), Md-2 (7), Cd14 (7), and MyD88 (8)] survive doses of LPS that are lethal to WT animals. In all these models, survival is associated with reduced cytokine production, appearing to confirm that a sepsis-induced cytokine storm is necessary and sufficient for inflammation associated mortality. This pathway and consequent cytokine production have been targeted for pharmacological intervention, but drugs that inhibit cytokines have limited clinical value (9).
Production of inflammatory mediators, e.g. TNF-α, is amplified by release of “priming cytokines” chiefly IFN-β, a type I IFN (10). The significance of type I IFN is demonstrated by survival of animals deficient in components of the type I IFN signaling cascade (Tyk2 and Ifn-β) when exposed to lethal doses of LPS (11). Stat2 mediates canonical signaling through the type I IFN receptor. IFN-α/β binds its receptor (Ifnar), which activates the intracellular members of the Janus-activated kinase (JAK) family of tyrosine kinases. Jak1 and Tyk2 phosphorylate and activate Stat1 and Stat2, which form the heterotrimeric IFN-stimulated gene factor 3 (ISGF3) transcriptional complex with IRF9 (12). Other signaling complexes (phosphorylated Stat1 homodimers and Stat-independent pathways) have been identified (13). Stat2 is essential to nuclear-cytoplasmic shuttling of the ISGF3 complex and necessary for expression of type I ISGs (14). Stat2 is protective in viral infection (15–17), but its role in other human diseases has received little attention. However, reduced levels of Stat2 have been reported in alcoholic hepatitis (18) and inflammatory bowel disease (19).
The present study tests the hypothesis that loss of Stat2 protects against LPS-induced sepsis, as reported in mice deficient in other components of type I IFN signaling. Unexpectedly, we find that loss of Stat2 leads to accelerated LPS-induced mortality resulting from increased cellular extravasation and inflammation with enhanced peritonitis and hepatitis. Contrary to the established view, we find that accelerated death occurs without surges in proinflammatory cytokines. These data reveal a regulatory role for Stat2 in responses to TLR stimulation and introduce an alternative mechanism of LPS-induced death that occurs in the absence of excessive cytokine production.
Results
Effect of LPS on Survival in Mice Deficient in Stat2 and Other Components of Type I IFN Signaling Cascade.
To examine effects of Stat2 loss on the inflammatory response, mice were treated with a lethal dose of i.p. LPS (70 mg/kg). Stat2−/− mice showed increased mortality compared with WT (Fig. 1A). Conversely, mice deficient in other components of type I IFN signaling, Ifnar1 (n = 6; P = 0.0084) and Tyk2 (n = 5, P = 0.049), were protected against LPS-induced lethality. Stat1−/− mice also showed reduced mortality, but this did not reach statistical significance (n = 5; P = 0.07).
Fig. 1.
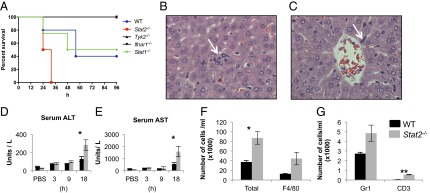
LPS-induced mortality in mice lacking IFN signaling mediators and sepsis-induced tissue inflammation in Stat2−/− mice. (A) Survival curves in WT, Tyk2−/−, Ifnar1−/−, Stat1−/−, and Stat2−/− mice following LPS challenge at a dose of 70 mg/kg. (B and C) Immune cells rapidly accumulate in the extravascular space of livers in Stat2−/− mice compared with WT following LPS challenge. H&E-stained sections of Stat2−/− mouse liver after 9 h LPS challenge (magnification of 600×) show signs of inflammation, microabscesses in liver parenchyma, and leukocytes within hepatic sinusoids (white arrows). (D and E) Serum concentrations (mean ± SEM) of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) from three to five animals following i.p. injection with PBS solution or LPS at time points indicated (*P < 0.05). (F and G) Number of cells in peritoneal lavage fluid of mice after 6 h of LPS (20 mg/kg). Total number (*P = 0.02), F4/80+, GR1+, and CD3+ (**P < 0.001).
Cardiovascular collapse is the hallmark of septic shock (20) with high mortality (21). No physiological changes that would account for the accelerated death in Stat2−/− mice were detected. No significant differences in the rate of myocardial depression (reduction in heart rate, stroke volume, peak velocity, aortic velocity time integral) or core temperature at baseline or after 18 h LPS stimulation were observed (Fig. S1).
We examined tissues from organs from Stat2−/− and WT animals at different times following LPS challenge. Loss of Stat2 led to increased neutrophilic hepatitis with microabscess formation at 9 h and 18 h compared with WT mice (Fig. 1 B and C). Serum alanine aminotransferase and aspartate aminotransferase (markers of liver injury) were significantly higher in Stat2−/− animals than WT at 18 h (P = 0.026; Fig. 1 D and E). We examined the possibility that loss of Stat2 may directly affect hepatocytes but did not find any increases in hepatocyte proliferation [minichromosome maintenance 4 (MCM4) positivity] or apoptosis (cleaved caspase 3 positivity), after LPS, suggesting that the intrahepatic effects of Stat2 loss are indirect (Fig. S2 A and B). Instead, we observed a reduction in hepatocyte apoptosis in Stat2−/− animals following LPS that probably reflects abrogated IFN signaling.
We found no other differences between WT and Stat2−/− mice to account for animal death in brain, heart, lung, thymus, kidney, adrenal gland, pancreas, or skin at any time (t = 3, 9, and 18 h) during LPS or PBS challenge (Fig. S2 C–J). Examination of the lungs revealed a similar increase in neutrophil infiltrates in WT and Stat2−/− mice following LPS (Fig. S2K), with no other signs of lung injury. At 18 h after LPS, more apoptosis was noted in the colonic epithelium of Stat2−/− mice, but this was not sufficient to account for death and culture of blood samples from mice showed no significant growth after aerobic and anaerobic culture (n = 5).
Increased peritonitis was detected in Stat2−/− mice after LPS administration. There were significantly greater numbers of leukocytes in peritoneal lavage fluid from Stat2−/− mice compared with WT (P = 0.02; 20 mg/kg; 6 h; Fig. 1F) that comprised mainly F4/80 macrophages (44,444 cells/mL vs. 12,963 cells/mL; Fig. 1F), Gr1+ granulocytes (5,016 cells/mL vs. 2,721 cells/mL; Fig. 1G), and CD3+ T cells (569 vs. 51 cells/mL; Fig. 1G), suggesting that Stat2 loss is associated with cellular egress in peritoneum and liver.
Loss of Stat2 Reduces LPS-Induced Production of Inflammatory Cytokines and Chemokines.
Current models of sepsis associate adverse outcomes with elevated production of inflammatory cytokines, so we assessed changes in the serum levels of proinflammatory cytokines. Contrary to expectation, reduced levels of TNF-α (2 h; P = 0.0286), IL-6 (4 h; P = 0.0286), IL-12p70 (2 h; P = 0.0286), MCP-1 (4 h and 7 h; P = 0.0286), and IL-10 (2 h; P = 0.0294) were detected in Stat2−/− mice compared with WT (Fig. 2 A–E). No differences in IFN-γ were found between the two strains (Fig. 2F). Increased mortality in Stat2−/− mice was not a result of susceptibility to TNF-α because WT and Stat2−/− mice were similarly resistant to i.p. administration of TNF-α (2 μg per mouse). Even when high-dose TNF-α (20 mg per mouse) was used, Stat2−/− mice showed no signs of sickness at any point. Conversely, WT mice showed piloerection and reduced movement at 10 h, indicating that Stat2−/− mice are resistant to TNF-α.
Fig. 2.

Effects of LPS-induced sepsis on cytokine production in Stat2−/− mice. Cytokine levels measured by ELISA in the serum of WT and Stat2−/− mice following i.p. LPS challenge (50 mg/kg).
To determine whether increased peritonitis in Stat2−/− animals following LPS challenge was associated with overexpression of chemokine(s), peritoneal lavage fluid of Stat2−/− and WT mice was evaluated for 32 chemokines and cytokines (Table S1). Of these, LPS stimulation led to an increase in the expression of 12 chemokines in WT mice (Fig. S3 A and B); nine of these were reduced in Stat2−/− mice. These results confirm that loss of Stat2 was associated with down-regulation of inflammatory cytokines and chemokines.
Loss of Stat2 in Endothelial/Stromal Compartment Modulates Cellular Migration.
Given that Stat2 deficiency reduced LPS-induced cytokine production and increased leukocyte egress, we hypothesized that Stat2 may be important in cell trafficking. Leukocyte–vessel wall interactions in Stat2−/− mice were investigated by intravital microscopy using the cremaster muscle model (22). Locally injected LPS into WT mice induced a significant increase in leukocyte adhesion (P < 0.05) and transmigration (P < 0.01) compared with saline solution-injected mice (Fig. 3A). Stat2−/− mice showed an adherence response comparable to WT mice, but had a significantly elevated transmigration response (P < 0.01; Fig. 3A).
Fig. 3.

LPS-induced death in Stat2−/− mice is mediated by cellular extravasation. (A) WT or Stat2−/− mice received intrascrotal injections of saline solution or LPS (300 ng). Leukocyte responses of adhesion (Left) and transmigration (Right) were quantified 4 h later in cremaster muscle by intravital microscopy (*P < 0.05, **P < 0.01, and ***P < 0.001, saline solution vs. LPS; ¶P < 0.001, WT vs. Stat2−/− mice). Tissue immunofluorescence microscopy for markers of blood vessels (α-smooth muscle actin; red) and neutrophils [myeloid related protein-14 (MRP-14); green]. A representative example of LPS-stimulated WT (Left) and Stat2−/− (Right) tissues. WT and Stat2−/− recipient animals received an adoptive transfer of singly fluorescently labeled primary WT and Stat2−/− bone marrow cells in equal amounts before i.p. administration of LPS (1 mg/kg). Peritoneal lavage was performed 18 h later, and the number and phenotype of WT and Stat2−/− immune cells within the lavage was quantified by FACS analysis. (B) Percentage of endogenous polymorphonuclear neutrophils (PMN). (C) Total number of WT or Stat2−/− fluorescent polymorphonuclear neutrophils retrieved from peritoneal cavities of WT or Stat2−/− mice. (D) WT and Stat2−/− mice received a single dose of ICAM-1 blocking antibody or isotype antibody control 18 h before i.p. LPS challenge (20 mg/kg). ICAM-1 inhibition reduced cellular egress into the peritoneum (*P = 0.026 and **P = 0.0001) and (E and F) survival of mice (*P = 0.02 and **P = 0.007). (G) WT and Stat2−/− mice (n = 6; n ∼ 6 vessels per animal) received LPS intrascrotally (300 ng) for 4 h and ICAM-1 expression was quantified (*P < 0.05). Examples of a maximum intensity projection depicting PECAM1 and ICAM-1 expression (and ICAM-1 isotype control) on WT and Stat2−/− venules are also shown. (Scale bar: 30 μm.)
To determine in which cellular compartment (leukocyte vs. stroma/endothelium) Stat2 affects leukocyte egress, equal number of bone marrow cells from WT and Stat2−/− mice were labeled with Calcein-AM (AM-acetoxymethylester) or PKH-26 (no differential effect of dye labeling was detected) and injected i.v. into WT or Stat2−/− animals before i.p. LPS injection. Peritoneal lavage fluid was collected 18 h later and analyzed by FACS to determine cell numbers, numbers of neutrophils, and number of labeled transferred cells. The main determinant of peritonitis was the genotype of the host animal, i.e., the stromal rather than the circulating leukocytes (Fig. 3 B and C). Analysis of inflamed tissue in the intravital microscopy model demonstrated significantly greater endothelial expression of ICAM-1 protein in Stat2−/− mice compared with WT following LPS stimulation (P < 0.05; Fig. 3G). There were no differences in platelet endothelial cell adhesion molecule-1 (PECAM-1) expression.
In vivo and in vitro migration assays showed equal responses of WT and Stat2−/− immortalized macrophages to a range of chemoattractants, further confirming that the migrating cell is unaffected by loss of Stat2 (Fig. S3 C and D).
Inhibition of Endothelial Cell Adhesion Reverses Inflammatory Extravasation and Prevents Mortality.
To test the hypothesis that increased cellular egress caused the death of LPS-treated Stat2−/− mice, animals received a single dose of anti–ICAM-1 blocking antibody [a global inhibitor of cellular egress (23)] 18 h before i.p. LPS injection. Blockade of cellular egress was confirmed by a reduction in total cell numbers in the peritoneal lavage (Fig. 3D). Pretreatment of mice with anti–ICAM-1 blocking antibody prolonged the survival of LPS-challenged mice (Fig. 3 E and F).
Stat2 Is Needed for TLR-Mediated Cytokine Production.
Paradoxically, our data describe reduced inflammatory cytokine production in a model of accelerated LPS-induced death. The response to LPS is mediated, at least in part, by an amplification loop involving IFN-β production (24). We excluded the possibility that the increase in mortality was attributable to enhanced type I IFN-induced responses as reported in macrophages from Stat2−/− mice (25). We did not observe mortality or signs of distress in WT and Stat2−/− mice injected with low- or high-dose IFN-β during 72 h. In addition, we confirmed that Stat2−/− mice did not respond to type I IFN. Hepatocyte expression of key IFN-stimulated genes (Ifit1, Ifit2, and Rsad2) was impaired in Stat2−/− mice injected with IFN-β (Fig. S4).
To confirm that Stat2 is required for inflammatory cytokine production, immortalized WT and Stat2−/− bone-marrow derived macrophage (BMDM) cell lines were established and tested for LPS responses. Expression of Stat2, TLR4, and macrophage markers (CD80 and F4/80) were confirmed (Fig. S5 A and B), and we found no differences in TLR4 expression between WT and Stat2−/− splenocytes (Fig. S5C).
Immortalized WT BMDMs produced large amounts of TNF-α and RANTES after LPS stimulation, whereas immortalized Stat2−/− BMDM produced significantly less over 12 h (P < 0.0001; Fig. 4 A and B). LPS stimulation for shorter periods did not reveal any early peaks in production of nitrite or TNF-α (Fig. S6 A–C).
Fig. 4.

TLR triggered cytokine production is diminished in the absence of Stat2. WT and Stat2−/− immortalized BMDMs were stimulated with (A and B) LPS (1 μg/mL) or (C and D) Poly I:C (25 μg/mL). Secretion of TNF-α and RANTES was measured by ELISA [mean (±SEM) of three replicates; representative of at least two experiments]. (E–H) Quantitative PCR analysis of LPS-responsive genes is shown as mean fold change relative to unstimulated cells following normalization to β-actin (mean ± SEM of two to four experiments). Quantitative PCR analysis (I) of Stat2, (J) TNF-α, and (K) RANTES in Stat2−/− and Stat2−/− reconstituted with Stat2 immortalized BMDMs.
We next examined the effects of IL-1 and other TLR agonists: poly I:C, flagellin, and imiquimod. IL-1 induced TNF-α production in immortalized WT BMDMs, but not in Stat2−/− BMDMs (P = 0.0046; Fig. S6D). Treatment with poly I:C led to the production of TNF-α and RANTES in immortalized WT BMDMs, with only minimal production in Stat2−/− cells (P < 0.0001 and P = 0.0046, respectively; Fig. 4 C and D). Neither TNF-α nor RANTES was produced by either cell type in response to flagellin or imiquimod.
The transcription of a set of genes triggered by TLR stimulation was analyzed by quantitative RT-PCR. In response to LPS treatment, a reduction in mRNA expression of TNF-α, cyclooxygenase2 (COX2), LPS-induced TNF-α factor (LITAF), and early growth response 1 (EGR1) was detected in immortalized Stat2−/− BMDM compared with WT BMDMs (Fig. 4 E–H). MxA (an IFN-induced gene) was included as a positive control of IFN response (Fig. S6E). In addition, TNF-related apoptosis-inducing ligand (TRAIL) mRNA levels were measured following stimulation with poly I:C and LPS. Regardless of the stimulus used, normal gene induction in immortalized WT cells was impaired in Stat2−/− cells (P = 0.0021 and P = 0.03; Fig. S6 F and G).
To confirm that this phenotype was a result of deficiency in Stat2, we transiently restored Stat2 expression in immortalized Stat2−/− BMDMs (Fig. 4I). Rescue of Stat2 was effectively demonstrated by MxA mRNA expression in response to IFN-α stimulation (Fig. S6H). Stat2 reconstituted immortalized BMDM showed an increase in transcription of TNF-α and RANTES mRNAs following 6 h LPS stimulation (Fig. 4 J and K). These results demonstrate that loss of Stat2 impairs cytokine production at the transcriptional level.
Taken together, these data show that, although Stat2−/− animals have increased mortality following LPS injection, TLR-mediated cytokine production in the absence of Stat2 is significantly impaired, and this is a global defect seen with several TLR agonists.
Effects of Stat2 Loss on Cytokine Production Are Independent of Type I IFN Signaling.
To date, the only known role for Stat2 is as a transducer of IFN signaling. It is possible that the reduction in cytokine production in Stat2−/− cells may have been a result of loss of a type I IFN-mediated amplification loop. However, this seems unlikely given the distinction between in vivo response to LPS seen in Stat2−/− vs. Stat1−/− and Ifnar1−/− mice. To confirm that the phenotypes seen here were independent of type I IFN, primary BMDMs from Stat1−/− and Ifnar1−/− mice were stimulated with LPS in vitro. No reduction in LPS-induced cytokine production was observed (Fig. S7 A and B).
To evaluate a role for type I IFNs in the Stat2-mediated LPS response, we blocked type I IFN signaling using a neutralizing antibody against the Ifnar1 component of the type I IFN receptor (26). This led to impaired expression of the IFN target gene MxA (Fig. S8A). However, LPS induced TNF-α and RANTES production remained unaffected even when IFN signaling was blocked (Fig. S8 B and C).
Stat2 Is Needed for Timely Translocation of NF-κBp65 into the Nucleus.
The widespread reduction in TLR response in the absence of Stat2 suggests that MyD88-dependent and MyD88-independent signaling pathways are affected. The NF-κB family of transcription factors represent a convergence point in all the signaling pathways noted to be defective in Stat2−/− cells (27).
To determine the effect of Stat2 deletion on NF-κB–driven responses, we used an NF-κB luciferase reporter system. Following 6 h stimulation with TNF-α, immortalized Stat2−/− BMDM showed a marked reduction in TNF-α–driven luciferase activity. This activity was recovered when Stat2 expression was restored (Fig. 5A). Similarly, immortalized Stat2−/− BMDM reconstituted with a mutated Stat2 that lacked the classical Tyr-689 phosphorylation site (Y689F) showed restored TNF-α response. LPS-stimulation of WT cells in the presence of the anti-Ifnar1 antibody and of Ifnar1−/− and Tlr4−/− splenocytes did not lead to phosphorylation of Stat2 Y689 (Fig. S8 D–F). These results indicate that defective cytokine production in the absence of Stat2 is caused, at least in part, by impaired NF-κB–mediated transcription, and Y689 phosphorylation of Stat2 is not involved.
Fig. 5.

Stat2 deficiency impairs nuclear localization of NF-κB p65. (A) Measurement of NF-κB luciferase reporter activity in WT and Stat2−/− immortalized BMDMs reconstituted with Stat2 or mutant Stat2 (Y689F) and stimulated with TNF-α (10 ng/mL). Data are shown as mean luminescence units (magnification of 1,000×) from two experiments. (B) Immunoblot analysis of NF-κB p65 using nuclear and cytoplasmic extracts from immortalized BMDMs stimulated with LPS (1 μg/mL). (C) LPS induced nuclear translocation of NF-κB p65 (red) assessed by immunofluorescence microscopy. Nucleus is shown in blue. Images are representative of three independent experiments. (D) EMSA of nuclear extracts prepared from LPS-stimulated immortalized BMDMs. Lane 1 is the free probe, lane 2 the labeled probe, and lane 3 the labeled probe competed with 200× molar excess of unlabeled probe. Images shown are representative experiments using at least three independent extracts. (E) Quantitative ChIP assay performed at the indicated time points with antibody against p65 or irrelevant IgG as indicated. Specific quantitative PCR primers were used to amplify the proximal TNF-α promoter region.
Given the defective NF-κB transcriptional activity in the absence of Stat2, mediators of LPS signaling upstream of the NF-κB activation complex were evaluated. The protein level of MyD88 was comparable between immortalized WT and Stat2−/− BMDMs following stimulation with LPS, and TNF receptor-associated factor 6 (TRAF6)-dependent reduction in IRAK1 did not require Stat2 (Fig. S9A). A key step in NF-κB activation is its translocation to the nucleus, dependent on the degradation of IκBα in the cytoplasm. Cytoplasmic levels of phosphorylated IκBα increased in WT, but not in Stat2−/− cells (Fig. S9B) following LPS stimulation; however, there was no difference in the abundance of total IκBα protein or IκB kinase B (IKKB) that is largely responsible for IκBα phosphorylation (28). The basal increased expression of the IκB kinase A (IKKA) in immortalized Stat2−/− BMDMs was higher compared with immortalized WT BMDMs. These data suggest that loss of Stat2 is associated with aberrations in inducing or maintaining phosphorylated IκBα.
To determine the effect of this defect on NF-κB function, we assessed its localization to the nucleus and its binding to chromatin. A reduction in the amount of LPS-induced NF-κBp65 in the nucleus was confirmed by immunoblotting of nuclear and cytoplasmic extracts from immortalized BMDMs (Fig. 5B). Before stimulation, p65 was found predominantly in the cytoplasm; however, after 15 min LPS exposure, much less p65 was detected in the nuclei of Stat2−/− than in WT BMDMs. Nuclear accumulation of p65 in immortalized Stat2−/− BMDM was not increased later. Immunofluorescence microscopy confirmed that, in the absence of Stat2, p65 is not maintained in the nucleus (Fig. 5C). In immortalized WT BMDM, p65 persisted in the nucleus at 45 min and 60 min, but this was not the case in Stat2−/− BMDMs. EMSA and ChIP analyses confirmed these results, as they also showed a reduction in protein complexes binding to an NF-κB target sequence (Fig. 5 D and E) in immortalized Stat2−/− BMDM. These data indicate that Stat2 is needed for the timely nuclear accumulation of NF-κBp65.
Discussion
Here we show that Stat2 is a key regulator of LPS-induced sepsis, as loss of Stat2 leads to increased mortality caused by cellular extravasation, but paradoxically, without the expected surge in inflammatory cytokines classically associated with septic death. Our study reports the existence of an alternative mechanism of LPS-induced sepsis independent of an excessive inflammatory response that is limited by Stat2.
The pathophysiology of sepsis is poorly understood. Although cytokine surges accompany the septic phenotype, there is poor correlation between cytokine levels and clinical outcome. The Stat2−/− mouse model shows accelerated mortality and a reduction in cytokine production following a single dose of LPS. A similar response was seen in early studies of LPS tolerance; monocyte hyporesponsiveness is accompanied by increased lethality in mice pretreated and rechallenged with sublethal doses of Salmonella abortus-equi LPS (29).
In this i.p. toxin model (30, 31), death in Stat2−/− mice is accompanied by increased leukocyte migration; the most prominent pathological findings are hepatitis (histological and biochemical) and peritonitis. Significant bacteremia following LPS injection in Stat2−/− mice was not detected; however, we have not excluded the possibility that bacterial fragments (including endotoxin) might be present in the blood to promote an immune response. Reduced levels of Stat2 have been reported in inflammatory liver (18) and bowel (19) diseases, indicating that the inverse association of Stat2 and cellular migration is likely to be a global effect. We show that transmigration is increased in Stat2−/− mice and this is caused by a stromal defect rather than a leukocyte deficiency. It will be important to identify the role for Stat2 in the numerous cell types that constitute this microenvironment. Use of the anti–ICAM-1 blocking antibody as a global inhibitor of cellular egress (23) that reversed the mortality seen in Stat2−/− mice confirms our observation that, in the absence of a cytokine storm, cellular egress mediates death in this model of sepsis.
Decreased cytokine production in Stat2−/− cells correlates with reduced NF-κB promoter activity, and our results show that Stat2 regulates NF-κB signaling. Consistent differences between WT and Stat2−/− BMDMs lead us to conclude that this is an important functional difference. Stat2 was found to mediate release of p65 from IκB inhibitors and promote nuclear translocation of activated p65, subsequent retention of the activated complex in the nucleus, and binding to chromatin. Some of these traits have been reported to occur in virus infection. Measles virus V protein prevents NF-κB gene expression by retaining NF-κB in the cytoplasm (32), and, because V protein has been shown to bind Stat2 and inhibit IFN signaling (17), we speculate that inhibition of Stat2 may explain the effect on NF-κB–mediated gene expression. The precise nature of the role of Stat2 in NF-κB activation remains to be determined. Phosphorylation of IκBα is impaired in immortalized Stat2−/− BMDMs, suggesting that this may be the site of action of Stat2.
Loss of Stat2 did not completely abrogate cytokine production. We observe that, in the absence of Stat2, p65 nuclear entry is only reduced, not inhibited. Furthermore, in the absence of Stat2, p65 is not maintained in the nucleus. Timely nuclear entry of p65 is critical to normal function of NF-κB (33–35), however, release of inhibition from IκBα and nuclear translocation is not sufficient for p65 target gene expression (36). Active retention of p65 in the nucleus and the availability of specific associated proteins are necessary for transcriptional activity (37), and it is plausible that Stat2 [known to interact with p300/CBP (38)] may interact with p65 to maintain it in the nucleus.
This study shows that the effects of Stat2 on NF-κB–mediated cytokine production are independent of type I IFN signaling. The increase in TNF-α observed in Ifnar1−/− BMDMs is likely a result of loss of suppressor of cytokine signaling-1 (SOCS1)-mediated inhibition of TLR signaling pathways (39). Similarly, Tyk2−/− mice have defective type I IFN signaling and yet LPS-induced production of cytokines (including TNF-α) is also unaffected (11). We also show that Stat2 does not appear to require Tyr689 phosphorylation as the Y689F mutation restored NF-κB activity. Interest in noncanonical Stat signaling has increased; an ISGF3 complex containing nonphosphorylated Stat2 was identified as a significant mediator of IFN-γ antiviral effects (40) and alternative Stat2 posttranslational modifications may be required for normal NF-κB function.
In summary, our results show a role for Stat2 in determining outcome from LPS sepsis independent of excessive immune inflammatory response. Loss of Stat2 is associated with accelerated death and marked hepatitis and peritonitis. Increased inflammatory cell migration is a property of increased permissiveness to extravasation in the endothelial/stromal compartment. Paradoxically, a surge in inflammatory cytokines is not seen as a result of a disruption in the nuclear localization of NF-κB p65 and activity at its target promoter. These unexpected findings identify a role for Stat2 as a key mediator in the acute inflammatory response that is independent of its known functions as a transducer of IFN signaling.
Methods
Mice.
Stat2−/− mice in 129SvJ background were a gift from Christian Schindler (Columbia University, New York, NY). 129SvJ mice were purchased from National Cancer Institute (Frederick, MD) and used as WT controls. Stat1−/− mice were purchased from Taconic Farms. Tyk2−/− mice were a gift of Kazuya Shimoda (Miyasaki University, Miyasaki, Japan). These mice were then backcrossed seven times to 129SvE. Ifnar1−/− mice (41) were obtained from Brian Kelsall (National Institute of Allergy and Infectious Diseases, Bethesda, MD). Mice were housed in a conventional facility in individually ventilated cages and used at 6 to 10 wk of age. All animal studies were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee at Temple University. The UK Home Office and Local Ethics Committee approved the work done at Queen Mary, University of London.
Intravital microscopy was used to directly monitor leukocyte responses within mouse cremasteric venules as previously detailed (26). To investigate the contribution of leukocyte and endothelial Stat2 in leukocyte transmigration, a cell-transfer technique-modified version of bone marrow transfer from published methods was used (42).
Age- and sex-matched mice were injected i.p. with 200 μL of the indicated doses of toxins or agonists. Mice were monitored for survival for as long as 7 d. In some experiments, antibodies were injected i.v. 18 h before LPS challenge. In other experiments, mouse organs were harvested at different time points and formalin-fixed before paraffin embedding for histological assessment.
Mice were bled by cardiac puncture at the indicated times after i.p. LPS injection, and serum samples were kept at −80 °C. Sera were analyzed for cytokine levels and nitrite production.
In other experiments, mice were euthanized 6 h following i.p. injection with LPS or PBS solution. Five milliliters of cold PBS solution was injected into the peritoneal cavity. Peritoneal lavage fluid was collected for complete analysis of chemokines/cytokines by Milliplex magnetic bead assay according to the manufacturer’s instructions (Millipore).
Supplementary Material
Acknowledgments
We thank Dr. Christian Schindler for sharing Stat2−/− mice, Ms. Della Reynolds for assistance in establishing immortalized bone marrow-derived macrophage cell lines, Dr. Darren Baker (Biogen Idec, Cambridge, MA) for assistance and reagents, Mr. Tony Price for assistance with mouse experiments, and Dr. Richard Waite for assistance with bacterial cultures. This work was supported by a Starter Grant for Clinical Lecturers from the Academy of Medical Sciences (to W.A.) and a grant from the Pennsylvania Department of Health (to A.M.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221652110/-/DCSupplemental.
References
- 1.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 2.Namas R, et al. Sepsis: Something old, something new, and a systems view. J Crit Care. 2012;27(3):314.e1–314.e11. doi: 10.1016/j.jcrc.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salomao R, et al. Bacterial sensing, cell signaling, and modulation of the immune response during sepsis. Shock. 2012;38(3):227–242. doi: 10.1097/SHK.0b013e318262c4b0. [DOI] [PubMed] [Google Scholar]
- 4.Remick DG, et al. Role of tumor necrosis factor-alpha in lipopolysaccharide-induced pathologic alterations. Am J Pathol. 1990;136(1):49–60. [PMC free article] [PubMed] [Google Scholar]
- 5.Thiemermann C, Vane J. Inhibition of nitric oxide synthesis reduces the hypotension induced by bacterial lipopolysaccharides in the rat in vivo. Eur J Pharmacol. 1990;182(3):591–595. doi: 10.1016/0014-2999(90)90062-b. [DOI] [PubMed] [Google Scholar]
- 6.Hoshino K, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162(7):3749–3752. [PubMed] [Google Scholar]
- 7.Nagai Y, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3(7):667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11(1):115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 9.Wiersinga WJ. Current insights in sepsis: From pathogenesis to new treatment targets. Curr Opin Crit Care. 2011;17(5):480–486. doi: 10.1097/MCC.0b013e32834a4aeb. [DOI] [PubMed] [Google Scholar]
- 10.Zhang S-Y, et al. Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev. 2007;220(1):225–236. doi: 10.1111/j.1600-065X.2007.00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karaghiosoff M, et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol. 2003;4(5):471–477. doi: 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- 12.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 13.Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM. Interferon: Cellular executioner or white knight? Curr Med Chem. 2007;14(12):1279–1289. doi: 10.2174/092986707780597907. [DOI] [PubMed] [Google Scholar]
- 14.Leung S, Qureshi SA, Kerr IM, Darnell JE, Jr, Stark GR. Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol. 1995;15(3):1312–1317. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hahm B, Trifilo MJ, Zuniga EI, Oldstone MBA. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22(2):247–257. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Jones M, et al. Dengue virus inhibits α interferon signaling by reducing STAT2 expression. J Virol. 2005;79(9):5414–5420. doi: 10.1128/JVI.79.9.5414-5420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramachandran A, Parisien J-P, Horvath CM. STAT2 is a primary target for measles virus V protein-mediated α/β interferon signaling inhibition. J Virol. 2008;82(17):8330–8338. doi: 10.1128/JVI.00831-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen VA, Gao B. Expression of interferon alfa signaling components in human alcoholic liver disease. Hepatology. 2002;35(2):425–432. doi: 10.1053/jhep.2002.31169. [DOI] [PubMed] [Google Scholar]
- 19.Mudter J, et al. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am J Gastroenterol. 2005;100(1):64–72. doi: 10.1111/j.1572-0241.2005.40615.x. [DOI] [PubMed] [Google Scholar]
- 20.Dellinger RP. Cardiovascular management of septic shock. Crit Care Med. 2003;31(3):946–955. doi: 10.1097/01.CCM.0000057403.73299.A6. [DOI] [PubMed] [Google Scholar]
- 21.Angus DC, et al. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Thompson RD, et al. Platelet-endothelial cell adhesion molecule-1 (PECAM-1)-deficient mice demonstrate a transient and cytokine-specific role for PECAM-1 in leukocyte migration through the perivascular basement membrane. Blood. 2001;97(6):1854–1860. doi: 10.1182/blood.v97.6.1854. [DOI] [PubMed] [Google Scholar]
- 23.Kumasaka T, et al. Role of the intercellular adhesion molecule-1(ICAM-1) in endotoxin-induced pneumonia evaluated using ICAM-1 antisense oligonucleotides, anti-ICAM-1 monoclonal antibodies, and ICAM-1 mutant mice. J Clin Invest. 1996;97(10):2362–2369. doi: 10.1172/JCI118679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahieu T, Libert C. Should we inhibit type I interferons in sepsis? Infect Immun. 2007;75(1):22–29. doi: 10.1128/IAI.00829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao W, Cha EN, Lee C, Park CY, Schindler C. Stat2-dependent regulation of MHC class II expression. J Immunol. 2007;179(1):463–471. doi: 10.4049/jimmunol.179.1.463. [DOI] [PubMed] [Google Scholar]
- 26.Sheehan KC, et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res. 2006;26(11):804–819. doi: 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- 27.Génin P, Algarté M, Roof P, Lin R, Hiscott J. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J Immunol. 2000;164(10):5352–5361. doi: 10.4049/jimmunol.164.10.5352. [DOI] [PubMed] [Google Scholar]
- 28.Solt LA, May MJ. The IkappaB kinase complex: Master regulator of NF-kappaB signaling. Immunol Res. 2008;42(1-3):3–18. doi: 10.1007/s12026-008-8025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freudenberg MA, Salômao R, Sing A, Mitov I, Galanos C. Reconciling the concepts of endotoxin sensitization and tolerance. Prog Clin Biol Res. 1998;397:261–268. [PubMed] [Google Scholar]
- 30.Ghosn EEB, et al. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci USA. 2010;107(6):2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henderson RB, Hobbs JAR, Mathies M, Hogg N. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. 2003;102(1):328–335. doi: 10.1182/blood-2002-10-3228. [DOI] [PubMed] [Google Scholar]
- 32.Schuhmann KM, Pfaller CK, Conzelmann KK. The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J Virol. 2011;85(7):3162–3171. doi: 10.1128/JVI.02342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baltimore D. NF-κB is 25. Nat Immunol. 2011;12(8):683–685. doi: 10.1038/ni.2072. [DOI] [PubMed] [Google Scholar]
- 34.Ashall L, et al. Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science. 2009;324(5924):242–246. doi: 10.1126/science.1164860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309(5742):1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 36.Bergmann M, Hart L, Lindsay M, Barnes PJ, Newton R. IkappaBalpha degradation and nuclear factor-kappaB DNA binding are insufficient for interleukin-1beta and tumor necrosis factor-alpha-induced kappaB-dependent transcription. Requirement for an additional activation pathway. J Biol Chem. 1998;273(12):6607–6610. doi: 10.1074/jbc.273.12.6607. [DOI] [PubMed] [Google Scholar]
- 37.Zerfaoui M, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J Immunol. 2010;185(3):1894–1902. doi: 10.4049/jimmunol.1000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhattacharya S, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature. 1996;383(6598):344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- 39.Mansell A, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7(2):148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 40.Morrow AN, Schmeisser H, Tsuno T, Zoon KC. A novel role for IFN-stimulated gene factor 3II in IFN-γ signaling and induction of antiviral activity in human cells. J Immunol. 2011;186(3):1685–1693. doi: 10.4049/jimmunol.1001359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Müller U, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 42.Woodfin A, Voisin MB, Nourshargh S. PECAM-1: A multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27(12):2514–2523. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.