

Abstract
When cancer metastasizes to bone, considerable pain and deregulated bone remodelling occurs, greatly diminishing the possibility of cure. Metastasizing tumour cells mobilize and sculpt the bone microenvironment to enhance tumour growth and to promote bone invasion. Understanding the crucial components of the bone microenvironment that influence tumour localization, along with the tumour-derived factors that modulate cellular and protein matrix components of bone to favour tumour expansion and invasion, is central to the pathophysiology of bone metastases. Basic findings of tumour–bone interactions have uncovered numerous therapeutic opportunities that focus on the bone microenvironment to prevent and treat bone metastases.
Tumours are generally incurable once they have metastasized to bone. Devastating consequences of bone metastases include pathological bone fractures, pain, hypercalcaemia, and spinal cord and nerve-compression syndromes1. Bone metastases are a common complication of cancer and occur in 65–80% of patients with metastatic breast and prostate cancers2,3. The incidence of bone metastases is also increasing in other cancers, probably owing to improved tumour control at other disease sites4. Tumour invasion into bone is associated with osteoclast and osteoblast recruitment, resulting in the liberation of growth factors from the bone matrix, which can feed back to enhance tumour growth resulting in the ‘vicious cycle’ of bone metastases1,3,5,6. Indeed, the successful suppression of bone turnover with bisphosphonates in patients who had bone metastases that resulted in high levels of bone resorption markers was associated with improved survival7. Beyond the effects on osteoclasts and osteoblasts, tumours in the bone microenvironment recruit and modulate the function of platelets, myeloid cells, immune cells and nerve cells, and induce the formation of new blood vessels. The bone marrow also serves as a reservoir for dormant tumour cells that can resist chemotherapeutic attack, and these tumour cells can emerge later as full-blown metastases in bone or other organs8–10.
Drugs, such as bisphosphonates or receptor activator of NF-κB ligand (RANKL; also known as TNFSF11) antibodies, that target osteoclastogenesis significantly decrease the incidence of skeletal complications and are the current standard of care for patients with bone metastases1,11–13. There are emerging data that these anti-resorptive agents can also have direct antitumour effects. However, 30–50% of patients on such therapies still develop new bone metastases, skeletal complications and disease progression1, emphasizing the need for new therapies. Important advances in understanding the basic biology of bone remodelling, haematopoiesis, haematopoietic cell egress and homing to bone marrow have uncovered new therapeutic targets for the prevention and treatment of bone metastasis.
Bone resorption and formation
The bone microenvironment is comprised of a mineralized extracellular matrix and specific cell types that are under the control of local and systemic factors. This special milieu provides a fertile soil for many cancers to thrive (FIG. 1). Certain types of solid tumours metastasize to bone and induce destructive osteolytic and/or bone-forming osteoblastic lesions, with most solid tumours commonly producing both. Tumour cells secrete a vast array of proteins, many of which interact with resident cells in the bone marrow to induce the differentiation, recruitment and activation of osteoclasts and osteoblasts. During the process of bone resorption, stored growth factors and ionized calcium are released from the mineralized bone matrix, and these factors feed back to promote tumour cell growth and further production of osteolytic and osteoblastic factors. This vicious cycle can support tumour growth in bone3,14 (FIG. 2).
Figure 1. Bone remodelling.

The bone is a dynamic hard tissue that undergoes a continuous remodelling process to maintain skeletal strength and integrity, with 10% of the skeleton being replaced annually. In a finely balanced, coupled and sequential process (indicated by the dashed arrows), haematopoietic stem cell (HSC)-derived osteoclasts resorb bone (releasing growth factors and calcium) and mesenchymal stem cell (MSC)-derived osteoblasts replace the voids with new bone, a process that is dependent on osteoblast commitment, proliferation and differentiation coupled with osteoblast production of type I collagen and its subsequent mineralization to form the calcified matrix of bone. Osteocytes, which are terminally differentiated osteoblasts that are embedded in bone, sense mechanical strain, signal to osteoclasts and osteoblasts, and participate in the remodelling process206. Bone lining cells are osteoblastic in origin and have been proposed to form both a canopy over remodelling sites and a layer over bone surfaces, as well as a conduit to communicate with osteocytes207. The endosteum and periosteum (the lining on the inner and outer bone surfaces) contain a population of tissue macrophages, termed osteomacs, which are likely to have important roles in bone remodelling208. M-CSF, macrophage colony stimulating factor; RANK, receptor activator of NF-κB; RANKL, RANK ligand.
Figure 2. Cross-section of bone depicting stages of bone metastases.

Schematic representation of tumour cell interactions within the bone microenvironment during stages of tumour metastasis to bone — tumour cell homing, dormancy, colonization and expansion. Tumour cells home to and enter the bone marrow cavity and either remain quiescent or dormant or begin growth and colonization. Tumour-mediated recruitment and modulation of bone-residing cells (osteoclasts, osteoblasts, fibroblasts, blood vessels, mesenchymal stem cells, haematopoetic stem cells (HSCs), lymphocytes, macrophages, platelets, neurons and osteocytes) and bone matrix modifications alter the bone environment thus favouring tumour growth and invasion and resulting in pain, fracture and further tumour dissemination.
Osteoclasts are polarized, multinucleated myeloid lineage cells that adhere to the bone surface through αvβ3 integrin, form an actin ring, and secrete acid, collagenases and proteases that demineralize the bone matrix and degrade matricellular proteins such as type I collagen. Macrophage colony stimulating factor (M-CSF) and RANKL are important growth factors that support osteoclastogenesis, and they are primarily produced by osteoblasts. M-CSF and interleukin-34 (IL-34) both bind to the FMS receptor (also known as CSF1R) on myeloid cells and promote osteoclastogenesis209. RANKL binds to its cognate receptor, RANK, on osteoclast precursors, to induce osteoclastogenesis through the nuclear factor-κB (NF-κB), NFATc1 and JUN N-terminal kinase signalling pathways15. Osteoprotegerin (OPG; also known as TNFRSF11B) is an endogenous decoy receptor of RANKL that inhibits osteoclastogenesis. Deletion of RANK or RANKL, or overexpression of OPG, causes severe osteopetrosis, which is consistent with the central role of this pathway in osteoclastogenesis16,17.
Mesenchymal stem cells (MSCs) in the bone marrow are directed along the osteoblast lineage through local factors, such as transforming growth factor-β (TGFβ)18, bone morphogenetic proteins (BMPs) and WNT proteins (BOX 1). These pathways lead to the expression of three key transcriptional regulators of osteoblast function: RUNX2 (REF. 19), osterix20 and activating transcription factors (ATFs)21,22. The osteoblast-stimulating activity of metastatic tumour cells is thought to be due to the ability of these cells to express many of the factors listed above that can drive osteoblast formation and activation (FIGS 1, 3).
Box 1. WNT signalling in bone metastasis.
WNT proteins are secreted, highly post-translationally modified proteins that have a key role in osteogenesis199. The WNT–β-catenin signalling pathway is controlled by two cell surface receptors, Frizzled and the LRP5 and LRP6 co-receptors. Canonical WNT signalling activates bone formation, and among the many proteins downstream of WNT is RUNX2, which is required for osteoblast formation. The WNT signalling inhibitor, dikkopf 1 (DKK1) has a crucial role in osteolytic skeletal metastasis in which osteoblast formation is suppressed and osteoclast activity is enhanced60,200. DKK1 has been attributed to the support of osteolytic breast cancer metastasis, and serum DKK1 levels are increased in patients with multiple myeloma and in women with breast cancer skeletal metastasis200,201. In experimental models of prostate cancer, DKK1 expression is high during early carcinogenesis and decreases with skeletal progression, suggesting a molecular switch between osteolytic and osteoblastic disease60. Tumour-derived parathyroid hormone-related protein decreases DKK1 expression and maybe responsible for such a switch202. Endothelin 1, a tumour-derived factor, decreases osteoblast DKK1 expression to cause the deregulated formation of new bone, as observed in prostate cancer bone metastases181,183. It is likely that WNT signalling is central to the osteoblast-stimulating activity of metastatic tumours. In contrast to solid tumour metastases in bone, myeloma is largely associated with an inhibition of osteoblastic activity. Inhibitory factors secreted by myeloma cells include DKK1, secreted Frizzled-related protein 2, interleukin-7 and hepatocyte growth factor203–205.
Figure 3. Tumour–osteoblast interactions.

Tumours produce various factors that regulate bone formation at different levels of osteoblast development. Bone morphogenetic proteins (BMPs), WNTs and transforming growth factor-β (TGFβ) provide signals to mesenchymal stem cells (MSCs) to move to areas of bone formation and to differentiate to the osteoblast lineage. Osteoblast progenitors and pre-osteoblastic cells respond to positive osteoblastic factors that are produced by tumour cells, such as BMPs, endothelin 1 (ET1), insulin-like growth factors (IGFs), platelet-derived growth factor (PDGF), urinary plasminogen activator (uPA) and fibroblast growth factors (FGFs), as well as the negative regulator dickkopf 1 (DKK1). Osteoblast-associated transcription factors include RUNX2, osterix (OSX) and activating transcription factor 4 (ATF4). Once osteoblasts produce and mineralize a collagen matrix (shown in blue) they may undergo apoptosis, become lining cells or be sequestered in the bone matrix as terminally differentiated osteocytes. TGFβ can function at multiple stages that include recruiting stem cells and promoting stem cell renewal, coupling osteoclastic bone resorption to bone formation and inhibiting osteoblast differentiation. The BMP inhibitor, noggin, as well as endothelin A receptor antagonists, can block osteoblastic metastases. Little is known of the potential interactions between tumours and osteocytes.
The process of bone colonization
The pre-metastatic niche
The concept of a pre-metastatic niche has emerged as a means through which a primary tumour is able to prepare sites of metastasis. In preclinical melanoma and lung cancer models, for example, vascular endothelial growth factor receptor 1 (VEGFR1)-positive bone marrow-derived haematopoietic cells home to the sites of future metastasis and form cellular clusters that precede tumour cell arrival and increase fibronectin production in tumour target sites23. Inflammatory chemoattractants that are produced in the lung in a pulmonary metastasis model further support the concept of the pre-metastatic niche24. However, others have described a lack of effect of bone marrow-derived endothelial cell precursors on tumour growth25.
In the bone microenvironment, most evidence in support of a pre-metastatic niche is in the context of endocrine-like actions. Primary tumours may condition the bone marrow through the production of circulating factors that target cells in the bone microenvironment and thus render it conducive to tumour localization and colonization. Examples include heparanase that is produced by breast cancer cells increasing bone resorption26; osteopontin (OPN) that is secreted by tumour cells and/or senescent fibroblasts promoting bone marrow cell recruitment or tumour formation27–29; and matrix metalloproteinase (MMP) production from osteoclasts supporting prostate cancer skeletal metastasis30. Parathyroid hormone-related protein (PTHRP; also known as PTHLH) is produced by various tumours and can promote bone resorption5,14 and can enhance the production of local factors in the bone marrow, such as the chemokine CCL2 (REF. 31).
Homing to bone
Tumour cells preferentially adhere to the bone marrow endothelium, a potentially initial and key event in their introduction to the bone microenvironment32. Tumour cells that metastasize to bone can use the same physiological mechanisms as those used by haematopoietic stem cells (HSCs) homing to bone33–36 (FIG. 2). Osteoblasts and bone marrow stromal cells attract and regulate HSCs, and provide a niche through protein interactions that include integrins, such as α4β1–vascular cell adhesion molecule 1 (VCAM1); chemokines, such as CXCL12 (also known as SDF1)–CXCR4; BMPs, Notch, nestin and OPN33,34,37–43. Preclinical studies demonstrate that bone resorption can also regulate HSC mobilization and homing44,45. Metastatic prostate cancer and probably other cancers directly compete for the occupancy of the HSC niche during localization to the marrow. Once in the niche, they begin a process of either evicting HSCs into the peripheral blood or driving them into progenitor pools46.
CXCL12 is expressed at high levels by osteoblasts and bone marrow stromal cells, and expression of its receptor, CXCR4, on cancer cells has an important role in tumour cell homing to bone35,47–50. CXCL12 expression from bone marrow endothelial monolayers has been demonstrated to promote prostate cancer cell migration and upregulation of both MMP9 and αvβ3 on prostate cancer cells48,51. Kang and colleagues35 found that CXCR4 was highly overexpressed in subpopulations of a serially selected and highly metastatic human breast cancer cell line. The overexpression of CXCR4 along with other bone metastasis signature genes, such as IL11, connective tissue growth factor (CTGF) and MMP1, in the parental breast cancer cell lines increased their capacity to metastasize to bone35. IL-11 and MMP1 stimulate bone resorption by increasing osteoblast production of RANKL, and CTGF can stimulate osteoblast proliferation, as well as neoangiogenesis. When expressed together, these proteins can act cooperatively to cause osteolytic metastasis, but overexpression of individual proteins was insufficient to accelerate bone metastases35. Several groups have also demonstrated a direct role for CXCR4–CXCL12 in breast and prostate cancer cell proliferation50,52–54, suggesting that this pathway might also be required for tumour colonization in bone.
Tumour cell surface integrins interact with extracellular matrix (ECM) proteins that are expressed in the bone microenvironment. The αvβ3 integrin interacts with bone-derived OPN, fibronectin and vitronectin, and expression of αvβ3 by breast and prostate cancer cells is associated with higher rates of bone metastasis, tumour-associated osteolysis and colonization in bone55,56. Tumour cell expression of the β1 integrin family members α5β1, α2β1 and α4β1 — which are receptors for fibronectin, collagen I and VCAM1, respectively — has been implicated in the interactions of leukaemia, myeloma, prostate and breast cancer cells with bone marrow stroma, and can result in enhanced colonization and survival in bone57–64.
CXCR4 ligation increases αvβ3 expression on prostate cancer cells and α4β1 on myeloma cells, suggesting a crosstalk between CXCR4 and integrin expression that could both promote tumour cell recruitment to bone and colonization48,49,65. Definition of the temporal, anatomical and spatial pathways for tumour cell homing to and colonization in bone will be necessary to develop anti-homing and anti-dormancy therapies.
Invading bone
The types of cancer that are commonly associated with profound osteolysis include breast, lung and renal cancer, as well as multiple myeloma and adult T cell leukaemia. Clinical and experimental evidence indicates that bone resorption is also increased in osteoblastic metastases. Indeed, concentration of the bone resorption marker, N-telopeptide (NTX), is high in patients with prostate cancer with osteoblastic disease and is a strong predictor of morbidity and mortality66. Tumour-derived PTHRP was one of the first characterized mediators of local bone destruction to be associated with bone metastases. In mice with an absence of hypercalcaemia or detectable circulating levels of PTHRP, neutralizing antibodies to PTHRP could block breast tumour-associated bone loss and tumour growth in bone14. In patients with metastatic breast cancer, PTHRP expression levels are increased in bone metastases compared with the primary tumour67. Yin et al.68 showed that TGFβ in the bone microenvironment induced tumoural PTHRP production that resulted in enhanced bone resorption68. Subsequent studies, described below, showed that TGFβ in bone modulates many other pro-metastatic and osteolytic factors.
MMPs have been implicated in the general metastatic cascade, and more specifically in bone invasion and bone metastases through an increase in locally active RANKL. The effects can be direct, as exemplified by MMP7 cleavage of RANKL in prostate cancer30, or indirect, as shown by MMP1 and a disintegrin-like and metalloproteinase with thrombospondin motifs 1 (ADAMTS1), which proteolytically cleave epidermal growth factor (EGF)-like ligands to decrease osteoblast-derived OPG and hence favour osteoclastogenesis69. Moreover, MMP13 can activate MMP9 and TGFβ to increase local expression of RANKL at the breast cancer–bone interface70.
Other autocrine–paracrine mechanisms that promote tumour osteolysis involve the Jagged 1–Notch signalling pathway. Jagged 1 expressed in breast cancer cells mediates bone metastasis by activating the Notch pathway in bone cells resulting in increased IL-6, which confers a growth advantage to tumour cells. γ-secretase inhibitors reduce Jagged 1-mediated bone metastasis by disrupting the Notch pathway in stromal bone cells and so provide the rationale for targeting this pathway to treat bone metastases71.
Transcription factors, such as GLI2, RUNX2 and hypoxia-induced growth factor 1α (HIF1α) in tumour cells have been implicated in promoting tumour osteolysis. The Hedgehog signalling molecule GLI2 induces PTHRP expression and resultant osteolysis in metastatic human breast cancer cells72. GLI2 is also involved in TGFβ-mediated melanoma metastasis to bone73. The osteoblast transcription factor RUNX2 regulates MMP9 in bone metastatic cancer cells and controls cell invasion74. In support of this, impaired intranuclear trafficking of RUNX2 in breast cancer cells inhibits osteolysis in vivo75. Tumour expression of HIF1α inhibits osteoblast differentiation and promotes osteoclast differentiation, supporting HIF1α as a factor that promotes tumour osteolysis and tumour growth in bone76,77
Bone matrix and tumour growth
Beyond encountering cells that are resident in the bone marrow, there is an exchange of factors from the bone matrix that are released during resorption, the most notable being TGFβ, which affect tumour localization and growth. Calcium, which is abundant in the bone matrix, has a profound effect on tumour cells. Breast and prostate cancer cells express the calcium-sensing receptor (CASR) and respond to ionized calcium78 (FIG. 4). Calcium stimulation of these cells leads to an inhibition of apoptosis and a stimulation of proliferation79. In addition, ionized calcium leads to increased PTHRP secretion by tumour cells and hence induces further resorption and calcium release80,81. The CASR has been shown to be central to prostate cancer skeletal metastasis, as short hairpin RNA knockdown of this receptor in prostate cancer cells reduced tumour localization in bone82. Ionized calcium can be a potent chemoattractant to breast cancer cells and could support bone localization in addition to tumour cell proliferation83. Calcilytics, which are antagonists of the CASR, are under intensive investigation for the treatment of autosomal dominant hypocalcaemia, which results from activating mutations in the CASR84. Agents such as these could represent a strategy to restrict cancer growth in bone.
Figure 4. Mechanisms of tumour-associated osteolysis.
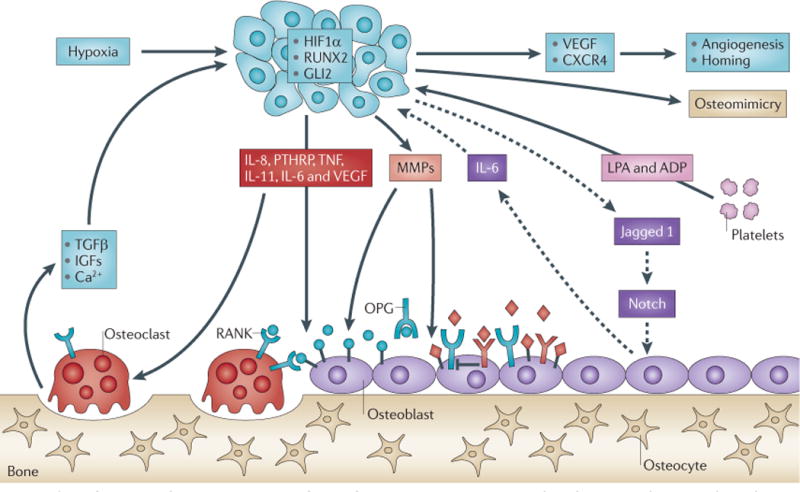
Tumours secrete osteolytic factors (such as, parathyroid hormone-related protein (PTHRP), interleukin-11 (IL-11), IL-6, IL-8, vascular endothelial growth factor (VEGF), tumour necrosis factor (TNF), Jagged 1 and epidermal growth factor (EGF)-like ligands) that stimulate osteoclastic bone resorption either directly (indicated by solid arrows) or indirectly (indicated by dashed arrows) by increasing the ratio of receptor activator of NF-κB ligand (RANKL) to osteoprotegerin (OPG). Osteoclastic bone resorption causes the release and activation of growth factors (transforming growth factor-β (TGFβ) and insulin-like growth factors (IGFs)) and ions (calcium) that are stored in mineralized bone matrix to further enhance the local milieu. Tumour-associated hypoxia and hypoxia-inducible factor 1α (HIF1α) in conjunction with TGFβ can increase tumour production of VEGF and the chemokine CXCR4 to increase angiogenesis and tumour homing. Tumour-produced matrix metalloproteinases (MMPs) can cleave membrane-bound RANKL (blue balls) or EGF-like growth factors (red diamonds), which can increase the ratio of RANKL to OPG to favour osteoclastogenesis. Platelet-derived lysophosphatidic acid (LPA) and ADP act on tumour cells to induce growth and the release of osteolytic factors IL-8 and IL-6.
Bone matricellular proteins that could affect tumour localization include OPN, which is also released by stromal and tumour cells, secreted protein acidic and rich in cysteine (SPARC), periostin, bone sialoprotein, dentin matrix acidic phosphoprotein 1 (DMP1), syndecan 1 and decorin. SPARC, which is produced by osteoblasts, leukocytes and cancer cells, induces cancer cell migration by interacting with αvβ5 (REF. 85) (FIG. 4). OPN, which is derived from bone matrix, stromal and tumour cells, has an important role in tumour metastasis. Experiments in mice deficient for OPN or overexpressing OPN revealed that levels of OPN correlate with skeletal metastatic potential86. Periostin, which is increased during the initial response of the bone marrow stroma to a tumour, has been shown to promote the growth of metastases in mouse models87. Bone sialoprotein has also been associated, at least in part, with increased migration and cell survival in breast and prostate cancer metastasis88,89. Differential expression of OPN and bone sialoprotein in breast and prostate cancers has been proposed as a potential switch between osteolytic versus osteoblastic presentation90. Other proteins of the ECM, including the proteoglycan syndecan 1, dentin sialophosphoprotein (DSPP) and DMP1 have been shown to be associated with breast and/or prostate cancer progression in bone91–93. By contrast, decorin suppresses bone metastasis in breast cancer models94.
TGFβ that is derived from resorbed bone matrix has a central role in most of the events leading to bone metastases and tumour expansion in bone through the regulation of osteolytic and pro-metastatic factors95 (FIGS 3, 4). Human breast cancer bone metastases show active TGFβ signalling by nuclear accumulation of phosphoSMAD2 (REF. 96). TGFβ signalling blockade by the stable expression of a dominant-negative TGFβ receptor 2 (DNTβRII) in MDA-MB-231 breast cancer cells inhibited TGFβ-induced tumour expression of PTHRP and suppressed bone metastasis in a mouse model68. Knockdown of SMAD4 also inhibited the development and the formation of bone metastases in a similar model96. In turn, overexpression of the inhibitory SMAD7 in 1205Lu melanoma cells reduced the formation of osteolytic lesions97.
TGFβ may interact with other microenvironmental factors in bone, such as hypoxia, to promote tumour growth. The bone marrow is a hypoxic microenvironment (with 1–7% O2) that enhances tumour metastasis and growth76 (FIG. 2). Interactions between HIF1α and TGFβ signalling pathways in breast cancer cells have additive responses in inducing the expression of vascular endothelial growth factor (VEGF) and CXCR4 in vitro and in vivo77. In a mouse model, the inhibition of either pathway in the tumour cells through mRNA knockdown decreased bone metastasis, and the loss of both did not have additional effects. By contrast, combined treatment with pharmacological pathway inhibitors decreased bone metastases more than either treatment alone, resulting in less osteoclastic bone resorption and tumour growth. These data indicate that hypoxia and TGFβ signalling drive, in parallel, bone metastases and also that they regulate a common set of tumour genes; small-molecule inhibitors by acting on both tumour cells and the bone microenvironment, thus additively decrease tumour burden77. Of all the growth factors that are present in mineralized bone matrix, the TGFβ pathway dominates and promotes bone metastases of many different tumour types through the mechanisms described above. Abundant preclinical evidence supports blocking the TGFβ pathway to treat bone metastases and that the major source of TGFβ in bone metastases is that released from the bone matrix as a consequence of osteoclastic bone resorption98.
Resorption, formation and metastatic growth
Tumour metastasis to bone can alter normal bone physiology, resulting in uncoupled bone remodelling (FIG. 5). Enhanced osteoclast activity exacerbates the growth and progression of bone metastases (FIGS 4, 5), and preclinical data suggest that modulation of bone resorption prevents the development of bone metastases. Mice with increased osteoclast activity that is induced through vitamin D deficiency, oestrogen or androgen deprivation, or through the administration of granulocyte colony stimulating factor (G-CSF), GM-CSF or parathyroid hormone (PTH), develop increased osteolytic tumour burden99–102. Furthermore, Cxcr4-/- mice, which have enhanced osteoclast activity, have a higher tumour burden in bone103. By contrast, the administration of osteoclast inhibitors, such as bisphosphonates, OPG, RANKL antagonists and β3 integrin antagonists104, before tumour inoculation diminishes the growth of solid tumour bone metastases and myeloma tumour burden in bone105–107. Likewise, mice with genetically defective osteoclast function as a result of Src-/-, Cd47-/-, Opn-/- or Itgb3-/- deletion have reduced skeletal tumour burden108–110. Thus, osteoclastic changes are essential for both tumour cell colonization and growth in bone. Together, these data indicate that the state of the bone microenvironment before bone metastasis can modulate tumour growth and the subsequent behaviour of tumours in bone and thus it represents an important target for metastasis prevention.
Figure 5. Clinical presentations of bone metastases.

Bone metastases can be detected or indicated by various approaches. The presence of disseminated tumour cells in the bone marrow (shown in part a by immunohistochemical staining for cytokeratin in a bone marrow smear taken from a patient with breast cancer) is associated with an increased risk of bone metastasis. Post-mortem examination (part b) also clearly shows osteoblastic lesions, in this example in the vertebral bodies from a patient who died of mestatatic prostate cancer. A bone biopsy (part c) stained with haematoxylin and eosin from a patient with metastatic breast cancer clearly shows the invasion of the tumour cells into the bone and the presence of osteoclasts (OCs) and osteoblasts (OBs). Computerized tomography (CT) scans (part d) can clearly show the different types of bone lesions: a lytic metastasis present in a vertebral body from a patient with metastatic lung cancer; a blastic metastasis (deposition of new bone) in the pelvis of a patient with metastatic prostate cancer; and a scan that shows a patient with metastatic breast cancer who has both lytic and blastic metastases in the pelvis. Bone metastases can be extensive as indicated by the full-body bone scans (part e) from a patient with metastatic breast cancer. Metastases are clearly present in the skull, ribs, clavicles, spine, pelvis and the tops of the femurs. Positron emission tomography using radiolabelled [18F]-2-fluoro-deoxy-D-glucose combined with CT (part f) also clearly shows active bone metastases, in this case a sacral metastasis in a patient with metastatic renal cancer. BV, blood vessel. Part a courtesy of R. Aft, Washington University School of Medicine, USA. Part b courtesy of K. Pienta, University of Michigan School of Medicine, USA. Part c courtesy of D. Novack, Washington University School of Medicine, USA. Images in parts d–f courtesy of V. Reichert and J. Burkett, Washington University School of Medicine, USA.
Interestingly, reports suggest that cancer cells can fuse with macrophages111 or can induce the fusion of osteoclast precursors to promote multinucleated giant cells112, leading to mature osteoclasts with tumour cell nuclei and increased function113–116. This identification of tumour cells with osteoclast properties shows that tumour cells are able to behave like bone cells in some circumstances, and this is known as osteomimicry. How widespread such osteomimicry is in skeletal metastasis and how this can be optimized for therapeutic advantage has yet to be determined. Recent work in mouse models of breast cancer has shown that progesterone receptor-positive epithelial cancer cells express RANKL and that treatment with RANKL inhibitors inhibits their growth, which highlights their potential for inhibition through targeted anti-RANKL strategies117.
Another form of osteomimicry involving osteoblasts has been identified in breast and prostate cancer cells. The osteoblastic nature of prostate cancer skeletal metastases (FIGS 3, 5) results in bone that is of a poor quality, immature and woven type118. Various mechanisms for a tumour-associated increase in bone tissue have been proposed. Tumour cells in the metastatic prostate lesion may transdifferentiate to become mesenchymal cells that are capable of osteoblastic activity, cancer cells may induce resident cells in the marrow microenvironment to enter the osteoblast lineage, and prostate cancer cells may induce the proliferation and/or differentiation of osteoblast lineage cells. The ability of prostate cancer to undergo epithelial–mesenchymal transition (EMT) and hence osteomimicry has been substantiated in animal models and humans119–122. RUNX2 has been implicated in the osteomimicry that is attributed to breast and prostate skeletal metastasis123–125. Osteoblasts are a vital component in certain aspects of tumour localization in bone. Tumours that display the phenotypic ‘osteoblastic’ response are thought to be dependent on osteoblasts in the bone microenvironment for their continued growth and survival, resulting in a co-dependent cycle126,127.
Other cell types that prepare and feed the soil
Bone marrow endothelial and haematopoietic cells
Cancer cells preferentially adhere to bone marrow endothelial cells rather than to endothelial cells that are derived from other organs128. Such adherent cancer cells may extravasate through the bone marrow endothelium to take up residence in the bone microenvironment. Subsequently, tumour neovascularization is essential for establishing micrometastases and for enabling their expansion in the bone. Bone metastasis and bone-residing tumours, such as myeloma, modify and recruit endothelial cells to enhance neoangiogenesis129. Targeting of stromal and endothelial cells in the bone marrow with the multitargeted tyrosine kinase inhibitor sunitinib resulted in bone marrow vessel leakage, stromal apoptosis and disrupted lung cancer cell colonization and osteolysis210. Increasing evidence supports myeloid cell participation in angiogenesis, but the ability to effectively measure the vascular supply to a tumour in bone is challenging130. Haematopoietic cells that reside in the marrow influence cell communication by releasing pro-angiogenic factors or by creating permissive conditions in the tumour microenvironment that favour the growth of blood vessels131. The haemangioblast is the progenitor cell for endothelial and haematopoietic cells and has been reported to express CD34 and CD133. Beyond their role during development, these cells continue to exist in adults in the bone marrow and peripheral blood as stem-like cells, which could be implicated in tumour vasculogenesis. Endothelial progenitor cells (EPCs) have been implicated in numerous different tumours, but their identification and precise functions remain elusive (FIGS 2, 6).
Figure 6. Overlapping benefits of targeting tumour and stromal cells for bone metastases.

The growth and survival of metastatic cancer cells in bone require the support of numerous cells and molecules within the bone microenvironment. Many of the therapies targeted to cancer cell signalling pathways (part a) have overlapping effects on bone stromal components (part b), which can enhance the antitumour effects. Likewise, stromal-targeted therapies can also target tumour cells. Many therapies target more than these two cell types, such as receptor tyrosine kinase (RTK) inhibitors. Anti-resorptive therapies target genes and proteins that are involved in functional osteoclast development from pre-osteoclasts and haematopoietic progenitor cells, but most of these therapies also have direct antitumour effects. Targeting pro-tumour and antitumour immunity that is mediated by myeloid cells and lymphocytes and the proteins that they secrete can disrupt tumour growth in bone and in other sites. Inhibition of platelet aggregation and activation can limit the release of platelet-derived pro-tumour and pro-angiogenic molecules and can disrupt tumour–platelet aggregates, reducing adhesion to bone vessels. Targeting acid-sensing and sympathetic neurons can decrease bone pain. A reduction in pain levels can improve quality of life, enhance mobility (which can strengthen bones and prevent fracture) and improve nutrition, which can promote improved survival. Anti-angiogenic therapies decrease tumour burden in bone and potential metastasis of metastases. Bone-targeted radiation not only targets tumour cells but can also disrupt fibroblast–stromal support of tumour cells. Numerous protein factors (such as transforming growth factor-β (TGFβ) and bone morphogenetic proteins (BMPs)) that are stored within the bone matrix, as well as calcium, are released during bone metastases and can enhance tumour growth and promote the activation and release of other bone-derived, pro-tumour growth factors. Targeting of osteoblasts through endothelin 1, chemokine disruption, anabolic effects of proteosomes or RTK and SRC inhibition can decrease fractures and decrease the release of tumour chemoattractants and osteoclast growth factors. MMP, matrix metalloproteinase; XRT, radiotherapy.
Adipose stem cells
Recently, adipose stem cells that are present in the bone marrow microenvironment, which develop from the same MSC lineage as osteoblasts, were shown to promote prostate tumour growth132. Bone marrow-derived MSCs have also been implicated in tumour growth and metastasis through their ability to promote and protect tumour-initiating cancer stem cells133. Interestingly, the potential of MSCs to interact with tumour cells has prompted investigations into whether MSCs can be used for the targeted delivery of antitumour agents to cancer cells134. More work is clearly needed in this area and is limited at least in part by difficulties in the standardization of MSC identification135.
Myeloid and immune cells
Bone marrow-derived myeloid cells (including, macrophages, monocytes, myeloid-derived suppressor cells, myeloid dendritic cells and osteoclasts) and lymphocytes are recruited to tumours and areas of hypoxia and neoangiogenesis and can either promote tumour growth or enhance antitumour immune responses136–138 (FIGS 2, 6). Recent evidence suggests that tumour-associated myeloid cells can be influenced by anti-resorptive targeted therapies (through either direct or indirect mechanisms), which could represent an osteoclast-independent mechanism of action of the anti-resorptives.
Myeloid-derived suppressor cells (MDSCs) are a subpopulation of immature myeloid cells that are characterized by GR1 expression and by the αMβ2 (CD11b) integrin adhesion marker139. MDSCs from myeloma-bearing mice had a greater capacity to become bone-resorbing cells compared with MDSCs from control mice136. Recent reports that mice with osteoclast defects and immune defects were protected from tumour-associated bone loss but did not have decreased tumour burden in bone140 highlight the role of immune cells in bone metastasis. Bisphosphonates decrease MDSC numbers and reduce MMP secretion, which may represent an osteoclast-independent mechanism of action141. Indeed, zoledronic acid decreased tumour burden in murine bone with severely defective osteoclasts142. The regulation of MDSC differentiation, recruitment from the bone marrow to tumour sites and MDSC function in tumour biology is under intensive investigation.
Although T lymphocytes have a key role in influencing general tumour growth143 and bone remodelling144, many animal models of bone metastasis are carried out in immunocompromised mice that lack T cells, thus their role in regulating tumour growth within the bone microenvironment remains largely unstudied. Of note, pro-inflammatory CD4+ TH17 cells secrete RANKL, tumour necrosis factor (TNF) and TGFβ, all of which activate osteoclasts and promote bone resorption144,145 and thus may enhance tumour growth in bone through the enhancement of the vicious cycle. It is anticipated that future investigations will reveal an important role for T cell subsets in regulating bone metastasis.
Platelets
In addition to effects on haemostasis, activated platelets are an important source of pro-angiogenic (VEGF) and anti-angiogenic (thrombospondin 1 (TSP1)) factors, the aggregation of which at tumour sites affects tumour growth146 (FIGS 2, 6). Tumour cells engineered to respond to platelet-derived lysophosphatidic acid (LPA) have enhanced bone metastatic potential in mice partly through platelet-derived LPA-mediated induction of osteolytic factors such as IL-6 and IL-8 (REF. 147). Likewise, genetic and pharmacological inhibition of platelet-specific integrins (αIIbβ3) or platelet activation decreased osteolytic bone metastases in mice110,148. Targeting platelets is a promising therapeutic approach for inhibiting bone metastasis, in particular to prevent metastasis or to decrease tumour burden in bone.
Conversely, bone marrow megakaryocytes, from which platelets are derived, inhibit prostate cancer tumour growth in bone149 and also inhibit bone resorption by inhibiting osteoclast formation150. The negative effect of megakaryocytes on bone resorption might be partly mediated through the production of the osteoclast inhibitory factor OPG151. Megakaryocytes might also influence bone remodelling and resorption through effects on osteoblast proliferation that are mediated by the α3β1, α5β1 and glycoprotein IIb integrins152. As mature megakaryocytes are located at vascular sinusoids, they are also among the first cells to encounter cancer cells as they enter the bone marrow environment, so direct action involving integrin-mediated signal transduction could be involved. Interestingly, bisphosphonates increase megakaryocyte proliferation and increase the platelet concentration of the anti-angiogeneic integrin ligand TSP1 (REFS 153,154), which suggests that bisphosphonates could have non-osteoclast mechanisms that decrease tumour growth in bone. Thus, platelets and their megakaryocytic precursors interact with cancer cells before, during and after metastasis to bone.
Tumour cell dormancy
Paradoxically, metastasis is an inefficient process with only 0.001–0.02% of cancer cells that are experimentally introduced into the circulation actually forming metastatic foci99,155,156. Preclinical evidence suggests that metastatic tumour cells can home to and localize in the HSC niche and survive in a dormant state. The skeleton is the preferred site for many tumour cells to reside, and they can remain there in a dormant state for long periods of time.
The presence of disseminated tumour cells (DTCs) in the bone mar row of patients with primary cancer reveals the housing potential of the skeleton (FIGS 1, 5). Patients with bone marrow DTCs at diagnosis are at a higher risk of both skeletal and extraskeletal metastasis. Evidence exists that DTCs can persist in the bone marrow for years in a quiescent state, and are resistant to cancer therapies8,55,157. Myeloma cells interact with bone marrow stroma partly through integrin α4 (also known as VLA4) and VCAM1 to facilitate quiescence and protection from apoptosis, providing resistance to chemotherapy158. Of patients with prostate cancer who have had a radical prostatectomy, 72% have DTCs in the bone marrow159; and 30% of patients with localized breast cancer have bone marrow DTCs at diagnosis9,160. These DTCs are strong predictors of biochemical recurrence of disease and do not bode well for outcomes161. Molecular characterization of disseminated breast cancer cells in the bone marrow has revealed the specific expression of a subgroup of transcripts, including the metastasis regulator TWIST1, and SRC activation162,163. Breast cancer cells that have active SRC are associated with an increased risk of bone metastases in humans163. This was attributed to a cell-autonomous pro-survival role of SRC, and was linked to the pro-survival effects of CXCL12, as well as resistance to the pro-apoptotic effects of TRAIL. Increasing clinical evidence suggests that DTCs are influenced by the bone microenvironment. The treatment of women with localized breast cancer with the bisphosphonate zelondronic acid modulated the presence of DTCs in the bone marrow8,164.
Tumour cells might mimic HSC cells, and evidence for factors that induce HSC dormancy could explain tumour cell dormancy in bone marrow niches. Prostate cancer cells bind to osteoblast annexin II, which induces the expression of growth arrest-specific 6 (GAS6) receptors. GAS6 receptors are well-known inducers of dormancy in HSCs and were found to reduce cell cycle progression in prostate cancer cells, thus implicating osteoblasts as facilitators for tumour dormancy in bone165. Understanding tumour cell dormancy within the bone marrow is probably key to inducing long-term remissions and overcoming resistance to cancer therapies.
Therapeutic targeting of bone metastases
Targeting osteoclasts
Osteoclastic bone resorption inhibitors are the standard of care for patients with established bone metastases, and the therapeutic targeting of osteoclasts is an area of intense clinical investigation (FIGS 4, 6). Bone matrix-targeted bisphosphonates, such as pamidronate, zoledronic acid and ibandronate, are amino-bisphosphonates that block farnesyl pryrophosphate synthase and that disrupt protein prenylation166. In addition to the effects on bone resorption by osteoclasts, amino-bisphosphonates can affect other cell types that promote bone metastases, including γδ-T cells, monocytes, MDSCs and endothelial cells, as well as tumour cells. Clinical data suggest that bisphosphonates can limit the progression of breast cancer both in bone and in other tissues8,167. The therapeutic disruption of RANKL with a subcutaneously administered humanized neutralizing antibody, denosumab, results in significant decreases in skeletal complications and reduces bone pain168. In addition, denosumab may be effective in directly targeting subtypes of breast and prostate cancers that express RANKL117. Other osteoclast-targeted therapies under clinical evaluation for the treatment of bone metastasis include cathepsin K inhibitors, SRC inhibitors and αvβ3 inhibitors169–171; interestingly, these therapies also affect tumour cells and stromal components that participate in bone metastases (FIG. 6).
Targeting TGFβ and bone matrix proteins
Several strategies to inhibit TGFβ signalling are being applied to cancer172. The different classes of TGFβ inhibitors that have been used in preclinical models and clinical trials of bone metastases include monoclonal neutralizing TGFβ antibodies that prevent TGFβ ligand–receptor interactions; small molecules that inhibit TβRI (and TβRI II) kinase activity, preventing the activation TGFβ R-SMADs; and a natural product derivative, halofuginone (HFG), which inhibits TGFβ, although the exact mechanism remains to be investigated (FIGS 4, 6).
Although the effects of TGFβ on osteoclasts and osteoblasts are dose- and context-dependent in vitro, genetically modified mice showed that increased levels of TGFβ in bone promoted osteoclastogenesis and bone resorption173. In support of this, systemic TGFβ inhibition with an orally active TβRI kinase inhibitor SD-208, or a pan-neutralizing TGFβ antibody, causes increased bone mass and increased bone mineralization by inhibiting osteoclastic bone resorption and stimulating new bone formation174,175. BMP7, an inhibitor of TGFβ signalling, inhibits the formation of bone metastases in preclinical models176,177 and is a potent inducer of bone formation; it is currently approved for clinical orthopaedic fracture treatments178.
As a result of its wide variety of effects on numerous cell types and pathways, blockade of TGFβ or its downstream signalling could have undesirable consequences for wound healing and immune function179. However, human dose escalation studies with the small molecule TGFβ inhibitor, LY2157299, for patients with advanced metastatic melanoma, pheochromocytoma, and colon, prostate and breast cancer demonstrated that the drug is well tolerated with minimal toxicity.
Other bone matrix proteins that are released during osteoclastic resorption include insulin-like growth factor 1 (IGF1), platelet-derived growth factors (PDGFs) and BMPs, as well as calcium. These proteins and mineral elements have been shown to increase the growth of certain tumour types, and represent druggable (and in most cases currently available) anti-bone metastasis targets. Bisphosphonates and other diphosphonate molecules have strong avidity to bone matrix with long bone half-lives and can be used to target compounds to the bone matrix.
Targeting osteoblasts
One of the promising osteoblast targets investigated in preclinical and clinical trials has been endothelin 1. Endothelin 1 is a vasoconstrictive agent that stimulates osteoblast proliferation and enhances osteoblast differentiation180,181. Strong support of its ability to target prostate cancer skeletal metastases in animal models promoted its investigation in human clinical trials128,182,183. Atrasentan was the first endothelin receptor subtype A antagonist to be investigated in patients with prostate cancer, and zibotentan is a more selective and promising inhibitor that is currently in clinical trials184 (FIGS 3, 6).
The bone stroma and osteoblast compartments can interact with tumour cells to promote resistance to cytotoxic chemotherapy. An exciting new approach takes advantage of the fact that cancer cells use CXCR4 and VLA4 to home to and engraft in the marrow. Cancer cells in the bone marrow are often resistant to chemotherapy because they are held in G0 phase of the cell cycle by contact with bone marrow stromal cells. HSC mobilizing agents such as AMD3100 and anti-VLA4-targeted agents mobilize leukaemia and myeloma cells into the blood, which leads to increased sensitivity to chemotherapy in mice185,186. This approach is now being tested in clinical trials in the treatment of acute myeloid leukaemia and multiple myeloma.
Targeting neoangiogenesis
Anti-angiogenic therapies have been of intense interest and promise in cancer therapy in general, but of less focus in skeletal metastasis. Cutting the blood supply to a tumour regardless of its location has clear merit; however, the feasibility and strategies needed to achieve this in the vascular-rich site of the bone marrow are challenging and the biology is poorly understood. In a model of skeletal metastasis, the anti-angiogenic agent avastin was found to inhibit tumour growth, albeit through the indirect targeting of osteoclasts187. It is not clear what the exact target of anti-angiogenic therapy is, as the actions of such agents on circulating endothelial progenitor cells have been inconclusive188. Combining an anti-angiogenic agent with cytotoxic chemotherapy produced promising results in an animal model of breast cancer skeletal metastasis189.
Bone marrow-derived endothelial progenitors are under investigation as potential targets for anti-angiogenic strategies190. Experimental strategies using endothelial progenitor cell markers such as ID1 are emerging, and they provide valuable new avenues for therapeutic development191. Difficulty in this area has stemmed from the lack of a reliable endothelial progenitor cell marker that can be used in the differing locations that these cells reside in, such as the bone marrow, peripheral blood and tumours.
Metastasis-associated protein 1 (MTA1) is a pro-angiogenic factor that is associated with prostate cancer progression192. Silencing MTA1 effectively limited the expression of pro-angiogenic factors such as VEGF by prostate cancer cells and thus represents a potential new target of angiogenic events in skeletal metastases in patients with prostate cancer.
Treating bone pain
Cancer-associated bone pain is extremely difficult to treat and is often fairly resistant to opioids193. Bone is densely innervated by primary sensory afferent and sympathetic neurons that are located in the periosteum and the intramedullary bone194. Bone resorption inhibitors substantially decrease pain that is produced by the growth of bone metastases161. Honore et al.195 found that the acidic environment that is produced by resorbing osteoclasts activates the acid-sensing transient receptor potential vanilloid type 1 (TRPV1) pain neurons195 (FIG. 2). The RANKL antagonist OPG successfully prevented activation of pain fibres and decreased pain behaviour in mice with bone metastases. Nakanishi et al.196 demonstrated that acid activation of TRPV1 neurons upregulates calcitonin gene-related peptide in the dorsal root ganglia and is associated with inflammatory-mediated pain196. Likewise, bone-residing tumour cells secrete factors, such as nerve growth factor (NGF) and endothelin 1, that activate pain neurons. Invasively expanding tumours in bone may also compress or directly induce nerve injury. Inhibitors of bone resorption, TRPV1, endothelin A receptor and NGF all represent therapeutic targets for cancer-induced bone pain194. Another approach to pain relief in patients with bone metastases has been the use of radiopharmaceuticals. Strontium, samarium and radium have strong avidity to the calcified matrix of bone. These agents can exert a small antitumour effect through localized radiation, but have substantial effects on bone pain and are primarily indicated to provide pain relief197.
Future directions
Bone metastases are common and have devastating effects on cancer patients. The bone microenvironment is a unique and fertile soil for cancer metastasis, and tumour cells modify the bone microenvironment during cancer invasion and expansion through the recruitment and modulation of osteoclasts, osteoblasts, immune cells, vascular elements, bone matrix and neuronal processes. Better characterization of this tumour-modified stroma and the identification of the molecules that affect tumour expansion and dormancy in bone is essential moving forwards. In particular, the role of immune cells (T cells, natural killer cells, macrophages, MDSCs and dendritic cells) is crucial for understanding tumour expansion in bone and represents an area of great therapeutic promise. Metastasis to bone has proved a challenging process to model in vitro and in mice because various cell types are required in distinct temporal stages. Current animal models are appropriate for evaluating tumour expansion in bone using aggressive tumour cell lines. However, these models use highly unstable and selected cell lines developed in different strains or species inoculated into immunocompromised (or non-strain matched) animals, thus it is unclear how representative these models are of physiological bone metastases. Also, the effects of immune responses on all stages of the metastatic process are difficult to evaluate. Current animal models of cancer develop spontaneous metastasis to bone too infrequently to readily facilitate the study of tumour homing to and dormancy within bone. Animal models of osteoblastic metastases are also challenging. There is a need to develop better animal models to study tumour localization and tumour cell dormancy in bone. The enigma of why some tumour cells lie dormant in bone for decades before progressing to symptomatic disease requires comprehensive and detailed clarification.
Massively parallel DNA sequencing technologies have the potential to transform our understanding of cancer biology. Beyond the somatic mutations in cancer cells, germline DNA variants can affect any stromal cell element and can probably contribute to cancer metastasis in ways that will be uncovered as we turn our attention away from the fractured and unstable cancer genomes and towards the analysis of stromal cell genomes. Genetically unstable tumour cells are exposed to clonal selection during the colonization of distant sites198, but cells of the host tumour microenvironment represent underused, genetically stable therapeutic targets. Great efforts are underway to better understand the epigenome of cancer cells, including the function of microRNAs; however, epigenetic changes in tumour stromal cells probably have important roles in cancer biology and may be amenable to therapeutic intervention. The identification and characterization of genetic and epigenetic changes in the key stromal elements that are involved in skeletal metastasis could lead to valuable and wide-reaching therapeutics for preventing the fatal attraction and devastating consequences of skeletal metastasis.
Powerful imaging technologies are rapidly evolving, and it will be of great interest to use these technologies to evaluate how the different stromal cell elements that are involved in bone metastasis may be visualized in live animals. Nanotechnology applications in medicine are in their infancy, and efforts are underway to use our knowledge of the bone microenvironment to target nanoparticles to tumour sites within bone. Stromal-targeted nanoparticles can have applications that are diagnostic, therapeutic or both: such agents have been dubbed ‘theranostics’.
We hope that advances in understanding the basic biology of bone remodelling, biomechanics and haematopoiesis, coupled with the advances in cancer genetics, will continue to yield new and exciting therapeutic targets and insights into cancer metastasis in bone.
At a glance.
Bone metastases are a common complication of cancer and are generally incurable. They cause considerable pain, pathological bone fractures and hypercalcaemia. Up to 50% of patients prescribed anti-resorptive drugs to treat bone metastases develop new bone metastases, skeletal complications and disease progression, emphasizing the need for new therapies.
Tumour invasion into bone is associated with osteoclast and osteoblast recruitment. Osteoclasts secrete acid, collagenases and proteases that demineralize the bone matrix and degrade matricellular proteins. Macrophage colony stimulating factor and receptor activator of NF-κB ligand (RANKL) are important growth factors that support osteoclastogenesis, and they are primarily produced by osteoblasts. Osteoprotegerin is an endogenous decoy receptor of RANKL that inhibits osteoclastogenesis.
Bone marrow mesenchymal stem cells are directed along the osteoblast lineage through local factors, such as transforming growth factor-β (TGFβ), bone morphogenetic proteins (BMPs) and WNT proteins. These pathways lead to the expression of three key transcriptional regulators of osteoblast function, including RUNX2.The osteoblast-stimulating activity of metastatic tumour cells is thought to be due to the ability of these cells to express many of the factors that can drive osteoblast formation.
Osteoblasts and bone marrow stromal cells may attract metastatic tumour cells to bone and provide a niche through protein interactions that include integrins, such as α4β 1–vascular cell adhesion molecule 1; chemokines, such as CXCL12–CXCR4; BMPs; Notch; nestin; and osteopontin. These mechanisms are similar to the physiological recruitment of haematopoietic stem cells.
The invasion and growth of metastatic tumour cells in the bone involves the modulation of a large number of genes and proteins that include matrix metalloproteinases, parathyroid hormone-related protein, TGFβ, interleukin-6, Jagged 1–Notch, GLI2, RUNX2, hypoxia-induced growth factor 1α, calcium and the calcium-sensing receptor.
Beyond the effects on osteoclasts and osteoblasts, tumours in the bone microenvironment recruit and modulate the function of platelets, myeloid cells, immune cells and nerve cells, and induce the formation of new blood vessels. These changes all help to ensure the growth and survival of metastatic tumour cells in bone and represent important therapeutic targets.
Drugs, such as bisphosphonates or RANKL antibodies, that target osteoclastogenesis decrease the incidence of skeletal complications and are the current standard of care for patients with bone metastases. These anti-resorptive agents might also have direct antitumour effects.
Advances in our understanding of the basic biology of bone remodelling, biomechanics and haematopoiesis, coupled with the advances in cancer genetics and tumour imaging should yield new therapeutic targets and insights into cancer metastasis in bone.
Acknowledgments
Dedicated in memory of G. Mundy. The authors would like to thank P. Ross, M. Tomasson, C. Hall, D. Novack, S. Amend, J. Schneider, M. Hurchla and C. Winkeler for feedback on this manuscript. The authors are grateful to K. Pienta (University of Michigan School of Medicine, USA) for providing the gross autopsy specimen from vertebral body involved with prostate cancer, V. Reichert and J. Burkett (Washington University Medical School, St. Louis, USA) for providing the radiological images of bone metastases, R. Aft (Washington University School of Medicine, USA) for providing the photograph of bone marrow disseminated tumour cells, and D. Novack (Washington University School of Medicine) for providing the histological slide from bone metastasis biospy. K.N.W. is supported by the US National Institutes of Health (NIH) (R01-CA52152 and P01-CA100730) and the US Department of Defense (W81XWH-01-1-360). T.A.G. is supported by R01CA69158, R01DK065837, R01DK067333, U01CA143057, V-Foundation and Indiana Economic Development Grant. L.K.M. is supported by the US National Institutes of Health (NIH) (PO1-CA093900, RO1- DK53904), the US Department of Defense (W81XWH-08-1-0037) and Centocor Inc. The authors regret that there are many other important studies that they were unable to include owing to space limitations.
Footnotes
Competing interests statement
The authors declare competing financial interests. See Web version for details.
FURTHER INFORMATION
Katherine N. Weilbaecher's homepage: http://hematology.im.wustl.edu/faculty/weilbaecher/weilbaecherBio.html
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 2.Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001;27:165–176. doi: 10.1053/ctrv.2000.0210. [DOI] [PubMed] [Google Scholar]
- 3.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nature Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 4.Fukutomi M, et al. Increased incidence of bone metastases in hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2001;13:1083–1088. doi: 10.1097/00042737-200109000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Guise TA, et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res. 2006;12:6213s–6216s. doi: 10.1158/1078-0432.CCR-06-1007. [DOI] [PubMed] [Google Scholar]
- 6.Kingsley LA, Fournier PG, Chirgwin JM, Guise TA. Molecular biology of bone metastasis. Mol Cancer Ther. 2007;6:2609–2617. doi: 10.1158/1535-7163.MCT-07-0234. [DOI] [PubMed] [Google Scholar]
- 7.Lipton A, et al. Normalization of bone markers is associated with improved survival in patients with bone metastases from solid tumors and elevated bone resorption receiving zoledronic acid. Cancer. 2008;113:193–201. doi: 10.1002/cncr.23529. [DOI] [PubMed] [Google Scholar]
- 8.Aft R, et al. Effect of zoledronic acid on disseminated tumour cells in women with locally advanced breast cancer: an open label, randomised, phase 2 trial. Lancet Oncol. 2010;11:421–428. doi: 10.1016/S1470-2045(10)70054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pantel K, et al. Detection and clinical implications of early systemic tumor cell dissemination in breast cancer. Clin Cancer Res. 2003;9:6326–6334. [PubMed] [Google Scholar]
- 10.Braun S, et al. Lack of effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high-risk breast cancer patients. J Clin Oncol. 2000;18:80–86. doi: 10.1200/JCO.2000.18.1.80. [DOI] [PubMed] [Google Scholar]
- 11.Hirbe A, Morgan EA, Uluçkan Ö, Weilbaecher K. Skeletal complications of breast cancer therapies. Clin Cancer Res. 2006;12:6309s–6314s. doi: 10.1158/1078-0432.CCR-06-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Body JJ, et al. Effects of denosumab in patients with bone metastases, with and without previous bisphosphonate exposure. J Bone Miner Res. 2009;25:440–446. doi: 10.1359/jbmr.090810. [DOI] [PubMed] [Google Scholar]
- 13.Fizazi K, et al. Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J Clin Oncol. 2009;27:1564–1571. doi: 10.1200/JCO.2008.19.2146. [DOI] [PubMed] [Google Scholar]
- 14.Guise TA, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544–1549. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nature Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- 16.Dougall WC, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suda T, et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 18.Tang Y, et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nature Med. 2009;15:757–765. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komori T, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 20.Nakashima K, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. J Biol Chem. 2004;279:47109–47114. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- 22.Xiao G, et al. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J Biol Chem. 2005;280:30689–30696. doi: 10.1074/jbc.M500750200. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiratsuka S, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nature Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 25.Purhonen S, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci USA. 2008;105:6620–6625. doi: 10.1073/pnas.0710516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly T, et al. Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res. 2005;65:5778–5784. doi: 10.1158/0008-5472.CAN-05-0749. [DOI] [PubMed] [Google Scholar]
- 27.Pazolli E, et al. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res. 2009;69:1230–1239. doi: 10.1158/0008-5472.CAN-08-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAllister SS, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med. 2006;14:2037–2044. doi: 10.1111/j.1582-4934.2010.01115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch CC, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–496. doi: 10.1016/j.ccr.2005.04.013. Microarray analysis identified MMP7 as being upregulated at the tumor–bone interface in mice with prostate cancer. Osteoclast-produced MMP7 processed RANKL to a soluble form that promoted osteoclast activation. MMP7-deficient mice had reduced prostate tumour-induced osteolysis and RANKL processing. [DOI] [PubMed] [Google Scholar]
- 31.Li X, et al. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009;69:1685–1692. doi: 10.1158/0008-5472.CAN-08-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehr JE, Pienta KJ. Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. J Natl Cancer Inst. 1998;90:118–123. doi: 10.1093/jnci/90.2.118. [DOI] [PubMed] [Google Scholar]
- 33.Brenner S, et al. CXCR4-transgene expression significantly improves marrow engraftment of cultured hematopoietic stem cells. Stem Cells. 2004;22:1128–1133. doi: 10.1634/stemcells.2003-0196. [DOI] [PubMed] [Google Scholar]
- 34.Kahn J, et al. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood. 2004;103:2942–2949. doi: 10.1182/blood-2003-07-2607. [DOI] [PubMed] [Google Scholar]
- 35.Kang Y, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. This study shows the multigenic nature of tumour tropism to bone: tumour cells must express a ‘toolbox’ of genes that act on the bone microenvironment in a cooperative way to promote bone metastases. [DOI] [PubMed] [Google Scholar]
- 36.Yoneda T. Cellular and molecular basis of preferential metastasis of breast cancer to bone. J Orthop Sci. 2000;5:75–81. doi: 10.1007/s007760050012. [DOI] [PubMed] [Google Scholar]
- 37.Stier S, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. 2005;201:1781–1791. doi: 10.1084/jem.20041992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christopher MJ, Liu F, Hilton MJ, Long F, Link DC. Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood. 2009;114:1331–1339. doi: 10.1182/blood-2008-10-184754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvi LM, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 41.Papayannopoulou T. Mechanisms of stem-/progenitor-cell mobilization: the anti-VLA-4 paradigm. Semin Hematol. 2000;37:11–18. doi: 10.1016/s0037-1963(00)90084-2. [DOI] [PubMed] [Google Scholar]
- 42.Hidalgo A, Peired AJ, Weiss LA, Katayama Y, Frenette PS. The integrin αMβ2 anchors hematopoietic progenitors in the bone marrow during enforced mobilization. Blood. 2004;104:993–1001. doi: 10.1182/blood-2003-10-3702. [DOI] [PubMed] [Google Scholar]
- 43.Mendez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kollet O, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nature Med. 2006;12:657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- 45.Mendez-Ferrer S, Frenette PS. Hematopoietic stem cell trafficking: regulated adhesion and attraction to bone marrow microenvironment. Ann N Y Acad Sci. 2007;1116:392–413. doi: 10.1196/annals.1402.086. [DOI] [PubMed] [Google Scholar]
- 46.Shiozawa Y, et al. Prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in marrow. J Clin Invest. 2011;121:1298–1312. doi: 10.1172/JCI43414. Metastatic prostate cancer cells directly compete for occupancy of the HSC niche and once there begin a process of either evicting HSCs into the peripheral blood or driving HSCs into progenitor cell pools. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller A, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 48.Sun YX, et al. Expression and activation of αvβ3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate. 2007;67:61–73. doi: 10.1002/pros.20500. [DOI] [PubMed] [Google Scholar]
- 49.Sun YX, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res. 2005;20:318–329. doi: 10.1359/JBMR.041109. [DOI] [PubMed] [Google Scholar]
- 50.Smith MC, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 51.Chinni SR, et al. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate. 2006;66:32–48. doi: 10.1002/pros.20318. [DOI] [PubMed] [Google Scholar]
- 52.Lapteva N, Yang AG, Sanders DE, Strube RW, Chen SY. CXCR4 knockdown by small interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther. 2005;12:84–89. doi: 10.1038/sj.cgt.7700770. [DOI] [PubMed] [Google Scholar]
- 53.Sun YX, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003;89:462–473. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- 54.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 55.Clezardin P. Integrins in bone metastasis formation and potential therapeutic implications. Curr Cancer Drug Targets. 2009;9:801–806. doi: 10.2174/156800909789760348. [DOI] [PubMed] [Google Scholar]
- 56.Schneider JG, Amend SR, Weilbaecher KN. Integrins and bone metastasis: integrating tumor cell and stromal cell interactions. Bone. 2010;48:54–65. doi: 10.1016/j.bone.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korah R, Boots M, Wieder R. Integrin α5β1 promotes survival of growth-arrested breast cancer cells: an in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004;64:4514–4522. doi: 10.1158/0008-5472.CAN-03-3853. [DOI] [PubMed] [Google Scholar]
- 58.Liesveld JL, Dipersio JF, Abboud CN. Integrins and adhesive receptors in normal and leukemic CD34+ progenitor cells: potential regulatory checkpoints for cellular traffic. Leuk Lymphoma. 1994;14:19–28. doi: 10.3109/10428199409049647. [DOI] [PubMed] [Google Scholar]
- 59.Lang SH, Clarke NW, George NJ, Testa NG. Primary prostatic epithelial cell binding to human bone marrow stroma and the role of α2β1 integrin. Clin Exp Metastasis. 1997;15:218–227. doi: 10.1023/a:1018465213641. [DOI] [PubMed] [Google Scholar]
- 60.Hall CL, et al. Type I collagen receptor (α2β1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia. 2008;10:797–803. doi: 10.1593/neo.08380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall CL, Dai J, van Golen KL, Keller ET, Long MW. Type I collagen receptor (α2β1) signaling promotes the growth of human prostate cancer cells within the bone. Cancer Res. 2006;66:8648–8654. doi: 10.1158/0008-5472.CAN-06-1544. [DOI] [PubMed] [Google Scholar]
- 62.Mori Y, et al. Anti-α4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood. 2004;104:2149–2154. doi: 10.1182/blood-2004-01-0236. [DOI] [PubMed] [Google Scholar]
- 63.Matsuura N, et al. Induction of experimental bone metastasis in mice by transfection of integrin α4β1 into tumor cells. Am J Pathol. 1996;148:55–61. [PMC free article] [PubMed] [Google Scholar]
- 64.Michigami T, et al. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and α4β1-integrin enhances production of osteoclast-stimulating activity. Blood. 2000;96:1953–1960. [PubMed] [Google Scholar]
- 65.Parmo-Cabanas M, et al. Integrin α4β1 involvement in stromal cell-derived factor-1α-promoted myeloma cell transendothelial migration and adhesion: role of cAMP and the actin cytoskeleton in adhesion. Exp Cell Res. 2004;294:571–580. doi: 10.1016/j.yexcr.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Coleman RE, et al. Predictive value of bone resorption and formation markers in cancer patients with bone metastases receiving the bisphosphonate zoledronic acid. J Clin Oncol. 2005;23:4925–4935. doi: 10.1200/JCO.2005.06.091. [DOI] [PubMed] [Google Scholar]
- 67.Henderson MA, et al. Parathyroid hormone-related protein localization in breast cancers predict improved prognosis. Cancer Res. 2006;66:2250–2256. doi: 10.1158/0008-5472.CAN-05-2814. [DOI] [PubMed] [Google Scholar]
- 68.Yin JJ, et al. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu X, et al. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 2009;23:1882–1894. doi: 10.1101/gad.1824809. This is the first preclinical evidence that blocking TGFβ signalling decreases the development and progression of breast cancer bone metastases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nannuru KC, et al. Matrix metalloproteinase (MMP)-13 regulates mammary tumor-induced osteolysis by activating MMP9 and transforming growth factor-β signaling at the tumor-bone interface. Cancer Res. 2010;70:3494–3504. doi: 10.1158/0008-5472.CAN-09-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19:192–205. doi: 10.1016/j.ccr.2010.12.022. The tumour-derived Notch ligand Jagged1 promotes tumour growth in bone by stimulating IL-6 release from osteoblasts and subsequent osteoclastogenesis. Therefore, secretase inhibitors of the Notch pathway, could prove to be valuable therapeutic drugs for bone metastases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sterling JA, et al. The hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 2006;66:7548–7553. doi: 10.1158/0008-5472.CAN-06-0452. [DOI] [PubMed] [Google Scholar]
- 73.Alexaki VI, et al. GLI2-mediated melanoma invasion and metastasis. J Natl Cancer Inst. 2010;102:1148–1159. doi: 10.1093/jnci/djq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pratap J, et al. Ectopic runx2 expression in mammary epithelial cells disrupts formation of normal acini structure: imimplications for breast cancer progression. Cancer Res. 2009;69:6807–6814. doi: 10.1158/0008-5472.CAN-09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Javed A, et al. Impaired intranuclear trafficking of Runx2 (AML3/CBFA1) transcription factors in breast cancer cells inhibits osteolysis in vivo. Proc Natl Acad Sci USA. 2005;102:1454–1459. doi: 10.1073/pnas.0409121102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hiraga T, Kizaka-Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67:4157–4163. doi: 10.1158/0008-5472.CAN-06-2355. [DOI] [PubMed] [Google Scholar]
- 77.Dunn LK, et al. Hypoxia and TGF-β drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE. 2009;4:e6896. doi: 10.1371/journal.pone.0006896. Hypoxia and TGFβ signalling in parallel drive the development of tumour bone metastases and regulate a common set of tumour genes VEGF and CXCR4. Small-molecule inhibitors, by acting on both tumour cells and the bone microenvironment, additively decrease tumour burden, while improving skeletal quality. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanders JL, et al. Extracellular calcium-sensing receptor expression and its potential role in regulating parathyroid hormone-related peptide secretion in human breast cancer cell lines. Endocrinology. 2000;141:4357–4364. doi: 10.1210/endo.141.12.7849. [DOI] [PubMed] [Google Scholar]
- 79.Yamaguchi T, et al. Expression of extracellular calcium-sensing receptor in human osteoblastic MG-63 cell line. Am J Physiol Cell Physiol. 2001;280:C382–C393. doi: 10.1152/ajpcell.2001.280.2.C382. [DOI] [PubMed] [Google Scholar]
- 80.Yano S, et al. Calcium-sensing receptor activation stimulates parathyroid hormone-related protein secretion in prostate cancer cells: role of epidermal growth factor receptor transactivation. Bone. 2004;35:664–672. doi: 10.1016/j.bone.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 81.Mamillapalli R, Van Houten J, Zawalich W, Wysolmerski J. Switching of G-protein usage by the calcium-sensing receptor reverses its effect on parathyroid hormone-related protein secretion in normal versus malignant breast cells. J Biol Chem. 2008;283:24435–24447. doi: 10.1074/jbc.M801738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liao J, Schneider A, Datta NS, McCauley LK. Extracellular calcium as a candidate mediator of prostate cancer skeletal metastasis. Cancer Res. 2006;66:9065–9073. doi: 10.1158/0008-5472.CAN-06-0317. [DOI] [PubMed] [Google Scholar]
- 83.Saidak Z, et al. Extracellular calcium promotes the migration of breast cancer cells through the activation of the calcium sensing receptor. Exp Cell Res. 2009;315:2072–2080. doi: 10.1016/j.yexcr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 84.Letz S, et al. Novel activating mutations of the calcium-sensing receptor: the calcilytic NPS-2143 mitigates excessive signal transduction of mutant receptors. J Clin Endocrinol Metab. 2010;95:E229–E233. doi: 10.1210/jc.2010-0651. [DOI] [PubMed] [Google Scholar]
- 85.Sangaletti S, et al. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008;68:9050–9059. doi: 10.1158/0008-5472.CAN-08-1327. [DOI] [PubMed] [Google Scholar]
- 86.Shevde LA, Das S, Clark DW, Samant RS. Osteopontin: an effector and an effect of tumor metastasis. Curr Mol Med. 2010;10:71–81. doi: 10.2174/156652410791065381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Contie S, Voorzanger-Rousselot N, Litvin J, Clezardin P, Garnero P. Increased expression and serum levels of the stromal cell-secreted protein periostin in breast cancer bone metastases. Int J Cancer. 2010;128:352–360. doi: 10.1002/ijc.25591. [DOI] [PubMed] [Google Scholar]
- 88.Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nature Rev Cancer. 2008;8:212–226. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sharp JA, Waltham M, Williams ED, Henderson MA, Thompson EW. Transfection of MDA-MB-231 human breast carcinoma cells with bone sialoprotein (BSP) stimulates migration and invasion in vitro and growth of primary and secondary tumors in nude mice. Clin Exp Metastasis. 2004;21:19–29. doi: 10.1023/b:clin.0000017167.17065.61. [DOI] [PubMed] [Google Scholar]
- 90.Carlinfante G, et al. Differential expression of osteopontin and bone sialoprotein in bone metastasis of breast and prostate carcinoma. Clin Exp Metastasis. 2003;20:437–444. doi: 10.1023/a:1025419708343. [DOI] [PubMed] [Google Scholar]
- 91.Chaplet M, et al. Expression of dentin sialophosphoprotein in human prostate cancer and its correlation with tumor aggressiveness. Int J Cancer. 2006;118:850–856. doi: 10.1002/ijc.21442. [DOI] [PubMed] [Google Scholar]
- 92.Bucciarelli E, et al. Low dentin matrix protein 1 expression correlates with skeletal metastases development in breast cancer patients and enhances cell migratory capacity in vitro. Breast Cancer Res Treat. 2007;105:95–104. doi: 10.1007/s10549-006-9436-0. [DOI] [PubMed] [Google Scholar]
- 93.Kelly T, Suva LJ, Nicks KM, MacLeod V, Sanderson RD. Tumor-derived syndecan-1 mediates distal cross-talk with bone that enhances osteoclastogenesis. J Bone Miner Res. 2010;25:1295–1304. doi: 10.1002/jbmr.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Araki K, et al. Decorin suppresses bone metastasis in a breast cancer cell line. Oncology. 2009;77:92–99. doi: 10.1159/000228253. [DOI] [PubMed] [Google Scholar]
- 95.Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nature Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 96.Kang Y, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA. 2005;102:13909–13914. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Javelaud D, et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007;67:2317–2324. doi: 10.1158/0008-5472.CAN-06-3950. [DOI] [PubMed] [Google Scholar]
- 98.Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-β signaling dynamics and therapeutic response in breast cancer bone metastasis. Nature Med. 2009;15:960–966. doi: 10.1038/nm.1943. This is the first direct evidence that a major source of active TGFβ signalling to tumour cells is that released from mineralized bone matrix as a consequence of osteoclastic bone resorption. [DOI] [PubMed] [Google Scholar]
- 99.Schneider A, et al. Bone turnover mediates preferential localization of prostate cancer in the skeleton. Endocrinology. 2005;146:1727–1736. doi: 10.1210/en.2004-1211. [DOI] [PubMed] [Google Scholar]
- 100.Ooi LL, et al. Vitamin D deficiency promotes human breast cancer growth in a murine model of bone metastasis. Cancer Res. 2010;70:1835–1844. doi: 10.1158/0008-5472.CAN-09-3194. Vitamin D deficiency working at least in part via alterations in the bone microenvironment promotes the growth of human breast cancer cells in bone. [DOI] [PubMed] [Google Scholar]
- 101.Hirbe AC, et al. Granulocyte colony-stimulating factor enhances bone tumor growth in mice in an osteoclast-dependent manner. Blood. 2007;109:3424–3431. doi: 10.1182/blood-2006-09-048686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Park BK, et al. NF-κB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nature Med. 2007;13:62–69. doi: 10.1038/nm1519. [DOI] [PubMed] [Google Scholar]
- 103.Hirbe AC, et al. Disruption of CXCR4 enhances osteoclastogenesis and tumor growth in bone. Proc Natl Acad Sci USA. 2007;104:14062–14067. doi: 10.1073/pnas.0705203104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takayama S, et al. The relationship between bone metastasis from human breast cancer and integrin αvβ3 expression. Anticancer Res. 2005;25:79–83. [PubMed] [Google Scholar]
- 105.Boissier S, et al. Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases. Cancer Res. 2000;60:2949–2954. [PubMed] [Google Scholar]
- 106.Croucher PI, et al. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood. 2001;98:3534–3540. doi: 10.1182/blood.v98.13.3534. [DOI] [PubMed] [Google Scholar]
- 107.Gao L, et al. HTLV-1 Tax transgenic mice develop spontaneous osteolytic bone metastases prevented by osteoclast inhibition. Blood. 2005;106:4294–4302. doi: 10.1182/blood-2005-04-1730. This paper describes a spontaneous animal model of hypercalcemia and tumour-associated osteolysis and demonstrates that the viral oncogene Tax is responsible for cellular transformation and induces the expression of osteoclast-activating factors resulting in bone loss and hypercalcemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Desai B, Rogers MJ, Chellaiah MA. Mechanisms of osteopontin and CD44 as metastatic principles in prostate cancer cells. Mol Cancer. 2007;6:18. doi: 10.1186/1476-4598-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nemoto H, et al. Osteopontin deficiency reduces experimental tumor cell metastasis to bone and soft tissues. J Bone Miner Res. 2001;16:652–659. doi: 10.1359/jbmr.2001.16.4.652. [DOI] [PubMed] [Google Scholar]
- 110.Bakewell SJ, et al. Platelet and osteoclast β3 integrins are critical for bone metastasis. Proc Natl Acad Sci USA. 2003;100:14205–14210. doi: 10.1073/pnas.2234372100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rachkovsky M, et al. Melanoma x macrophage hybrids with enhanced metastatic potential. Clin Exp Metastasis. 1998;16:299–312. doi: 10.1023/a:1006557228604. [DOI] [PubMed] [Google Scholar]
- 112.Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nature Rev Cancer. 2008;8:377–386. doi: 10.1038/nrc2371. [DOI] [PubMed] [Google Scholar]
- 113.Andersen TL, et al. Osteoclast nuclei of myeloma patients show chromosome translocations specific for the myeloma cell clone: a new type of cancer-host partnership? J Pathol. 2007;211:10–17. doi: 10.1002/path.2078. [DOI] [PubMed] [Google Scholar]
- 114.Vignery A. Macrophage fusion: are somatic and cancer cells possible partners? Trends Cell Biol. 2005;15:188–193. doi: 10.1016/j.tcb.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 115.Lu X, Kang Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc Natl Acad Sci USA. 2009;106:9385–9390. doi: 10.1073/pnas.0900108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abe M, et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood. 2004;104:2484–2491. doi: 10.1182/blood-2003-11-3839. [DOI] [PubMed] [Google Scholar]
- 117.Gonzalez-Suarez E, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010;468:103–107. doi: 10.1038/nature09495. RANKL and RANK are essential for mammary gland development in mice and mediate progesterone-induced proliferation in the mammary gland. This paper highlights the bone-independent role of RANKL that may provide new targeted approaches for breast cancer therapy. [DOI] [PubMed] [Google Scholar]
- 118.Clarke NW, McClure J, George NJ. Morphometric evidence for bone resorption and replacement in prostate cancer. Br J Urol. 1991;68:74–80. doi: 10.1111/j.1464-410x.1991.tb15260.x. [DOI] [PubMed] [Google Scholar]
- 119.Roudier MP, et al. Histopathological assessment of prostate cancer bone osteoblastic metastases. J Urol. 2008;180:1154–1160. doi: 10.1016/j.juro.2008.04.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39:246–261. doi: 10.1002/(sici)1097-0045(19990601)39:4<246::aid-pros5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 121.Dudley AC, et al. Calcification of multipotent prostate tumor endothelium. Cancer Cell. 2008;14:201–211. doi: 10.1016/j.ccr.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Odero-Marah VA, et al. Receptor activator of NF-κB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008;18:858–870. doi: 10.1038/cr.2008.84. [DOI] [PubMed] [Google Scholar]
- 123.Brubaker KD, Vessella RL, Brown LG, Corey E. Prostate cancer expression of runt-domain transcription factor Runx2, a key regulator of osteoblast differentiation and function. Prostate. 2003;56:13–22. doi: 10.1002/pros.10233. [DOI] [PubMed] [Google Scholar]
- 124.Leong DT, et al. Cancer-related ectopic expression of the bone-related transcription factor RUNX2 in non-osseous metastatic tumor cells is linked to cell proliferation and motility. Breast Cancer Res. 2010;12:R89. doi: 10.1186/bcr2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Akech J, et al. Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene. 2010;29:811–812. doi: 10.1038/onc.2009.389. RUNX2, traditionally known to be osteoblast specific, was found to be highly expressed in metastatic prostate cancer cells and associated with tumour growth in bone. This highlights the osteomimicry of tumour cells that metastasize to bone and provides a new target to consider for therapeutic development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chackal-Roy M, Niemeyer C, Moore M, Zetter BR. Stimulation of human prostatic carcinoma cell growth by factors present in human bone marrow. J Clin Invest. 1989;84:43–50. doi: 10.1172/JCI114167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gleave M, Hsieh JT, Gao CA, von Eschenbach AC, Chung LW. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Res. 1991;51:3753–3761. [PubMed] [Google Scholar]
- 128.Cooper CR, et al. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97:739–747. doi: 10.1002/cncr.11181. [DOI] [PubMed] [Google Scholar]
- 129.De Palma M, Naldini L. Role of haematopoietic cells and endothelial progenitors in tumour angiogenesis. Biochim Biophys Acta. 2006;1766:159–166. doi: 10.1016/j.bbcan.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 130.Stockmann C, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–818. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Silva R, D’Amico G, Hodivala-Dilke KM, Reynolds LE. Integrins: the keys to unlocking angiogenesis. Arterioscler Thromb Vasc Biol. 2008;28:1703–1713. doi: 10.1161/ATVBAHA.108.172015. [DOI] [PubMed] [Google Scholar]
- 132.Prantl L, et al. Adipose tissue-derived stem cells promote prostate tumor growth. Prostate 70, 1709–1715. 2010 doi: 10.1002/pros.21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Klopp AH, et al. Mesenchymal stem cells promote mammosphere formation and decrease E-cadherin in normal and malignant breast cells. PLoS ONE. 2010;5:e12180. doi: 10.1371/journal.pone.0012180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Studeny M, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96:1593–1603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 135.Bianco P, Robey PG, Saggio I, Riminucci M. “Mesenchymal” stem cells in human bone marrow (skeletal stem cells): a critical discussion of their nature, identity, and significance in incurable skeletal disease. Hum Gene Ther. 2010;21:1057–1066. doi: 10.1089/hum.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang L, Edwards CM, Mundy GR. Gr-1+CD11b+ myeloid-derived suppressor cells: formidable partners in tumor metastasis. J Bone Miner Res. 2010;25:1701–1706. doi: 10.1002/jbmr.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 138.Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol. 2006;80:1183–1196. doi: 10.1189/jlb.0905495. [DOI] [PubMed] [Google Scholar]
- 139.Gabrilovich DI, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. author reply 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vaira S, et al. RelB is the NF-κB subunit downstream of NIK responsible for osteoclast differentiation. Proc Natl Acad Sci USA. 2008;105:3897–3902. doi: 10.1073/pnas.0708576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007;67:11438–11446. doi: 10.1158/0008-5472.CAN-07-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hirbe AC, et al. The bisphosphonate zoledronic acid decreases tumor growth in bone in mice with defective osteoclasts. Bone. 2009;44:908–916. doi: 10.1016/j.bone.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nature Rev Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 144.Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nature Rev Rheumatol. 2009;5:667–676. doi: 10.1038/nrrheum.2009.217. [DOI] [PubMed] [Google Scholar]
- 145.Sato K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nature Rev Cancer. 2011;11:123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Boucharaba A, et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest. 2004;114:1714–1725. doi: 10.1172/JCI22123. This paper shows that lysophosphatidic acid derived from activated platelets can promote tumour growth and osteolysis in a mouse model of bone metastases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Uluçkan Ö, et al. APT102, a novel adpase, cooperates with aspirin to disrupt bone metastasis in mice. J Cell Biochem. 2008;104:1311–1323. doi: 10.1002/jcb.21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li X, et al. Inhibitory effects of megakaryocytic cells in prostate cancer skeletal metastasis. J Bone Miner Res. 2010;26:125–134. doi: 10.1002/jbmr.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Beeton CA, Bord S, Ireland D, Compston JE. Osteoclast formation and bone resorption are inhibited by megakaryocytes. Bone. 2006;39:985–990. doi: 10.1016/j.bone.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 151.Chollet ME, et al. Evidence of a colocalisation of osteoprotegerin (OPG) with von Willebrand factor (VWF) in platelets and megakaryocytes α granules. Studies from normal and grey platelets. Br J Haematol. 2009;148:805–807. doi: 10.1111/j.1365-2141.2009.07989.x. [DOI] [PubMed] [Google Scholar]
- 152.Lemieux JM, Horowitz MC, Kacena MA. Involvement of integrins α3β1 and α5β1 and glycoprotein IIb in megakaryocyte-induced osteoblast proliferation. J Cell Biochem. 2010;109:927–932. doi: 10.1002/jcb.22468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bernasconi S, et al. Effects of granulocyte-monocyte colony-stimulating factor (GM-CSF) on expression of adhesion molecules and production of cytokines in blood monocytes and ovarian cancer-associated macrophages. Int J Cancer. 1995;60:300–307. doi: 10.1002/ijc.2910600304. [DOI] [PubMed] [Google Scholar]
- 154.Zaslavsky A, et al. Platelet-derived thrombospondin-1 is a critical negative regulator and potential biomarker of angiogenesis. Blood. 2010;115:4605–4613. doi: 10.1182/blood-2009-09-242065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fidler IJ. Metastasis: guantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2′-deoxyuridine. J Natl Cancer Inst. 1970;45:773–782. [PubMed] [Google Scholar]
- 156.Luzzi KJ, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Braun S, et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med. 2000;342:525–533. doi: 10.1056/NEJM200002243420801. [DOI] [PubMed] [Google Scholar]
- 158.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 159.Morgan TM, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res. 2009;15:677–683. doi: 10.1158/1078-0432.CCR-08-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Diel IJ, et al. Micrometastatic breast cancer cells in bone marrow at primary surgery: prognostic value in comparison with nodal status. J Natl Cancer Inst. 1996;88:1652–1658. doi: 10.1093/jnci/88.22.1652. [DOI] [PubMed] [Google Scholar]
- 161.Coleman RE, et al. Advancing treatment for metastatic bone cancer: consensus recommendations from the Second Cambridge Conference. Clin Cancer Res. 2008;14:6387–6395. doi: 10.1158/1078-0432.CCR-08-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Watson MA, et al. Isolation and molecular profiling of bone marrow micrometastases identifies TWIST1 as a marker of early tumor relapse in breast cancer patients. Clin Cancer Res. 2007;13:5001–5009. doi: 10.1158/1078-0432.CCR-07-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang XH, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rack B, et al. Effect of zoledronate on persisting isolated tumour cells in patients with early breast cancer. Anticancer Res. 2010;30:1807–1813. [PubMed] [Google Scholar]
- 165.Shiozawa Y, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12:116–127. doi: 10.1593/neo.91384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rogers MJ. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des. 2003;9:2643–2658. doi: 10.2174/1381612033453640. [DOI] [PubMed] [Google Scholar]
- 167.Gnant M, et al. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N Engl J Med. 2009;360:679–691. doi: 10.1056/NEJMoa0806285. [DOI] [PubMed] [Google Scholar]
- 168.Stopeck AT, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol. 2010;28:5132–5139. doi: 10.1200/JCO.2010.29.7101. [DOI] [PubMed] [Google Scholar]
- 169.Jensen AB, et al. The cathepsin K inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: results of a 4-week, double-blind, randomized, controlled trial. Clin Breast Cancer. 2010;10:452–458. doi: 10.3816/CBC.2010.n.059. [DOI] [PubMed] [Google Scholar]
- 170.Rucci N, Susa M, Teti A. Inhibition of protein kinase c-Src as a therapeutic approach for cancer and bone metastases. Anticancer Agents Med Chem. 2008;8:342–349. doi: 10.2174/187152008783961905. [DOI] [PubMed] [Google Scholar]
- 171.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature Rev Cancer. 2009;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Korpal M, Kang Y. Targeting the transforming growth factor-β signalling pathway in metastatic cancer. Eur J Cancer. 2010;46:1232–1240. doi: 10.1016/j.ejca.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 173.Balooch G, et al. TGF-β regulates the mechanical properties and composition of bone matrix. Proc Natl Acad Sci USA. 2005;102:18813–18818. doi: 10.1073/pnas.0507417102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mohammad KS, et al. Pharmacologic inhibition of the TGF-β type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS ONE. 2009;4:e5275. doi: 10.1371/journal.pone.0005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Edwards JR, et al. Inhibition of TGF-β signaling by 1D11 antibody treatment increases bone mass and quality in vivo. J Bone Miner Res. 2010;25:2419–2426. doi: 10.1002/jbmr.139. [DOI] [PubMed] [Google Scholar]
- 176.Buijs JT, et al. Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res. 2007;67:8742–8751. doi: 10.1158/0008-5472.CAN-06-2490. [DOI] [PubMed] [Google Scholar]
- 177.Buijs JT, et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am J Pathol. 2007;171:1047–1057. doi: 10.2353/ajpath.2007.070168. In normal epithelial cells, BMP7 and TGFβ prevent EMT. Loss of BMP7 expression in tumours results in reduced E-cadherin expression, EMT and metastases to bone. This model re-emphasizes the ‘double-edged sword’ properties and the complexity of TGFβ superfamily members in cancer biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gautschi OP, Frey SP, Zellweger R. Bone morphogenetic proteins in clinical applications. ANZ J Surg. 2007;77:626–631. doi: 10.1111/j.1445-2197.2007.04175.x. [DOI] [PubMed] [Google Scholar]
- 179.Prud’homme GJ. Pathobiology of transforming growth factor β in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest. 2007;87:1077–1091. doi: 10.1038/labinvest.3700669. [DOI] [PubMed] [Google Scholar]
- 180.Nelson JB, et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nature Med. 1995;1:944–949. doi: 10.1038/nm0995-944. [DOI] [PubMed] [Google Scholar]
- 181.Yin JJ, et al. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci USA. 2003;100:10954–10959. doi: 10.1073/pnas.1830978100. This is the first preclinical evidence that endothelin receptor blockade reduces the development and progression of osteoblastic bone metastases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Carducci MA, Jimeno A. Targeting bone metastasis in prostate cancer with endothelin receptor antagonists. Clin Cancer Res. 2006;12:6296s–6300s. doi: 10.1158/1078-0432.CCR-06-0929. [DOI] [PubMed] [Google Scholar]
- 183.Clines GA, et al. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol Endocrinol. 2007;21:486–498. doi: 10.1210/me.2006-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shepard DR, Dreicer R. Zibotentan for the treatment of castrate-resistant prostate cancer. Expert Opin Investig Drugs. 2010;19:899–908. doi: 10.1517/13543784.2010.491822. [DOI] [PubMed] [Google Scholar]
- 185.Nervi B, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206–6214. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Azab AK, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Peyruchaud O, Serre CM, NicAmhlaoibh R, Fournier P, Clezardin P. Angiostatin inhibits bone metastasis formation in nude mice through a direct anti-osteoclastic activity. J Biol Chem. 2003;278:45826–45832. doi: 10.1074/jbc.M309024200. [DOI] [PubMed] [Google Scholar]
- 188.Twardowski PW, et al. Biologic markers of angiogenesis: circulating endothelial cells in patients with advanced malignancies treated on phase I protocol with metronomic chemotherapy and celecoxib. Cancer Invest. 2008;26:53–59. doi: 10.1080/07357900701681541. [DOI] [PubMed] [Google Scholar]
- 189.Merz M, Komljenovic D, Zwick S, Semmler W, Bauerle T. Sorafenib tosylate and paclitaxel induce anti-angiogenic, anti-tumour and anti-resorptive effects in experimental breast cancer bone metastases. Eur J Cancer. 2010;47:277–286. doi: 10.1016/j.ejca.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 190.Gao D, et al. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 191.Mellick AS, et al. Using the transcription factor inhibitor of DNA binding 1 to selectively target endothelial progenitor cells offers novel strategies to inhibit tumor angiogenesis and growth. Cancer Res. 2010;70:7273–7282. doi: 10.1158/0008-5472.CAN-10-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kai L, et al. Targeting prostate cancer angiogenesis through metastasis-associated protein 1 (MTA1) Prostate. 2011;71:268–280. doi: 10.1002/pros.21240. [DOI] [PubMed] [Google Scholar]
- 193.Meuser T, et al. Symptoms during cancer pain treatment following WHO-guidelines: a longitudinal follow-up study of symptom prevalence, severity and etiology. Pain. 2001;93:247–257. doi: 10.1016/S0304-3959(01)00324-4. [DOI] [PubMed] [Google Scholar]
- 194.Goblirsch MJ, Zwolak PP, Clohisy DR. Biology of bone cancer pain. Clin Cancer Res. 2006;12:6231s–6235s. doi: 10.1158/1078-0432.CCR-06-0682. [DOI] [PubMed] [Google Scholar]
- 195.Honore P, et al. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nature Med. 2000;6:521–528. doi: 10.1038/74999. Bone pain that is associated with bone metastasis is difficult to treat. This report demonstrates that osteoclastic resorption at the site of a tumour deposit in bone recruits pain fibres and changes the spinal chord neurochemical composition to promote bone pain. Blockade of osteoclastic resorption reduced pain, spinal chord reorganziation and nerve recruitment associated with bone tumours. [DOI] [PubMed] [Google Scholar]
- 196.Nakanishi M, et al. Acid activation of Trpv1 leads to an up-regulation of calcitonin gene-related peptide expression in dorsal root ganglion neurons via the CaMK-CREB cascade: a potential mechanism of inflammatory pain. Mol Biol Cell. 2010;21:2568–2577. doi: 10.1091/mbc.E10-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.McEwan AJ. Use of radionuclides for the palliation of bone metastases. Semin Radiat Oncol. 2000;10:103–114. doi: 10.1016/s1053-4296(00)80047-8. [DOI] [PubMed] [Google Scholar]
- 198.Ding L, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hall CL, Kang S, MacDougald OA, Keller ET. Role of Wnts in prostate cancer bone metastases. J Cell Biochem. 2006;97:661–672. doi: 10.1002/jcb.20735. [DOI] [PubMed] [Google Scholar]
- 200.Voorzanger-Rousselot N, et al. Increased Dickkopf-1 expression in breast cancer bone metastases. Br J Cancer. 2007;97:964–970. doi: 10.1038/sj.bjc.6603959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tian E, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. This paper shows that the osteoblast inhibitor DKK1 is expressed by myeloma cells and is detected at higher levels in patients with myeloma who have osteolytic lesions. The discovery that myeloma cells express osteoblast inhibitors explains why bone loss is so severe in this disease. [DOI] [PubMed] [Google Scholar]
- 202.Dai J, et al. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res. 2005;65:8274–8285. doi: 10.1158/0008-5472.CAN-07-6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Giuliani N, et al. Production of Wnt inhibitors by myeloma cells: potential effects on canonical Wnt pathway in the bone microenvironment. Cancer Res. 2007;67:7665–7674. doi: 10.1158/0008-5472.CAN-06-4666. [DOI] [PubMed] [Google Scholar]
- 204.Hjertner O, et al. Hepatocyte growth factor (HGF) induces interleukin-11 secretion from osteoblasts: a possible role for HGF in myeloma-associated osteolytic bone disease. Blood. 1999;94:3883–3888. [PubMed] [Google Scholar]
- 205.Oshima T, et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood. 2005;106:3160–3165. doi: 10.1182/blood-2004-12-4940. [DOI] [PubMed] [Google Scholar]
- 206.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hauge EM, Qvesel D, Eriksen EF, Mosekilde L, Melsen F. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J Bone Miner Res. 2001;16:1575–1582. doi: 10.1359/jbmr.2001.16.9.1575. [DOI] [PubMed] [Google Scholar]
- 208.Pettit AR, Chang MK, Hume DA, Raggatt LJ. Osteal macrophages: a new twist on coupling during bone dynamics. Bone. 2008;43:976–982. doi: 10.1016/j.bone.2008.08.128. [DOI] [PubMed] [Google Scholar]
- 209.Baud’huin M, et al. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J Pathol. 2010;221:77–86. doi: 10.1002/path.2684. [DOI] [PubMed] [Google Scholar]
- 210.Catena R, et al. PDGFR signalling blockade in marrow stroma impairs lung cancer bone metastasis. Cancer Res. 2011;71:164–174. doi: 10.1158/0008-5472.CAN-10-1708. [DOI] [PubMed] [Google Scholar]