

Abstract
Fibroblasts play a major role in normal cardiac physiology and in the response of the heart to injury and disease. Cardiac electrophysiological research has primarily focused on the mechanisms of remodeling that accompany cardiac disease with an emphasis on myocyte electrophysiology. Recently, there has been increasing interest in the potential role of fibroblasts in cardiac electrophysiology. This review focuses on the arrhythmia mechanisms involving interactions between myocytes and fibroblasts. We also discuss the available evidence supporting the contribution of intracardiac and extracardiac sources to the fibroblast and myofibroblast populations in diseased hearts.
Keywords: Bone marrow-derived cells, Cardiac arrhythmia, Electrophysiology, Endothelial cells, Endothelial-to-mesenchymal transition, Epicardial cells, Epithelial-to-mesenchymal transition, Fibroblasts, Myofibroblasts
Introduction
Cardiovascular disease remains a major cause of death in the United States. The lifetime risk of developing coronary artery disease after the age of 40 is 49 % for men and 32 % for women [1]. Most of these deaths are associated with the development of malignant ventricular arrhythmias, yet the underlying mechanisms responsible for initiation and maintenance of ventricular arrhythmias remain poorly understood. Cardiac electrophysiological research has primarily focused on the mechanisms of remodeling that accompany cardiac disease with an emphasis on myocyte electrophysiology. Recently, there has been increasing interest in the potential contribution of the non-excitable cell populations to cardiac electrophysiology and arrhythmia mechanisms [2–17]. Fibroblasts are the major non-myocyte cell type and contribute to the structural, mechanical, and electrophysiological properties of the heart [18–20]. Fibrosis is present in a variety of cardiac diseases associated with a high incidence of arrhythmias. The predominant cellular mechanism of fibrosis involves the emergence of activated fibroblasts or myofibroblasts. Traditionally thought to originate from proliferation and activation of resident fibroblasts, recent studies have demonstrated myofibroblasts can also originate from other intracardiac and extracardiac sources [21–42]. Fibroblasts are known to be a heterogeneous population of cells, and it is currently unknown if phenotypic differences are associated with these different origins [43–50]. This review focuses on the contribution of fibroblasts to cardiac electrophysiology and arrhythmia mechanisms. We also discuss available evidence supporting the contribution of resident and non-resident sources to the fibroblast and myofibroblast populations in diseased hearts.
Cardiac Fibroblast Functions
Fibroblasts are highly responsive to the chemical and mechanical environment of the heart. Figure 1 summarizes the primary factors affecting fibroblasts and their major functional responses. The primary function of fibroblasts in the normal heart is maintenance of the extracellular matrix which requires tight regulation of synthesis and degradation pathways [51]. Fibroblasts synthesize fibrillar collagen as a precursor polypeptide which is further processed after cellular export and crosslinked to form mature collagen. Fibroblast collagen synthesis is transcriptionally regulated by fibrogenic growth factors including transforming growth factor β (TGFβ) [51]. Fibroblasts also coordinate degradation of collagen through secretion of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases.
Fig. 1.
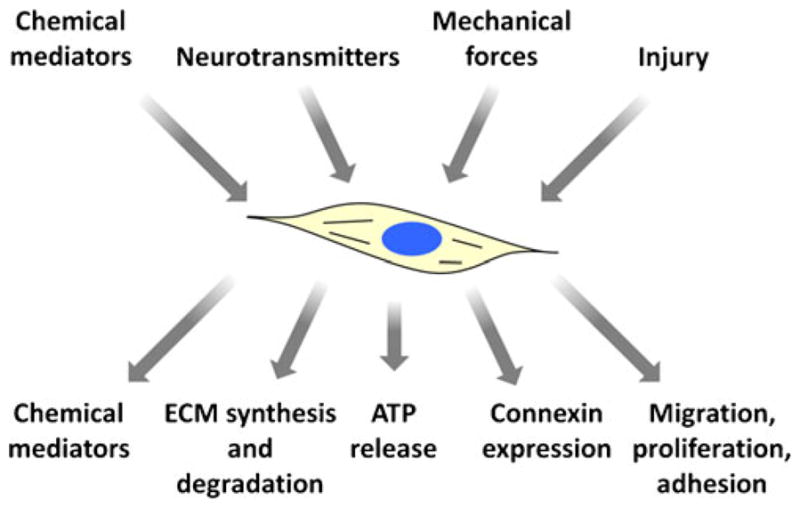
Functions of cardiac fibroblasts. Fibroblast function is affected by a number of stimuli including chemical mediators, neurotransmitters, mechanical forces, and injury. Fibroblasts respond to stimuli by releasing chemical mediators and altering the synthesis and degradation of extracellular matrix (ECM) proteins, ATP release, connexin expression, migration, proliferation, and adhesion properties
Cardiac myocytes have limited regenerative capabilities in response to cardiac disease and injury as dead and apoptotic myocytes are replaced with fibrotic tissue. Fibroblasts play a major role in the response to cardiac injury and the development of fibrosis. Critical to the injury response is the emergence of activated fibroblasts or myofibroblasts which are not present in the normal cardiac muscle. The emergence of myofibroblasts is strongly promoted by TGF-β1 [52]. Myofibroblasts have characteristics that are intermediate between fibroblasts and smooth muscle cells, express α-smooth muscle actin (α-SMA) and contractile proteins, and have higher rates of cellular proliferation and extracellular matrix deposition compared to fibroblasts [53–55]. The ultrastructural features of myofibroblasts include myofilaments, well-developed rough endoplasmic reticulum, and extensive cell–matrix contacts [56–58]. During wound healing, myofibroblasts strengthen injured tissue by providing additional extracellular collagen.
Cardiac tissue repair in response to myocardial infarction is a dynamic process and includes homeostasis, infiltration of immune and inflammatory cells, degradation and phagocytosis of dying cells and debris, repopulation of the zone of injury with fibroblasts and myofibroblasts, extracellular matrix remodeling, and formation of a mature scar [51, 59]. Fibroblasts and myofibroblasts are essential during all phases of tissue repair. During the initial injury response, fibroblasts and myofibroblasts secrete matrix metalloproteinases and other cytokines to facilitate recruitment of other fibroblasts to the injured region and facilitate the degradation of extracellular matrix. Recent studies have demonstrated that cardiac fibroblasts release ATP into the extracellular space through connexin hemichannels that could further activates profibrotic cellular responses through the MAPK pathway [60]. Following the initial injury response phase, fibroblasts infiltrate the injured region and secrete extracellular matrix proteins to maintain the structural integrity of the affected area and facilitate the formation of scar tissue. Termination of the injury response occurs following the establishment of a mature scar through apoptosis of activated fibroblasts. Unlike the injury response in other tissues, a small number of cardiac myofibroblasts are still present in the scar region several years after injury [61–63].
Origin of Fibroblasts
During heart development, cardiac fibroblasts originate from mesenchymal cells which are principally derived from the embryonic epicardium [64–68]. Other sources of fibroblasts have been proposed including endocardial cushion, epithelial-to-mesenchymal transformation [69], and postnatal recruitment of circulating bone marrow cells [70]. Fibroblasts and myofibroblasts in injured hearts originate from intracardiac and extracardiac sources including expansion and activation of resident fibroblasts, epicardial cells that undergo epithelial-to-mesenchymal transition, endothelial cells that undergo transformation to mesenchymal cells, and circulating bone marrow-derived progenitor cells [24, 25, 27, 28, 30, 31, 40, 71–73] (see Fig. 2).
Fig. 2.
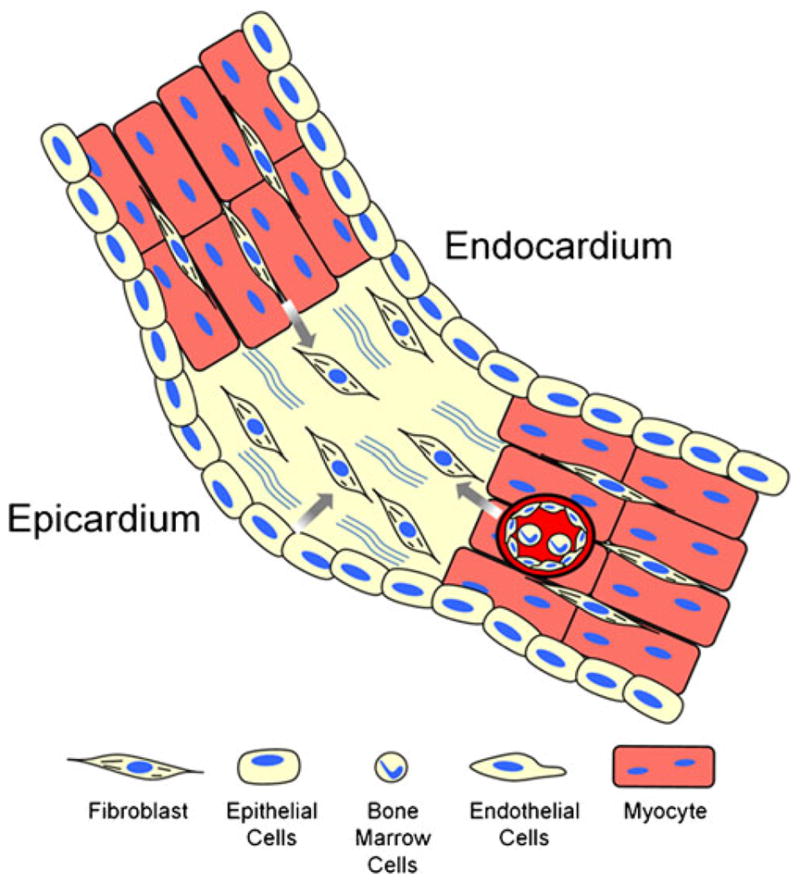
Cellular sources that contribute to the cardiac fibroblast population following injury. In response to cardiac injury, resident cardiac fibroblasts proliferate and migrate to the site of injury. Fibroblasts in injured sites are also derived from transformed epicardial, endothelial, and bone marrow sources
Intracardiac Sources of Fibroblasts
Resident Fibroblasts
It has been widely believed that a significant number of myofibroblasts present in fibrotic hearts are derived from the resident fibroblast population. Initial studies investigating the contribution of non-resident fibroblast sources supported this idea [22]. However, recently, several studies have challenged this concept by demonstrating that multiple cellular lineages contribute to a significant percentage of the cardiac fibroblast population under pathological conditions [24, 25, 27, 28, 30, 31, 40, 71–73]. The ability to determine the fate and contribution of resident cardiac fibroblasts under pathological conditions has been limited by the lack of specific markers for resident fibroblasts. Evidence supporting resident fibroblast contribution to the cardiac fibroblast population under pathological conditions has been obtained from studies of chronic cardiac transplant rejection [26, 29]. In a rat heart transplantation model, fibroblasts from allografts undergoing chronic rejection were shown to be derived from recipient (extracardiac) and donor (intracardiac) sources [29]. More recently, studies using endomyocardial biopsy samples from areas of increased cardiac fibrosis in heart transplant patients have indicated that fibroblasts are mainly derived from intracardiac sources [26]. These studies did not differentiate between fibroblast and myofibroblast populations. There were several important differences between these studies including the use of immunosuppression drugs in human patients and the use of antibodies specific to macrophages to better differentiate those cells from fibroblasts. Importantly, these studies did not exclude the possibility that a subpopulation of cardiac fibroblasts may have originated from the intracardiac sources discussed below.
Epithelial-to-Mesenchymal Transition-Derived Fibroblasts
The epicardium is the outermost layer of the heart and is composed of epithelial cells. During development, epicardial cells undergo epithelial-to-mesenchymal transition and give rise to multiple cell types including fibroblasts, smooth muscle cells, and endothelial cells. In the adult normal heart, epicardial cells are quiescent and do not undergo epithelial-to-mesenchymal transformation [41, 42]. However, recent studies have indicated that the epicardium is responsive to cardiac injury and plays an important role in cardiac repair [35, 39, 41, 42]. In vitro studies have shown that human epicardial cells spontaneously undergo epithelial-to-mesenchymal transition and the antigen profile of the transformed cells resembles subepithelial fibroblasts [39]. More recently, studies using animal models of myocardial infarction have shown that cardiac injury leads to epicardial cell proliferation and stimulates the formation of epicardium derived cells [35, 41, 42]. Epicardium-derived cells form a thick layer on the surface of the heart over the injured region and adopt fibroblast, myofibroblast, and smooth muscle cell phenotypes. The epicardial expansion and epithelial-to-mesenchymal transition have been shown to be dependent on Wnt1 signaling [35]. Additional studies are needed to determine whether Wnt1 signaling differentially affects the epithelial-to-mesenchymal transition-derived fibroblast and myofibroblast populations.
Endothelial-to-Mesenchymal Transition-Derived Fibroblasts
Endothelial cells line the interior surface of blood vessels and are in direct contact with the circulating cells of the blood. During development, endothelial cells undergo endothelial-to-mesenchymal transition and give rise to the atrioventricular cushion, the primordial of the valves and septa [74]. In the normal adult heart, endothelial cells do not significantly contribute to the cardiac fibroblast population [32, 33]. However, in response to cardiac disease or injury, endothelial cells undergo endothelial-to-mesenchymal transition and give rise to fibrosis and the accumulation of a significant number of fibroblasts and myofibroblasts [31, 32, 37, 40].
The percentage of total fibroblasts derived from endothelial sources may be dependent on the pathological stimulus. In a pressure overload model of heart failure, between 27 % and 35 % of the total fibroblast population is derived from endothelial cells [31]. A smaller percentage of fibroblasts of endothelial origin, 15 % to 20 % of all fibroblasts, were reported in a streptozotocin-induced diabetes model [40]. Differences in the number of myofibroblast derived from endothelial sources have also been reported. In pressure-overloaded hearts, approximately 75 % of myofibroblasts have endothelial origin [31]. In animal models of myocardial infarction, between 35 % and 40 % of α-SMA positive cells, some of which are likely to be myofibroblasts, are derived from endothelial cells [32].
Endothelial-to-mesenchymal transition to fibroblasts and myofibroblasts has been shown to be induced by TGF-β1 in a Smad3-dependent manner and inhibited by bone morphogenetic protein 7 and hepatocyte growth factor [31, 37]. Endothelin-1 can also stimulate endothelial cells to undergo endothelial-to-mesenchymal transition through inhibition of TGF-β1 signaling [40]. Activation of Wnt signaling causes endothelial cells to undergo endothelial-to-mesenchymal transformation, and the transformed cells can be identified by Wnt activity [32].
Extracardiac Sources of Fibroblasts
Bone Marrow-Derived Fibroblasts
Bone marrow-derived cells are recruited to the heart following cardiac injury and play a critical role in inflammation and cardiac repair [23–25, 27, 28, 30]. Several studies have demonstrated that a significant number of fibroblast and myofibroblasts in the infarcted myocardium are derived from bone marrow cells [23–25, 27, 28, 30]. In animal models of myocardial infarction, bone marrow-derived fibroblasts and myofibroblasts densities peak between 1 to 3 weeks after injury. During this period, bone marrow-derived fibroblasts represent between 25 % and 30 % of the fibroblast population, while bone marrow-derived myofibroblasts represent between 24 % and 57 % of the myofibroblast population in the injured region [24, 25, 28]. Studies have shown that the percent of fibroblast that are bone marrow-derived remains relatively constant for up to 4 months [25]. On the other hand, the percent of bone marrow-derived myofibroblasts decreases after 3 weeks and remains constant for at least 4 months [24, 25]. Bone marrow-derived fibroblasts and myofibroblasts are absent from areas remote from the infarct or perinfarcted regions [24, 28]. Recent studies have demonstrated that bone marrow-derived cells are actively replaced in the infarcted heart, and this process continues for several months after injury [27]. Specifically, bone marrow-derived fibroblasts in the injured region are turned over within 2 weeks after infarction [27]. Bone marrow-derived myofibroblasts participate actively in scar formation by producing collagen I in the injured region [28]. Significant numbers of bone marrow-derived fibroblasts and myofibroblasts have also been identified in senescent animals and other models of cardiac disease [23, 31, 34, 38].
Arrhythmogenic Potential of Cardiac Fibroblasts
Fibrosis leads to changes in the mechanical properties of the heart, disrupts electrical connectivity between myocytes, and impairs myocyte oxygen availability. Fibrosis and interactions between fibroblasts and myocytes can alter cardiac electrophysiology and contribute to arrhythmia formation through a number of mechanisms illustrated in Fig. 3. Traditionally, fibroblasts and fibrosis are considered to alter cardiac electrophysiology by mechanically separating myocytes and creating barriers to propagation of the electrical impulse (Fig. 3a) [75–77]. Such conduction disturbances can produce conduction delay or block that leads to the formation of functional reentry.
Fig. 3.
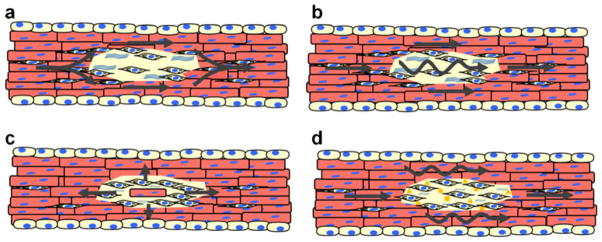
Arrhythmogenic mechanisms of fibroblasts myocyte interactions. a Fibrotic regions act as physical or functional barriers to electrical conduction. Electrical wavefronts that encounter such regions break at the proximal side and reconnect on the distal side. b Fibroblast–myocyte electrical coupling allows for slow electrotonic conduction through fibrotic regions. c Partial electrical isolation of myocytes can lead to successful electrical conduction of impulses originating from ectopic foci. d Chemical mediators released from fibroblasts can exert paracrine effects on myocytes leading to slow conduction
There is increasing evidence that fibroblasts may also actively contribute to cardiac electrophysiology and arrhythmogenesis through direct electrical coupling and paracrine mechanisms [2–17]. Fibroblasts are electrically passive cells that do not express sodium or other excitatory currents [50]. In addition, fibroblasts have high input impedances and more depolarized resting membrane potentials compared to cardiac myocytes [50]. In vitro and in vivo evidence suggests that fibroblasts and myocytes can electrically couple and that fibroblasts can affect myocyte resting potential and action potential parameters [6–9, 78, 79]. Together, these findings indicate that fibroblasts can directly modulate cardiac electrophysiology and surrounding myocytes through electrotonic mechanisms. These interactions can result in a number of conduction disturbances. Myocyte–fibroblast interactions facilitate conduction by allowing electrotonic propagation of the electrical activation between physically separated myocytes (Fig. 3b). Increasing densities of fibroblasts favor conduction slowing and eventually conduction block [6–8]. Recent studies have suggested that pharmacological disruption of the actin cytoskeleton can decrease these effects [3]. Fibroblasts can also contribute to arrhythmia initiation by influencing automaticity [5, 9, 80]. Partial electrical isolation of spontaneously active cells by coupling to fibroblasts can facilitate successful conduction from ectopic foci (Fig. 3c). Recent studies have also demonstrated that fibroblast–myocyte interactions can result in depolarization-induced automaticity in surrounding myocytes. Fibroblast can also increase automaticity by increasing the effect of ionic and oxidative stress on myocytes [12].
Studies have shown that fibroblasts can also alter myocyte electrophysiological properties through paracrine mechanisms (Fig. 3d). It is well-established cardiac fibroblasts release factors that contribute to pathological remodeling, and it is becoming increasing clear that these factors can also contribute to arrhythmogenesis [10, 51, 81, 82]. Paracrine factors released by cardiac fibroblasts have been shown to modulate myocyte action potential parameters and conduction properties [8, 10]. In addition, studies from our laboratory have demonstrated that the paracrine effects of fibroblasts on myocyte electrophysiology are enhanced following cardiac injury [8]. Fibroblast paracrine effects are dose dependent and partially reversible. Gene expression studies have indicated that these changes are the result of a reduced expression of fast sodium current, inward rectifier current, and the transient outward potassium current in myocytes treated with fibroblast paracrine factors [10]. Myocyte connexin43 (Cx43) expression, phosphorylation, and function are unaffected by fibroblast paracrine factors [10].
Traditionally, fibroblasts have been considered to be a homogenous population of cells. However, recent data support the idea that fibroblasts are phenotypically distinct depending on developmental stage, organ, and physiological conditions [8, 43–50]. This heterogeneity, particularly potential differences in ionic currents and connexin expression, has important implications with regards to modulation of the arrhythmogenic substrate. Studies of fibroblast connexin expression during pathological conditions have shown that fibroblasts in ventricular infarct scar tissue express Cx43 or connexin45 (Cx45) with spatially and temporally distinct patterns [83]. Connexin 40 (Cx40) has not been identified in these cells. Fibroblasts expressing Cx45 infiltrate damaged tissue within the first few hours after infarction, reach their peak density within 6 days, and decrease thereafter. The number of Cx43 expressing fibroblasts starts increasing 6 days after infarction and continues to rise until at least the fourth week. These data suggest that Cx45 may be responsible for electrical coupling between fibroblasts and myocytes during the acute remodeling process, while Cx43 may be involved at later stages. It is currently unknown whether these cells represent a single fibroblast population that initially expresses Cx45 and then Cx43, or if there are multiple fibroblast populations involved. The latter is supported by evidence showing that Cx45 and Cx43 do not colocalize. Recent studies have demonstrated that connexin levels and coupling to myocytes are elevated following cardiac injury which increases the potential of fibroblasts to influence myocyte electrophysiology [8, 84]. It is intriguing to speculate that the populations may correspond to quiescent or activated fibroblasts with different lineages. A better understanding of the origin and function of the different fibroblast populations may provide valuable insight into possible arrhythmogenic mechanisms and therapeutic approaches.
Conclusions
In this review we have discussed the role of cardiac fibroblasts in healthy and diseased hearts and described how fibroblasts contribute to the arrhythmogenic substrate. We have also reviewed the available evidence demonstrating fibroblasts and myofibroblasts originate from a number of lineages in diseased hearts. Fibroblasts and myofibroblasts originate from epithelial-to-mesenchymal transition, endothelial-to-mesenchymal transition, and bone marrow-derived sources. Due to the lack of specific markers, the relative contribution of resident fibroblasts in cardiac injury and disease is currently unclear. Studies have also indicated the relative contribution of these populations changes with time after injury and possibly with pathological stimuli. Further experimentation is needed to determine whether populations of fibroblasts derived from different sources are phenotypically distinct and the potential functional roles of each of these populations in cardiac electrophysiology. These studies would have wide-ranging implications for the treatment of cardiac arrhythmias and may significantly contribute to our understanding of the basic principles that govern electrophysiology in healthy and injured hearts.
Acknowledgments
This work was supported by a NIH grant to GEM (HL076751).
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB. Heart disease and stroke statistics 2012 update. Circulation. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohr S. Arrhythmogenic implications of fibroblast–myocyte interactions. Circulation. Arrhythmia and Electrophysiology. 2012;5(2):442–452. doi: 10.1161/CIRCEP.110.957647. [DOI] [PubMed] [Google Scholar]
- 3.Rosker C, Salvarani N, Schmutz S, Grand T, Rohr S. Abolishing myofibroblast arrhythmogeneicity by pharmacological ablation of alpha-smooth muscle actin containing stress fibers. Circulation Research. 2011;109(10):1120–1131. doi: 10.1161/CIRCRESAHA.111.244798. [DOI] [PubMed] [Google Scholar]
- 4.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm. 2009;6(6):848–856. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 5.Miragoli M, Salvarani N, Rohr S. Myofibroblasts induce ectopic activity in cardiac tissue. Circulation Research. 2007;101(8):755–758. doi: 10.1161/CIRCRESAHA.107.160549. [DOI] [PubMed] [Google Scholar]
- 6.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circulation Research. 2006;98(6):801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 7.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circulation Research. 2003;93(5):421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 8.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast–myocyte interactions in response to cardiac injury. Circulation Research. 2010;107(8):1011–1020. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McSpadden LC, Nguyen H, Bursac N. Size and ionic currents of unexcitable cells coupled to cardiomyocytes distinctly modulate cardiac action potential shape and pacemaking activity in micropatterned cell pairs. Circulation. Arrhythmia and Electrophysiology. 2012 doi: 10.1161/CIRCEP.111.969329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedrotty DM, Klinger RY, Kirkton RD, Bursac N. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovascular Research. 2009;83(4):688–697. doi: 10.1093/cvr/cvp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zlochiver S, Munoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophysical Journal. 2008;95(9):4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen TP, Xie Y, Garfinkel A, Qu Z, Weiss JN. Arrhythmogenic consequences of myofibroblast–myocyte coupling. Cardiovascular Research. 2012;93(2):242–251. doi: 10.1093/cvr/cvr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie Y, Garfinkel A, Weiss JN, Qu Z. Cardiac alternans induced by fibroblast–myocyte coupling: mechanistic insights from computational models. American Journal of Physiology - Heart and Circulatory Physiology. 2009;297(2):H775–H784. doi: 10.1152/ajpheart.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Y, Garfinkel A, Camelliti P, Kohl P, Weiss JN, Qu Z. Effects of fibroblast–myocyte coupling on cardiac conduction and vulnerability to reentry: a computational study. Heart Rhythm. 2009;6(11):1641–1649. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacquemet V, Henriquez CS. Modulation of conduction velocity by nonmyocytes in the low coupling regime. IEEE Transactions on Biomedical Engineering. 2009;56(3):893–896. doi: 10.1109/TBME.2008.2006028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacquemet V, Henriquez CS. Loading effect of fibroblast–myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. American Journal of Physiology - Heart and Circulatory Physiology. 2008;294(5):H2040–H2052. doi: 10.1152/ajpheart.01298.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacquemet V, Henriquez CS. Modelling cardiac fibroblasts: interactions with myocytes and their impact on impulse propagation. Europace. 2007;9(Suppl 6):vi29–vi37. doi: 10.1093/europace/eum207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28(109):41–61. [PubMed] [Google Scholar]
- 19.Zak R. Development and proliferative capacity of cardiac muscle cells. Circulation Research. 1974;35(2 suppl II):17–26. [PubMed] [Google Scholar]
- 20.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. American Journal of Physiology—Heart and Circulatory Physiology. 2007;293(3):H1883–H1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 21.Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circulation Research. 2010;107(11):1304–1312. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yano T, Miura T, Ikeda Y, Matsuda E, Saito K, Miki T, et al. Intracardiac fibroblasts, but not bone marrow derived cells, are the origin of myofibroblasts in myocardial infarct repair. Cardiovascular Pathology. 2005;14(5):241–246. doi: 10.1016/j.carpath.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S, et al. Heart-infiltrating prominin-1+/CD133 + progenitor cells represent the cellular source of transforming growth factor beta-mediated cardiac fibrosis in experimental auto-immune myocarditis. Circulation Research. 2009;105(5):462–470. doi: 10.1161/CIRCRESAHA.109.196287. [DOI] [PubMed] [Google Scholar]
- 24.Mollmann H, Nef HM, Kostin S, von Kalle C, Pilz I, Weber M, et al. Bone marrow-derived cells contribute to infarct remodelling. Cardiovascular Research. 2006;71(4):661–671. doi: 10.1016/j.cardiores.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Odorfer KI, Walter I, Kleiter M, Sandgren EP, Erben RG. Role of endogenous bone marrow cells in long-term repair mechanisms after myocardial infarction. Journal of Cellular and Molecular Medicine. 2008;12(6B):2867–2874. doi: 10.1111/j.1582-4934.2008.00511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pichler M, Rainer PP, Schauer S, Hoefler G. Cardiac fibrosis in human transplanted hearts is mainly driven by cells of intracardiac origin. Journal of the American College of Cardiology. 2012;59(11):1008–1016. doi: 10.1016/j.jacc.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 27.Sato D, Otani H, Enoki C, Fujita M, Minato N, Iwasaka T. Phenotypic modulation and turnover of bone marrow-derived cells after myocardial infarction in rats. Cardiovascular Pathology. 2011;20(3):146–155. doi: 10.1016/j.carpath.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 28.van Amerongen MJ, Bou-Gharios G, Popa E, van Ark J, Petersen AH, van Dam GM, et al. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. The Journal of Pathology. 2008;214(3):377–386. doi: 10.1002/path.2281. [DOI] [PubMed] [Google Scholar]
- 29.Wu GD, Tuan TL, Bowdish ME, Jin YS, Starnes VA, Cramer DV, et al. Evidence for recipient derived fibroblast recruitment and activation during the development of chronic cardiac allograft rejection. Transplantation. 2003;76(3):609–614. doi: 10.1097/01.TP.0000066362.37931.6D. [DOI] [PubMed] [Google Scholar]
- 30.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature Medicine. 2007;13(8):952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 32.Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Disease Models & Mechanisms. 2011;4(4):469–483. doi: 10.1242/dmm.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, et al. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Developmental Dynamics. 2006;235(3):759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 34.Chu PY, Mariani J, Finch S, McMullen JR, Sadoshima J, Marshall T, et al. Bone marrow-derived cells contribute to fibrosis in the chronically failing heart. American Journal of Pathology. 2010;176(4):1735–1742. doi: 10.2353/ajpath.2010.090574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO Journal. 2012;31(2):429–442. doi: 10.1038/emboj.2011.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hocht-Zeisberg E, Kahnert H, Guan K, Wulf G, Hemmerlein B, Schlott T, et al. Cellular repopulation of myocardial infarction in patients with sex-mismatched heart transplantation. European Heart Journal. 2004;25(9):749–758. doi: 10.1016/j.ehj.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Okayama K, Azuma J, Dosaka N, Iekushi K, Sanada F, Kusunoki H, et al. Hepatocyte growth factor reduces cardiac fibrosis by inhibiting endothelial–mesenchymal transition. Hypertension. 2012;59(5):958–965. doi: 10.1161/HYPERTENSIONAHA.111.183905. [DOI] [PubMed] [Google Scholar]
- 38.Szardien S, Nef HM, Troidl C, Willmer M, Voss S, Liebetrau C, et al. Bone marrow-derived cells contribute to cell turnover in aging murine hearts. International Journal of Molecular Medicine. 2012;30(2):283–287. doi: 10.3892/ijmm.2012.995. [DOI] [PubMed] [Google Scholar]
- 39.van Tuyn J, Atsma DE, Winter EM, van der Veldevan DI, Pijnappels DA, Bax NA, et al. Epicardial cells of human adults can undergo an epithelial-to-mesenchymal transition and obtain characteristics of smooth muscle cells in vitro. Stem Cells. 2007;25(2):271–278. doi: 10.1634/stemcells.2006-0366. [DOI] [PubMed] [Google Scholar]
- 40.Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121(22):2407–2418. doi: 10.1161/CIRCULATIONAHA.110.938217. [DOI] [PubMed] [Google Scholar]
- 41.Zhou B, Honor LB, He H, Ma Q, Oh JH, Butterfield C, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. Journal of Clinical Investigation. 2011;121(5):1894–1904. doi: 10.1172/JCI45529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou B, Pu WT. Epicardial epithelial-to-mesenchymal transition in injured heart. Journal of Cellular and Molecular Medicine. 2011;15(12):2781–2783. doi: 10.1111/j.1582-4934.2011.01450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fries KM, Blieden T, Looney RJ, Sempowski GD, Silvera MR, Willis RA, et al. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clinical Immunology and Immunopathology. 1994;72(3):283–292. doi: 10.1006/clin.1994.1144. [DOI] [PubMed] [Google Scholar]
- 44.Lekic PC, Pender N, McCulloch CA. Is fibroblast heterogeneity relevant to the health, diseases, and treatments of periodontal tissues? Critical Reviews in Oral Biology and Medicine. 1997;8(3):253–268. doi: 10.1177/10454411970080030201. [DOI] [PubMed] [Google Scholar]
- 45.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(20):12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burstein B, Libby E, Calderone A, Nattel S. Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation. 2008;117(13):1630–1641. doi: 10.1161/CIRCULATIONAHA.107.748053. [DOI] [PubMed] [Google Scholar]
- 47.Flack EC, Lindsey ML, Squires CE, Kaplan BS, Stroud RE, Clark LL, et al. Alterations in cultured myocardial fibroblast function following the development of left ventricular failure. Journal of Molecular and Cellular Cardiology. 2006;40(4):474–483. doi: 10.1016/j.yjmcc.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 48.Squires CE, Escobar GP, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, et al. Altered fibroblast function following myocardial infarction. Journal of Molecular and Cellular Cardiology. 2005;39(4):699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 49.Jarvis MD, Rademaker MT, Ellmers LJ, Currie MJ, McKenzie JL, Palmer BR, et al. Comparison of infarct-derived and control ovine cardiac myofibroblasts in culture: response to cytokines and natriuretic peptide receptor expression profiles. American Journal of Physiology—Heart and Circulatory Physiology. 2006;291(4):H1952–H1958. doi: 10.1152/ajpheart.00764.2005. [DOI] [PubMed] [Google Scholar]
- 50.Vasquez C, Benamer N, Morley GE. The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. Journal of Cardiovascular Pharmacology. 2011;57(4):380–388. doi: 10.1097/FJC.0b013e31820cda19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annual Review of Pharmacology and Toxicology. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 52.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. The Journal of Cell Biology. 1993;122(1):103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sappino AP, Schurch W, Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Laboratory Investigation. 1990;63(2):144–161. [PubMed] [Google Scholar]
- 54.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. American Journal of Physiology. 1999;277(1 Pt 1):C1–C9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 55.Petrov VV, Fagard RH, Lijnen PJ. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39(2):258–263. doi: 10.1161/hy0202.103268. [DOI] [PubMed] [Google Scholar]
- 56.Kondalenko VG, Babaev VR, Rukosuev VS. Myofibroblasts in a zone of myocardial infarction. Bulletin of Experimental Biology and Medicine. 1981;92(6):1727–1729. doi: 10.1007/bf00837725. [DOI] [Google Scholar]
- 57.Vracko R, Thorning D. Myofibroblasts and smooth muscle cells in human myocardial scars: possible origins and inductive factors. Cardiovascular Pathology. 1993;2(3):207–213. doi: 10.1016/1054-8807(93)90004-L. [DOI] [PubMed] [Google Scholar]
- 58.Eyden B. The myofibroblast: a study of normal, reactive and neoplastic tissues, with an emphasis on ultrastructure. Part 1—normal and reactive cells. Journal of Submicroscopic Cytology and Pathology. 2005;37(2):109–204. [PubMed] [Google Scholar]
- 59.Sun Y, Kiani MF, Postlethwaite AE, Weber KT. Infarct scar as living tissue. Basic Research in Cardiology. 2002;97(5):343–347. doi: 10.1007/s00395-002-0365-8. [DOI] [PubMed] [Google Scholar]
- 60.Lu D, Soleymani S, Madakshire R, Insel PA. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. The FASEB Journal. 2012;26(6):2580–2591. doi: 10.1096/fj.12-204677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovascular Research. 2000;46(2):250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 62.Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. American Journal of Pathology. 1994;145(4):868–875. [PMC free article] [PubMed] [Google Scholar]
- 63.Jugdutt BI. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Current Drug Targets Cardiovascular & Haematological Disorders. 2003;3(1):1–30. doi: 10.2174/1568006033337276. [DOI] [PubMed] [Google Scholar]
- 64.Wessels A, Perez-Pomares JM. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. The Anatomical Record. Part A, Discoveries in Molecular, Cellular, and Evolutionary Biology. 2004;276(1):43–57. doi: 10.1002/ar.a.10129. [DOI] [PubMed] [Google Scholar]
- 65.Wessels A, van den Hoff MJ, Adamo RF, Phelps AL, Lockhart MM, Sauls K, et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Developmental Biology. 2012;366(2):111–124. doi: 10.1016/j.ydbio.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mikawa T, Gourdie RG. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Developmental Biology. 1996;174(2):221–232. doi: 10.1006/dbio.1996.0068. [DOI] [PubMed] [Google Scholar]
- 67.Perez-Pomares JM, Carmona R, Gonzalez-Iriarte M, Atencia G, Wessels A, Munoz-Chapuli R. Origin of coronary endothelial cells from epicardial mesothelium in avian embryos. International Journal of Developmental Biology. 2002;46(8):1005–1013. [PubMed] [Google Scholar]
- 68.Lie-Venema H, van den Akker NM, Bax NA, Winter EM, Maas S, Kekarainen T, et al. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. The Scientific World Journal. 2007;7:1777–1798. doi: 10.1100/tsw.2007.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Potts JD, Runyan RB. Epithelial–mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor beta. Developmental Biology. 1989;134(2):392–401. doi: 10.1016/0012-1606(89)90111-5. [DOI] [PubMed] [Google Scholar]
- 70.Norris RA, Borg TK, Butcher JT, Baudino TA, Banerjee I, Markwald RR. Neonatal and adult cardiovascular pathophysiological remodeling and repair: developmental role of periostin. Annals of the New York Academy of Sciences. 2008;1123:30–40. doi: 10.1196/annals.1420.005. [DOI] [PubMed] [Google Scholar]
- 71.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. American Journal of Pathology. 2011;179(3):1074–1080. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fredj S, Bescond J, Louault C, Potreau D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. Journal of Cellular Physiology. 2005;202(3):891–899. doi: 10.1002/jcp.20197. [DOI] [PubMed] [Google Scholar]
- 73.Lucas JA, Zhang Y, Li P, Gong K, Miller AP, Hassan E, et al. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. American Journal of Physiology—Heart and Circulatory Physiology. 298(2):H424–432. doi: 10.1152/ajpheart.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circulation Research. 1995;77(1):1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- 75.Ursell PC, Gardner PI, Albala A, Fenoglio JJ, Jr, Wit AL. Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circulation Research. 1985;56(3):436–451. doi: 10.1161/01.res.56.3.436. [DOI] [PubMed] [Google Scholar]
- 76.Dillon SM, Allessie MA, Ursell PC, Wit AL. Influences of anisotropic tissue structure on reentrant circuits in the epicardial border zone of subacute canine infarcts. Circulation Research. 1988;63:182–206. doi: 10.1161/01.res.63.1.182. [DOI] [PubMed] [Google Scholar]
- 77.Gardner PI, Ursell PC, Fenoglio JJ, Jr, Wit AL. Electrophysiologic and anatomic basis for fractionated electrograms recorded from healed myocardial infarcts. Circulation. 1985;72(3):596–611. doi: 10.1161/01.cir.72.3.596. [DOI] [PubMed] [Google Scholar]
- 78.Rook MB, Jongsma HJ, de Jonge B. Single channel currents of homo- and heterologous gap junctions between cardiac fibroblasts and myocytes. Pflügers Archiv. 1989;414(1):95–98. doi: 10.1007/BF00585633. [DOI] [PubMed] [Google Scholar]
- 79.Rook MB, van Ginneken AC, de Jonge B, el Aoumari A, Gros D, Jongsma HJ. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. American Journal of Physiology. 1992;263(5 Pt 1):C959–C977. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 80.Fahrenbach JP, Mejia-Alvarez R, Banach K. The relevance of non-excitable cells for cardiac pacemaker function. The Journal of Physiology. 2007;585(Pt 2):565–578. doi: 10.1113/jphysiol.2007.144121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lafontant PJ, Burns AR, Donnachie E, Haudek SB, Smith CW, Entman ML. Oncostatin M differentially regulates CXC chemokines in mouse cardiac fibroblasts. American Journal of Physiology. Cell Physiology. 2006;291(1):C18–C26. doi: 10.1152/ajpcell.00322.2005. [DOI] [PubMed] [Google Scholar]
- 82.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovascular Research. 1998;40(2):352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 83.Camelliti P, Devlin GP, Matthews KG, Kohl P, Green CR. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovascular Research. 2004;62(2):415–425. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y, Kanter EM, Yamada KA. Remodeling of cardiac fibroblasts following myocardial infarction results in increased gap junction intercellular communication. Cardiovascular Pathology. 2010;19(6):E233–E240. doi: 10.1016/j.carpath.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]