

Abstract
The central histone H3/H4 chaperone Asf1 comprises a highly conserved globular core and a divergent C-terminal tail. While the function and structure of the Asf1 core are well known, the function of the tail is less well understood. Here, we have explored the role of the yeast (yAsf1) and human (hAsf1a and hAsf1b) Asf1 tails in Saccharomyces cerevisiae. We show, using a photoreactive, unnatural amino acid, that Asf1 tail residue 210 cross-links to histone H3 in vivo and, further, that loss of C-terminal tail residues 211 to 279 weakens yAsf1-histone binding affinity in vitro nearly 200-fold. Via several yAsf1 C-terminal truncations and yeast-human chimeric proteins, we found that truncations at residue 210 increase transcriptional silencing and that the hAsf1a tail partially substitutes for full-length yAsf1 with respect to silencing but that full-length hAsf1b is a better overall substitute for full-length yAsf1. In addition, we show that the C-terminal tail of Asf1 is phosphorylated at T270 in yeast. Loss of this phosphorylation site does not prevent coimmunoprecipitation of yAsf1 and Rad53 from yeast extracts, whereas amino acid residue substitutions at the Asf1-histone H3/H4 interface do. Finally, we show that residue substitutions in yAsf1 near the CAF-1/HIRA interface also influence yAsf1's function in silencing.
INTRODUCTION
The eukaryotic genome is packaged into chromatin, a nucleoprotein structure of which the nucleosome is the fundamental repeating unit. It is formed from a histone octamer core wrapped approximately two times with 147 bp of DNA. The octamer comprises a heterotetramer of two copies each of the histones H3 and H4 and two heterodimers of H2A and H2B (1). Histones are highly charged basic proteins that are shepherded in the cell by histone chaperones (2), which bind to histones, not only to prevent them from interacting nonspecifically with other proteins and DNA, but also to help regulate their proper deposition into nucleosomes (2).
Asf1 is a highly conserved eukaryotic histone H3/H4 chaperone. It was first identified as a gene that when overexpressed led to a loss of position-dependent transcriptional silencing (3, 4). Asf1 facilitates histone deposition onto (5) and removal from (6) chromatin, which helps to coordinate DNA-templated processes, such as transcription, replication, and repair. Deletion of Asf1 leads to sensitivity to DNA-damaging and replicational-stress-inducing agents (3, 5). Asf1 has a highly conserved N-terminal core and a divergent C-terminal tail. The conserved core of Asf1 is important for its function and is required for the interaction between Asf1 and histones H3 and H4 (7, 8) and the histone chaperone complexes CAF-1 (5, 9) and HIR (HIRA) (10). The structures of yeast Asf1 bound to histones H3 and H4 showed that the core of Asf1 binds to a heterodimer of histones H3/H4 by enveloping the dimerization interface of histone H3, physically blocking formation of the H3/H4 heterotetramer (7, 8).
In addition to binding histones, Asf1 also binds to Rad53, but not to both simultaneously (11), and this association may depend upon the phosphorylation of Asf1, as treatment of yeast extracts with lambda phosphatase disrupts the interaction of Asf1 and the forkhead-associated 1 (FHA1) domain of Rad53 (12, 13). Despite the N-terminal sequence similarity of Asf1 across all eukarya and its fundamental importance in most organisms, ASF1 is not an essential gene in Saccharomyces cerevisiae, making it an ideal system for the genetic manipulation of ASF1.
In contrast to the conserved N-terminal core, the C terminus of Asf1 is highly diverged. In mammals there are two distinct Asf1 isoforms, Asf1a and Asf1b (14), which are distinguishable by the amino acid sequences of their C-terminal tails. In S. cerevisiae, Schizosaccharomyces pombe, and Caenorhabditis elegans, the tail is highly negatively charged and resembles neither the Asf1a nor the Asf1b tail. The C terminus of Asf1 is apparently natively unfolded and is absent from the available Asf1 structures (15–17). Although the yeast-specific polyanionic stretch of Asf1 is not absolutely required for histone binding, nucleosome assembly, or antisilencing (18), it does impact function. For example, the C terminus of Asf1 contributes to the binding avidity for replication factor C, an interaction that also requires the N terminus of Asf1 (19). Furthermore, the C terminus of Asf1 contributes to the constitutive, untargeted association of Asf1 with chromatin (20). In addition, we have previously shown that the C terminus of Asf1 influences transcriptional silencing and potentially Asf1 binding to histones and Rad53 (21). Specifically, deletion of the C terminus of Asf1 in the absence of another chromatin assembly factor, CAF-1, led to a dominant enhanced-silencing, or “supersilencing,” phenotype and a reduction in the ability of Asf1 to coimmunoprecipitate histones H3 and H4, as well as Rad53. Chromatin immunoprecipitation (ChIP) analysis showed that, relative to the reduced H3 occupancy on chromatin in cac1Δ mutants (where CAC1 encodes a subunit of CAF-1), expression of the truncated asf1 genes restored H3 occupancy to wild-type (WT) levels (21). We showed that the increased histone occupancy on DNA upon deletion of the C terminus of Asf1 was not due to a defect in chromatin disassembly, leading us to propose that the reduced interaction between Asf1 and H3/H4 leads to more chromatin assembly (21). Further, despite highly conserved N termini, we found that the human Asf1a and Asf1b proteins suppressed distinct phenotypes of the yeast asf1Δ mutant, suggesting that their distinct tails might contribute to these phenotypes (22). Finally, in human cells, the C terminus of Asf1 is phosphorylated by the Tousled-like kinases (TLKs), which helps to regulate its stability (14, 23). All of these findings underscore potential functions of the Asf1 C-terminal tail.
Here, we explore the function of the Asf1 C terminus in yeast. We show that specific regions of the C terminus of Asf1 contribute to histone binding. Asf1 residue 210 cross-links to histone H3 in vivo when replaced with a photoreactive, unnatural amino acid. Biophysical analyses show that loss of the entire Asf1 C-terminal tail leads to a 200-fold-reduced affinity for H3/H4 in vitro. Progressive deletions of the C terminus reduce histone binding affinity, but only when Asf1 is truncated beyond residue 210, demonstrating that the N-terminal portion of the tail, between residues 156 and 210, promotes the binding of Asf1 to H3/H4. In addition, we test the functions of the human Asf1a and Asf1b protein (hAsf1a and hAsf1b) tails in yeast, in the contexts of both the yeast and human Asf1 N termini, and show that the tails can influence silencing. We also examine the role of the C terminus with respect to Rad53 binding and show that the tail of the S. cerevisiae Asf1 protein (yAsf1) is phosphorylated at T270 but that loss of this phosphorylation site does not prevent Asf1 and Rad53 from coimmunoprecipitating. However, residue substitutions in the histone interaction surface of Asf1 disrupt the Asf1-Rad53 interaction. Finally, we show that mutations in Asf1 surface residues that are not predicted to interact with histones can still influence silencing.
MATERIALS AND METHODS
Yeast strains and plasmids.
The strains and plasmids employed in this study are listed in Tables 1 and 2. All ASF1 genes integrated into the yeast genome for this study were derived from pFA6-13Myc-KAN-MX6 (26), resulting in a C-terminally Myc-tagged Asf1 protein marked with Kanr. All plasmid-borne wild-type and mutant yeast ASF1 genes made for this study, and expressed in yeast, were carried on derivatives of pRS314-Asf1-Myc.
Table 1.
Strains used in this study
| Strain | Genotypea | Parent, source, or referenceb |
|---|---|---|
| ROY1169 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100 | 5 |
| BKD094 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314] | ROY1169 |
| BKD095 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-Myc] | ROY1169 |
| MCY001 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-51] | ROY1169 |
| BKD237 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-51][pLH157-KAN] | ROY1169 |
| BKD257 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-Myc][pLH157-KAN] | ROY1169 |
| BKD259 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-140][pLH157-KAN] | ROY1169 |
| BKD263 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-149][pLH157-KAN] | ROY1169 |
| BKD241 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-210][pLH157-KAN] | ROY1169 |
| BKD243 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-231][pLH157-KAN] | ROY1169 |
| BKD245 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-246][pLH157-KAN] | ROY1169 |
| BKD247 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-249][pLH157-KAN] | ROY1169 |
| BKD249 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-257][pLH157-KAN] | ROY1169 |
| BKD251 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-260][pLH157-KAN] | ROY1169 |
| BKD261 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-267][pLH157-KAN] | ROY1169 |
| BKD255 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-UAG-275][pLH157-KAN] | ROY1169 |
| BKD103 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-D37R] | ROY1169 |
| BKD104 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-E39R] | ROY1169 |
| BKD106 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-E56R] | ROY1169 |
| BKD107 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-S98Q] | ROY1169 |
| BKD109 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-E105R] | ROY1169 |
| BKD110 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100[pRS314-Asf1-R108E] | ROY1169 |
| ROY1170 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 asf1::his5+ TELVIIL::URA3 HMRa::ADE2 can1-100 | 5 |
| ROY1171 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 | 5 |
| BKD185 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 ASF1::13MyckanMX6 | ROY1171 |
| BKD198 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 ASF1-W153-hasf1a::13MyckanMX6 | ROY1171 |
| BKD194 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 ASF1-W153-hasf1b::13MyckanMX6 | ROY1171 |
| BKD200 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 asf1::hAsf1a::13MyckanMX6 | ROY1171 |
| BKD235 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 asf1::hAsf1a-W153-hAsf1b::13MyckanMX6 | ROY1171 |
| BKD202 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 asf1::hAsf1b::13MyckanMX6 | ROY1171 |
| BKD196 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 cac1::LEU2 TELVIIL::URA3 HMRa::ADE2 can1-100 asf1::hAsf1b-W153-hAsf1a::13MyckanMX6 | ROY1171 |
| ROY1172 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 | 5 |
| MCY024 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100[pRS314] | ROY1172 |
| BKD120 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 ASF1-W153-hasf1a::13MyckanMX6 | ROY1172 |
| BKD121 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 ASF1-W153-hasf1a::13MyckanMX6 | ROY1172 |
| BKD124 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 ASF1-W153-hasf1b::13MyckanMX6 | ROY1172 |
| BKD125 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 ASF1-W153-hasf1b::13MyckanMX6 | ROY1172 |
| BKD187 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 ASF1::13MyckanMX6 | ROY1172 |
| BKD188 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 ASF1::13MyckanMX6 | ROY1172 |
| BKD135 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1a::13MyckanMX6 | ROY1172 |
| BKD136 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1a::13MyckanMX6 | ROY1172 |
| BKD130 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1b::13MyckanMX6 | ROY1172 |
| BKD131 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1b::13MyckanMX6 | ROY1172 |
| BKD144 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1a-W153-hAsf1b::13MyckanMX6 | ROY1172 |
| BKD146 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1a-W153-hAsf1b::13MyckanMX6 | ROY1172 |
| BKD149 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1b-W153-hAsf1a::13MyckanMX6 | ROY1172 |
| BKD150 | MATα ade2-1 leu2-3,112 his3-11 trp1-1 ura3-1 HMRa::ADE2 TELVIIL::URA3 can1-100 asf1::hAsf1b-W153-hAsf1a::13MyckanMX6 | ROY1172 |
| W1588-4a | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | R. Rothstein |
| W1588-4c | MAT a ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | R. Rothstein |
| MCY043 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ | W1588-4c |
| MCY045 | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ | W1588-4a |
| BKD305 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 HHT2(FLAG)3::hphMX4 | W1588-4c |
| BKD361 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6 | Segegrant from MCY045 × BKD305 |
| BKD378 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314-Asf1-Myc] [pLH157-KAN][pBKD51] | BKD361 |
| BKD382 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314-Asf1-UAG-51] [pLH157-KAN][pBKD51] | BKD361 |
| BKD386 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314-Asf1-UAG-210] [pLH157-KAN][pBKD51] | BKD361 |
| BKD390 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314] [pLH157-KAN][pBKD51] | BKD361 |
| BKD379 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314-Asf1-Myc] [pLH157-KAN][pBKD52] | BKD361 |
| BKD387 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314-Asf1-UAG-210] [pLH157-KAN][pBKD52] | BKD361 |
| BKD391 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314] [pLH157-KAN][pBKD52] | BKD361 |
| BKD383 | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 asf1::his5+ HHT2(FLAG)3::hphMX6[pRS314-Asf1-UAG-51] [pLH157-KAN][pBKD52] | BKD361 |
| JKT0200 | mataΔ::hisG hmlΔ::ADE1 hmrΔ::ADE1 leu2::leu2(Asp718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 asf1::KAN | 24 |
| HZY1161 | MATa ura3-52 trp1 leu2Δ1 his3Δ200 pep4::his3 prb1Δ1.612 can1 Rad53-6His-FLAG-tag::KAN | 25 |
Plasmids are in brackets; strains derived from the same parent are grouped together.
Unless otherwise noted, strains are from this study.
Table 2.
Plasmids used in this study
| Plasmid | Characteristics | Source or reference |
|---|---|---|
| pFA6a-KANMX6 | 26 | |
| pFA6a-his5+MX6 | 26 | |
| pFA6A-6×Gly-3×FLAG-hphMX4 | 27 | |
| pRS314 | CEN6 ARSH4 TRP1 | 28 |
| pRS414 | CEN6 ARSH4 TRP1 | 28 |
| pRS316 | CEN6 ARSH4 URA3 | 28 |
| pEMHE81 | pRS414 containing HHT2 and HHF2 | 29 |
| pRB294 | CSE4 plasmid bearing a NotI-3×HA-NotI cassette | Gift of Richard Baker |
| pBKD31 | SacI-KpnI fragment from pEMHE81 containing HHT2 and HHF2 with ADH1 terminator downstream of H4, inserted into SacI-KpnI sites of pRS316 | This study |
| pBKD51 | NotI site in polylinker of pBKD31 filled in with Klenow. A new NotI site was created at the N terminus of HHT2 followed by insertion of a 3×HA tagged NotI cassette from pRB294. HA-HHT2 was subcloned as a HindIII-XbaI fragment and ligated back into pRS316. This plasmid contains HA-H3 and no copy of H4. | This study |
| pBKD52 | NotI site in polylinker of pBKD31 filled in with Klenow. A new NotI site was created at the N terminus of HHF2, followed by insertion of a 3×HA-tagged NotI cassette from pRB294. HHT2 HA-HHF2 was subcloned as an XbaI-EcoRI fragment and ligated back into pRS316. This plasmid contains HA-H4 and untagged H3. | This study |
| pSup-BpaRS-6TRN | Contains tRNA synthetase (EcTyrRS) and tRNA (Ec tRNACUA) | 30 |
| pST39GSTAsf169Δ27H3H4 | 31 | |
| pST39BPA51GSTAsf1 | pST39GSTAsf169Δ27H3H4 with an amber stop codon replacing L51 | This study |
| pST39BPA140GSTAsf1 | pST39GSTAsf169Δ27H3H4 with an amber stop codon replacing L140 | This study |
| pST39BPA149GSTAsf1 | pST39GSTAsf169Δ27H3H4 with an amber stop codon replacing F149 | This study |
| pET-60-yAsf1FL | Saccharomyces cerevisiae ASF1 inserted between the GST tag and the His6 tag coding sequences. The r Asf1 −1 position (Pro) was changed to (Cys) for fluorophore attachment and production of yAsf1*532 | 11 |
| pET-60-yAsf1246 | pET-60-yAsf1FL modified to replace yAsf1 with yAsf1 C-terminally truncated at position 246 | This study |
| pET-60-yAsf1210 | pET-60-yAsf1FL modified to replace yAsf1 with yAsf1 C-terminally truncated at position 210 | This study |
| pET-60-yAsf1185 | pET-60-yAsf1FL modified to replace yAsf1 with yAsf1 C-terminally truncated at position 185 | This study |
| pET-60-yAsf1169 | pET-60-yAsf1FL modified to replace yAsf1 with yAsf1 C-terminally truncated at position 169 | This study |
| pET-60-yAsf1155 | pET-60-yAsf1FL modified to replace yAsf1 with yAsf1 C-terminally truncated at position 155 | This study |
| pLH157 | Contains tRNA synthetase (EcTyrRS) and tRNA (Ec tRNACUA) driven by the yeast polIII promoter N(GTT)PR CEN TRP1 | 32 |
| pLH157-KAN | pLH157 with the TRP1 marker replaced with KAN from pFA6a-KANMX6 | This study |
| pRS314-Asf1-Myc | pRS314 containing 13 C-terminally Myc-tagged Asf1 (pAsf1-Myc) | 7 |
| pRS314-Asf1-UAG-51-Myc | pRS314-Asf1-Myc with an amber stop codon replacing L51 | This study |
| pRS314-Asf1-UAG-140-Myc | pRS314-Asf1-Myc with an amber stop codon replacing L140 | This study |
| pRS314-Asf1-UAG-149-Myc | pRS314-Asf1-Myc with an amber stop codon replacing F149 | This study |
| pRS314-Asf1-UAG-210-Myc | pRS314-Asf1-Myc with an amber stop codon replacing E210 | This study |
| pRS314-Asf1-UAG-231-Myc | pRS314-Asf1-Myc with an amber stop codon replacing N231 | This study |
| pRS314-Asf1-UAG-246-Myc | pRS314-Asf1-Myc with an amber stop codon replacing V246 | This study |
| pRS314-Asf1-UAG-249-Myc | pRS314-Asf1-Myc with an amber stop codon replacing N249 | This study |
| pRS314-Asf1-UAG-257-Myc | pRS314-Asf1-Myc with an amber stop codon replacing R257 | This study |
| pRS314-Asf1-UAG-260-Myc | pRS314-Asf1-Myc with an amber stop codon replacing I260 | This study |
| pRS314-Asf1-UAG-267-Myc | pRS314-Asf1-Myc with an amber stop codon replacing I267 | This study |
| pRS314-Asf1-UAG-275-Myc | pRS314-Asf1-Myc with an amber stop codon replacing A275 | This study |
| pRS314-Asf1-Δ231-238-Myc | pRS314-Asf1-Myc lacking amino acids 231–238 | This study |
| pRS314-Asf1-Δ239-245-Myc | pRS314-Asf1-Myc lacking amino acids 239–245 | This study |
| pRS314-Asf1-188T (30P) | pRS314-Asf1-Myc truncated after amino acid 188 | 21 (see Materials and Methods) |
| pRS314-Asf1-153Ins (732) | pRS314-Asf1-Myc containing an in-frame insertion after amino acid 153 | 21 (see Materials and Methods) |
| pRS314-Asf1-L12A-Myc | pRS314-Asf1-Myc with the mutation L12A | This study |
| pRS314-Asf1-L12R-Myc | pRS314-Asf1-Myc with the mutation L12R | This study |
| pRS314-Asf1-N14A-Myc | pRS314-Asf1-Myc with the mutation N14A | This study |
| pRS314-Asf1-N14R-Myc | pRS314-Asf1-Myc with the mutation N14R | This study |
| pRS314-Asf1-E25R-Myc | pRS314-Asf1-Myc with the mutation E25R | This study |
| pRS314-Asf1-E29A-Myc | pRS314-Asf1-Myc with the mutation E29A | This study |
| pRS314-Asf1-E29R-Myc | pRS314-Asf1-Myc with the mutation E29R | This study |
| pRS314-Asf1-D37R-Myc | pRS314-Asf1-Myc with the mutation D37R | This study |
| pRS314-Asf1-E39R-Myc | pRS314-Asf1-Myc with the mutation E39R | This study |
| pRS314-Asf1-K41E-Myc | pRS314-Asf1-Myc with the mutation K41E | This study |
| pRS314-Asf1-E56R-Myc | pRS314-Asf1-Myc with the mutation E56R | This study |
| pRS314-Asf1-S98Q-Myc | pRS314-Asf1-Myc with the mutation S98Q | This study |
| pRS314-Asf1-S100Q-Myc | pRS314-Asf1-Myc with the mutation S100Q | This study |
| pRS314-Asf1-E105R-Myc | pRS314-Asf1-Myc with the mutation E105R | This study |
| pRS314-Asf1-R108E-Myc | pRS314-Asf1-Myc with the mutation R018E | This study |
| pRS314-Asf1-Y112A/T147E-Myc | pRS314-Asf1-Myc with the mutations Y112A/T147E | 7 |
| pRS314-Asf1-T147E/S45R-Myc | pRS314-Asf1-Myc with the mutations T147E/S45R | 7 |
| pRS314-Asf1-V94R-Myc | pRS314-Asf1-Myc with the mutation V94R | 7 |
| pRS314-Asf1-Y112A/V146L-Myc | pRS314-Asf1-Myc with the mutations Y112A/V146L | 7 |
| pRS314-Asf1-Y112A/R145E-Myc | pRS314-Asf1-Myc with the mutations Y112A/R145E | 7 |
| pRS314-Asf1-T147E/R145E-Myc | pRS314-Asf1-Myc with the mutations T147E/R145E | 7 |
| pRS314-Asf1-P144Y-Myc | pRS314-Asf1-Myc with the mutation P144Y | 7 |
| pRS314-Asf1-R145E-Myc | pRS314-Asf1-Myc with the mutation R145E | 7 |
| pRS314-Asf1-V146L-Myc | pRS314-Asf1-Myc with the mutation V146L | 7 |
| pRS314-Asf1-T147E-Myc | pRS314-Asf1-Myc with the mutation T147E | 7 |
| pRS314-Asf1-L6 M-Myc | pRS314-Asf1-Myc with the mutation L6 M | 7 |
| pRS314-Asf1-Y162A-Myc | pRS314-Asf1-Myc with the mutation Y162A | 7 |
| pRS314-Asf1-T265A-Myc | pRS314-Asf1-Myc with the mutation T265A | This study |
| pRS314-Asf1-T270A-Myc | pRS314-Asf1-Myc with the mutation T270A | This study |
| pRS314-Asf1-T270E-Myc | pRS314-Asf1-Myc with the mutation T270E | This study |
| pRS314-Asf1-T265A/T270A-Myc | pRS314-Asf1-Myc with the mutations T265A/T270A | This study |
| pRM102 | CEN4 ARS1 pGAL10-HHT2 pGAL1-HHF2 | 33 |
| pRM200 | CEN4 ARS1 TRP1 HHT2 HHF2 | 33 |
Yeast growth and manipulation.
Yeast extract-peptone-dextrose medium (YPD), synthetic complete medium (SC), and synthetic complete medium lacking one or more amino acids and/or bases (e.g., SC−Trp) with or without 5-fluoroorotic acid (5-FOA) have been described previously (34). Synthetic low-adenine medium contains 25% of the normal amount of adenine. The synthetic medium used for maintaining plasmids with the Kanr marker in yeast had 1 g/liter l-glutamic acid-monosodium salt substituted for ammonium sulfate as a nitrogen source.
Yeast transcriptional-silencing and drug sensitivity assays.
Equivalent numbers of logarithmically growing cells were harvested (optical density at 600 nm [OD600] = 1), resuspended, and serially diluted 5 or 10 times before plating, as described below and in the figure legends.
Creation of plasmid-borne Asf1 truncation, deletion, and other mutations.
Amber stop codons, small deletions, and other mutations were introduced into either pRS314-Asf1-Myc or pST39GSTAsf169Δ27H3H4 by site-directed mutagenesis (QuikChange; Invitrogen). The 153Ins mutation resulting from transposon mutagenesis was previously reported as 152I (21), but resequencing the plasmid showed that the transposon was inserted in frame following amino acid W153, replacing the native H154 with VFKHW. Similarly, the 185T mutation obtained from the same transposon mutagenesis screen has been renamed 188T here, as the transposon was inserted adjacent to amino acid D186, replacing E187 with D and V, followed by an in-frame stop codon. Both plasmids also encode E158G and D228G mutations. These mutations do not contribute to the silencing phenotype (data not shown). All plasmids used here have the same promoters and terminators, with the exception of 153Ins and 188T, which were described previously (21).
Fluorescence spectroscopy and binding affinity determination.
Preparation of Xenopus laevis H3/H4 from Escherichia coli was carried out as previously detailed, with mutations at H3 C110A and H4 T71C (35–37). C-terminal truncations of yAsf1 were cloned, expressed in E. coli, purified, and labeled with Alexa Fluor 532 as previously described (38, 39). Briefly, pET60-DEST-yAsf1 with an N-terminal glutathione S-transferase (GST) tag and a C-terminal 6×His tag, along with a P1C or I3C mutation for fluorophore labeling, was cloned, ending with residue 279, 246, 210, 185, 169, or 155. The plasmids were then expressed in E. coli Rosetta 2(DE3)(pLysS) (Novagen), grown at 37°C to an OD600 of 0.6, and induced with 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (27°C; 4 h). yAsf1 proteins were purified by incubation with glutathione-Sepharose 4B (GE Healthcare), followed by overnight cleavage with Precission protease (GE Healthcare) targeting a cleavage site upstream of the GST tag. The proteins were then incubated with Ni2+-nitrilotriacetic acid (NTA)-agarose beads and eluted through a 5- to 250-mM imidazole gradient.
For fluorophore labeling, yAsf1 was incubated with a 20-fold molar excess of Alexa Fluor 532 (Invitrogen) in 25 mM HEPES, 0.5 M NaCl, 0.5 mM Tris(2-carboxyethyl) phosphine (TCEP), 0.05% Brij, 10% glycerol, pH 7.25, for 2 h at room temperature (RT). The reaction was quenched by the addition of 20 mM β-mercaptoethanol, and protein was separated from free dye via a Superdex 200 column in buffer containing 10 mM Tris, 0.5 M NaCl, 0.5 mM TCEP, 10% glycerol, 0.05% Brij, pH 7.9.
For fluorescence spectroscopy, 1 nM Alexa Fluor 532-labeled yAsf1 was equilibrated in 3 ml of assay buffer (10 mM Tris, 150 mM KCl, 2 mM MgCl2, 1% glycerol, 0.5 mM TCEP, 0.05% Brij, pH 7.5) at 20°C using a Fluorolog-3 spectrofluorometer. H3/H4 was titrated from 200 pM to 900 nM. At each point, the reaction was allowed to equilibrate for 10 min before excitation at 528 nm (slit width, 5 nm) and scanning the emission spectra (slit width, 7 nm) from 538 to 558 nm.
To quantitate binding affinity, the raw fluorescence intensity at 548 nm of each H3/H4 titration point (Fo) was subtracted from background fluorescence and then normalized to yAsf1 fluorescence (Fi) by the formula (Fi − Fo)/Fi. The resulting fraction bound (Y) was determined relative to the binding saturation constant (Bmax) and plotted in GraphPad Prism (v. 5.0d). Binding affinities were analyzed by equation 1 for yAsf1 mutants 1-279, 1-246, and 1-210 and equation 2 for yAsf1 mutants 1-185, 1-169, and 1-155:
| (1) |
| (2) |
where Asf1 is the concentration of fluorophore-labeled yAsf1, x is the concentration of H3/H4 titrated, and KD is the dissociation constant.
BPA-substituted Asf1-histone cross-linking in E. coli.
The plasmids pST39GSTAsf169Δ27H3H4, pST39BPA51GSTAsf1, pST39BPA140GSTAsf1, and pST39BPA149GSTAsf1 were individually cotransformed with pSup-BpaRS-6TRN and grown in 2× yeast extract-tryptone medium with 50 μg/ml ampicillin, 34 μg/ml chloramphenicol, and 0.1% glucose. Expression of the GSTAsf169 protein (and amber codon mutants), H3, and H4 was induced by addition of 0.4 mM IPTG to exponentially growing E. coli Rosetta(pLys S) cells (Novagen) containing cotransformed mutant GSTAsf1 plasmid and the pSup-BpaRS-6TRN plasmid for 8 h at 27°C in the presence of 1 mM p-benzoyl-l-phenylalanine (BPA). Cell pellets from 1 liter of cells were resuspended in 50 ml of buffer 1 (20 mM Tris-HCl, pH 7.9, 10 mM EDTA, 0.5 M NaCl, 1 mM dithiothreitol [DTT], and Roche EDTA-free protease inhibitor cocktail tablets) and lysed by sonication. The extract was clarified by centrifugation (30 min; 14,502 × g; 4°C). A fraction of the soluble protein was UV irradiated at 350 nm for 2 h in a Rayonet RPR-100 photochemical reactor and compared to a nonirradiated extract via SDS-PAGE visualized by silver stain (Silverquest staining kit LC6070; Invitrogen) and immunoblotting. Proteins were transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Millipore) via semidry blotting. The membranes were blocked in 3% BSA in Tris-buffered saline plus 0.1% Tween (TBS-T) for GST detection (mouse monoclonal; University of Colorado Cancer Center) and in 5% nonfat dry milk in TBS-T for H3 detection (rabbit polyclonal; Abcam; ab1791). Signal was detected by enhanced chemiluminescence (ECL) (Millipore Immobilon) following incubation with horseradish peroxidase (HRP)-conjugated anti-mouse (GE; NXA931) and anti-rabbit (GE; NA934V) antibodies.
BPA-substituted Asf1 cross-linking in S. cerevisiae.
Plasmid pLH147 encoding the CUA amber suppressor tRNA and a Tyr tRNA synthetase modified for expression in yeast was the kind gift of S. Hahn (32). The plasmid was further modified by replacing the TRP1 marker on the plasmid with a Kanr marker by PCR-mediated gene replacement in yeast, creating pLH147-KAN. To maintain selection for both the tRNA and Asf1 plasmids, cells were grown in SC−Trp with ammonium sulfate replaced with 1 g/liter l-glutamic acid, with monosodium salt as a nitrogen source (40). Mid-log-phase cultures were diluted to an OD600 of 0.2 in 100 to 300 ml of medium containing BPA (PepTech Corp., Burlington, MA). BPA was dissolved in 1 N NaOH, filter sterilized, and added to the medium at a final concentration of 1 mM, followed by the addition of an equal volume of filter-sterilized 1 N HCl. Cells were harvested at an OD600 of ∼1, washed with water, and resuspended in 10 to 30 ml of medium lacking both glucose and BPA before transfer to petri dishes (10 ml/dish). The uncovered petri dishes were placed in a UV cross-linker (254 nm) and irradiated 4 times with 8,550 μJ of UV light. The cells were harvested, the wet weights of the cell pellets were determined, and they were stored at −80°C. Initial experiments were performed using yeast strain ROY1169. The experiments involving epitope-tagged proteins were performed in derivatives of W1588.
Cells were resuspended in 2 volumes (wt/vol, based on the wet weight of the cell pellet), of either lysis buffer I (20 mM Tris, pH 7.5, at 23°C, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 0.05% Triton X-100) or lysis buffer II (40 mM HEPES, pH 7.5, 150 mM KCl, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 0.05% NP-40) supplemented with a 2× protease inhibitor cocktail (Roche Complete without EDTA). Neither buffer appeared to confer an advantage in the immunoprecipitation reactions. Glass beads were added in a volume equal to the packed cell volume. The cells were lysed by vortexing 3 times for 8 min each time at 4°C. Following lysis, phenylmethylsulfonyl fluoride (PMSF) was added to 1 mM, and extracts were incubated on ice for 10 min. Beads and unopened cells were separated from cell lysates by centrifugation at 1,000 × g for 10 min at 4°C. The lysates were further cleared by centrifugation for 15 to 30 min at 10,000 × g, and protein concentrations were determined by Bradford assay. Myc-tagged Asf1 was purified onto anti-Myc agarose beads (EZ-View; Sigma, St. Louis, MO) preequilibrated in lysis buffer. Extracts containing 5 to 10 mg total protein were mixed with 25 μl anti-Myc agarose beads for 2 to 4 h and up to overnight. The beads were harvested by centrifugation for 5 min at 1,000 × g at 4°C, followed by washing with lysis buffer 3 times. The harvested beads were boiled for 5 min in 25 μl protein sample buffer prior to storage at −20°C. Samples were analyzed by SDS-PAGE, followed by immunoblotting. When required, blots were stripped with hot (65°C) 2% SDS, 62.5 mM Tris, pH 6.8, 100 mM β-mercaptoethanol, followed by extensive washing in TBS-T. The blots were reblocked and probed with secondary antibody (Ab) to ensure loss of signal prior to incubation with a second primary antibody. The antibodies used were anti-Myc (Santa Cruz sc-789) and anti-FLAG-M2 (Sigma F3165).
Construction and integration of yAsf1-Myc, hAsf1a, hAsf1b, and Asf1 chimeras and truncations.
All integrated Asf1 genes in this study were derived from pFA6-13Myc-KAN-MX6 (26), resulting in a C-terminally Myc-tagged Asf1 protein marked with Kanr. 13×Myc-tagged hAsf1a and hAsf1b were amplified from pFA6-Asf1a-13Myc-KAN-MX6 and pFA6-Asf1b-13Myc-KAN-MX6 (22) and integrated into the genome in place of ASF1. The tails of hAsf1a and hAsf1b were amplified from the same vectors and used to replace the endogenous yeast tail at W153. The hAsf1a-btail and hAsf1b-atail chimeras were created by megaprimer PCR (41, 42), followed by cloning. In the first megaprimer PCR step, the desired C terminus and the 13Myc-KAN moiety were amplified with a primer containing 5′-end sequence identical to that of the Asf1 isoform that the tail would be transplaced onto. In the second step, the products obtained from the first PCR step were used as 3′-end primers, along with new 5′-end primers to amplify the N-terminal portion of the desired Asf1 isoform. The resulting PCR products were cloned into PCR-4 TOPO (Invitrogen) and sequenced to ensure their integrity. Sequencing revealed a point mutation in hAsf1b originating from the hAsf1b cDNA clone. It creates a P168S mutation that naturally exists in the bovine (Bos taurus) Asf1b protein (GenBank accession number NM_001075453). The chimeras were subcloned from PCR-4 TOPO as SalI-PacI fragments into pFA6-13Myc-KAN-MX6, creating plasmids pBKD2-1 (hAsf1a with the hAsf1b tail) and pBKD2′-1 (hAsf1b with the hAsf1a tail). The chimeras were then amplified from pFA6-13Myc-KAN-MX6 as described above. yAsf1-Myc strains were created from ACN026 (22), which was itself created by insertion of a pFA6-13Myc-KAN-MX6 plasmid-derived PCR product into yeast Asf1.
Yeast integration events arising from transformation with the same PCR product had variable silencing phenotypes but not drug sensitivity phenotypes. To explore these differences, the integrated Asf1 variants were PCR amplified, and the coding regions were sequenced. After verifying the open reading frames (ORFs) for the integrated genes, the silencing assays were repeated using only isolates lacking point mutations in the coding regions.
Rad53 immunoprecipitations.
pRS314-Asf1-Myc or an empty pRS314 plasmid was transformed into either JKT0200 (asf1Δ) or HZY1161 (Rad53-FLAG ASF1). Logarithmically growing cells were harvested and resuspended in 1 ml of lysis buffer III (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.2% Triton X-100) plus protease and phosphatase inhibitors (0.5 mM sodium-metabisulfite, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 mM β-glycerophosphate, 0.5 mM dithiothreitol [DTT], 2 μg/ml aprotinin, 0.5 μg/ml leupeptin, 5 μg/ml pepstatin, 0.5 mM benzamidine, 50 mM NaF, and 1 mM NaVnO4). The cells were lysed by vortexing for 10 min with glass beads. The lysates were cleared by centrifugation at 12,000 × g in a microcentrifuge at 4°C for 10 min. Protein concentrations were determined by Bradford assay, and 8 to 15 mg total protein was used for immunoprecipitation with either 7.5 μl anti-c-Myc antibodies (Santa Cruz sc-789) or 6 μl anti-FLAG (Sigma F3165) overnight at 4°C. Immunocomplexes were captured with 60 μl of protein A-Sepharose beads (GE Healthcare) and washed 4 to 8 times in lysis buffer III. SDS protein buffer was added to the beads, and samples were boiled to elute and denature the proteins. Antibodies and dilutions were as follows: 1:500 anti-c-Myc (Santa Cruz sc-789), 1:5,000 anti-FLAG (Sigma F3165), 1:80,000 anti-Mouse HRP (Sigma A3682), 1:150,000 anti-rabbit HRP (Sigma A-1949), 1:500 antitubulin (Serotech NCMCA785), 1:2,000 anti-H3K56ac (Millipore-Upstate 07-677), and 1:80,000 anti-rat HRP (Sigma A5795). The detection kits used were ECL (GE Healthcare RPN2106) and ECL Plus (RPN 2132).
Phosphatase treatment.
Immunoprecipitations were performed as described above using 60-ml cultures grown to an OD600 of 1.0. Four milligrams of protein was immunoprecipitated with 7.5 μl anti-c-Myc antibodies (Santa Cruz sc-789). After the last wash, the supernatant was removed and 20 μl of beads was divided among three tubes. To the first sample, only 100 μl 1× λ protein phosphatase (PPase) buffer (2 mM MnCl2, 2.5 mM EDTA) was added. To a second sample, 100 μl 1× λ PPase buffer containing 200 U lambda PPase (NEB P0753S) was added. To the final sample, 100 μl 1× λ PPase buffer containing 200 U lambda PPase and phosphatase inhibitors 50 mM NaF and 1 mM NaVnO4 were added. The IP mixtures were incubated for 30 min, 25 μl of 5× SDS buffer was added, and the samples were boiled and analyzed by Mn2+ Phos-tag SDS-PAGE, followed by immunoblotting.
Mn2+ Phos-tag.
Standard Laemmli SDS-PAGE was performed, except that the Mn2+ acrylamide-pendant Phos-tag ligand and MnCl2 were added to the separating gel, prior to polymerization, at final concentrations of 100 μM and 200 μM, respectively (43). The Phos-tag was the kind gift of Dorothea Fielder (University of California, San Francisco [UCSF]).
Two-dimensional (2D) gel electrophoresis.
Cultures (60 ml) were grown in SC−Trp to an OD600 of 1.0. The cells were harvested and frozen in liquid nitrogen. The cell pellets were resuspended in 300 μl of 50 mM HEPES, pH 7, 1 mM MgCl2, 0.1 mM EDTA, 1 mM PMSF, 13 μM pepstatin, and phosphatase inhibitors: 1 mM NaF, 1 mM NaVnO4, and 10 mM β-glycerolphosphate. Following lysis with glass beads, the lysates were transferred to a new tube containing 800 μl of a second buffer (7 M urea, 2 M thiourea, 4% {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} [CHAPS], 1% DTT, and 1% Pharmalytes 3 to 10 [GE Healthcare]) and vortexed for 25 min at RT. The lysates were cleared by centrifugation, and the protein concentration was determined by Bradford assay. Fifty micrograms of each sample was applied by passive rehydration in an Immobiline DryStrip Reswelling Tray (24 cm) for ≥18 h. The immobilized pH gradient (IPG) strips were equilibrated in reduction and alkylation solutions, followed by isoelectric focusing (IEF) using an IPGPhor IEF unit according to the manufacturer's recommendations. SDS-PAGE in the second dimension was performed using 9 to 16% Tris-glycine gels (Jules Gels, Milford, CT) run at 20 W per gel on an Ettan Dalt system for 4 to 6 h. Proteins were transferred onto nitrocellulose (20 V overnight at 4°C). The membranes were stained with amido black and blocked with TBS-T plus 3% milk for 1 h and probed overnight at 4°C with a 1:500 dilution of anti-c-Myc antibody (Santa Cruz sc-789), followed by a 1:150,000 dilution of HRP-conjugated anti-rabbit secondary antibody (Sigma A-1949) and detected by ECL (GE Healthcare RPN2106).
Mass spectrometry.
Cultures (150 ml) were grown in SC−Trp to an OD600 of 1.0. Cells were harvested and frozen in liquid nitrogen. Immunoprecipitation was performed as described above in the presence (1 mM) of the phosphatase inhibitors NaF and NaVnO4. Proteins were resolved on a 7.5% SDS-PAGE gel and stained with Coomassie. Protein bands corresponding to the correct size for Asf1-Myc were excised and trypsinized as previously described (44, 45). Samples were analyzed on an LTQ-FT Ultra hybrid mass spectrometer (Thermo Fisher, Bremen, Germany) by the University of Colorado—Denver Proteomics Core. Raw distiller (UCSF, CA) was used to create deisotoped centroided peak lists from the raw spectra (.mgf format). These peak lists were searched against all yeast entries in databases using the Mascot server (version 2.2; Matrix Science) or Protein Prospector (38, 46) with mass tolerances of ±10 ppm for mass spectrometry (MS) peaks and ±0.6 Da for tandem MS (MS-MS) fragment ions. Trypsin specificity was used, allowing for 1 missed cleavage. The variable modifications of Met oxidation, protein N-terminal acetylation, peptide N-terminal Q pyroglutamic acid formation, and phosphorylation of T, Y, and S were allowed for in the search. An expect value (P value) of <0.05 was considered significant.
Asf1-T270 phosphoantibody.
Yeast cultures were grown in SC−Trp to an OD600 of 1.0, and 4 ml was harvested, washed 2 times with sterile H2O, and resuspended in 200 μl of SDS protein buffer containing 1 mM NaVnO4 and 50 mM NaF. Samples were boiled for 5 min, vortexed for 1 min, and clarified by centrifugation. The entire sample was loaded onto a 7.5% SDS-PAGE gel and transferred to nitrocellulose. Membranes were stained with amido black and blocked with TBS-T plus 3% milk. The blot was probed with various dilutions of T270ph-Asf1 antibodies (PhosphoSolns 7977) using a Mini Protean II multiscreen apparatus (Bio-Rad) overnight at 4°C, followed by incubation with a 1:150,000 dilution of HRP-conjugated anti-rabbit secondary antibody with detection by ECL (Visualizer; Upstate 64-202BP).
RESULTS
A region (amino acids [aa] 188 to 231) within the C terminus of Asf1 is required for its normal function in transcriptional silencing.
The C termini of yeast and mammalian Asf1 proteins are highly diverged, making it difficult to identify strongly conserved residues. Additionally, the S. pombe and S. cerevisiae Asf1 tails are highly polyanionic and therefore lack distinct sequence information. To identify features of the yAsf1 C terminus that might be important for function, rather than making uninformed point mutations, we made C-terminal truncations. We introduced single amber stop codons to replace residues 51, 140, and 149 (as controls that were expected to disrupt yAsf1 function) and 210, 231, 246, 249, 257, 260, 267, and 275, positioned throughout the C terminus of yAsf1 (Fig. 1A). All mutations were introduced onto the plasmid pRS314-Asf1-Myc (Table 2) and transformed into the asf1Δ cac1Δ strain ROY1169 (Table 1).
Fig 1.

Asf1 with an amber stop codon replacing residue 210 displays increased silencing over wild-type Asf1. (A) Alignment of human and yeast Asf1 proteins generated using multiple-sequence alignment (MSA) (61). The conserved globular core of Asf1 is shaded in gray. Conserved residues are shaded, and similar residues are colored the same, using blue (basic), red (acidic), yellow (hydrophobic), and green (polar). yAsf1 deletion endpoints later replaced with BPA are marked with cyan stars. All chimeric proteins used in this study contain the conserved core up to and including Trp153 (marked with a yellow star). (B) ROY1172, carrying an empty pRS314 vector (ASF1 CAC1), and ROY1169-based strains (asf1Δ cac1Δ), carrying either an empty pRS314 vector (asf1Δ cac1Δ) or a pRS314-Asf1-UAG-Myc plasmid encoding Asf1 proteins that are wild type or truncated at the indicated residues, were plated on the indicated media. (C) Summary of truncations and deletions with their silencing phenotypes, with minus signs indicating lack of silencing and plus signs indicating the relative strength of silencing.
Loss of Asf1 function confers sensitivity to DNA-damaging and replicational-stress-inducing agents (5). To assay the mutants for their responses to DNA damage and replicational stress, cells were plated on media containing hydroxyurea (HU), an agent that induces replicational stress; methyl methane sulfonate (MMS), an alkylating agent that induces single- and double-strand DNA breaks; and zeocin, a radiomimetic that induces double-strand DNA breaks. Consistent with previous work (15, 21), loss of the yAsf1 C terminus had little effect. Only those C-terminal truncations that encroached upon the Asf1 globular core exhibited sensitivity to HU, MMS, or zeocin (data not shown). This analysis also verifies the expression of functional yAsf1 protein from the C-terminal-truncation mutants.
In addition to its roles in DNA replication and repair, yAsf1 impacts transcriptional silencing (3–5). To examine position-dependent transcriptional silencing, we used a yeast reporter strain with copies of the URA3 and ADE2 genes located near the left arm of telomere VII and near the silenced mating-type locus, HMRa, respectively. At TELVIIL::URA3, a loss of silencing results in death on medium containing the uracil analog 5-FOA, whereas an increase in silencing allows growth on 5-FOA. The assay is also sensitive to overall upregulation of poorly expressed genes or changes in purine/pyrimidine metabolism (47).
At HMRa::ADE2, a reduction in silencing leads to whiter colonies, whereas an increase in silencing leads to redder colonies due to the accumulation of a by-product in the adenine synthetic pathway.
We examined the yAsf1 truncation mutants described above, as well as four internal-deletion mutants, Δ231-238, Δ239-245, Δ236-242, and Δ231-245, that removed regions of higher amino acid diversity than the surrounding acidic residues (Fig. 1A), for their effects on silencing in the sensitized cac1 genetic background, where CAC1 encodes a subunit of the chromatin assembly factor CAF-1. The mutants were compared to wild-type yAsf1 and the previously identified yAsf1 supersilencer mutants—188T, which lacks the C terminus, and 153Ins, which contains an insertion between the globular core and the C terminus—and had enhanced silencing compared to wild-type Asf1 (21) (Fig. 1B and data not shown). Here, truncations at amino acids 51, 140, and 149, which impact the conserved core of yAsf1, resulted in a loss of silencing, demonstrated by the inability to grow on 5-FOA and the appearance of white colonies on medium containing small amounts of adenine. In contrast, truncations at amino acid 210, and to a lesser extent 231, resulted in a gain of silencing, evidenced by an enhanced ability to grow on 5-FOA (both 210 and 231) and an increase in the red color of yeast grown on low-adenine plates (210) (Fig. 1B). Neither of these gain-of-function mutations was as striking as the 188T and 153Ins mutations. Truncations at residues 246, 257, and 260 and the internal deletions had wild-type silencing phenotypes. The gain-of-function supersilencing phenotype that occurs upon loss of the C terminus of Asf1 has been shown to correlate with a decrease in the ability of yAsf1 to coimmunoprecipitate histone H3 and an increase in histone H3 on chromatin, suggesting that the C-terminal tail of yAsf1 is important for interaction with histones (21). As such, these data (Fig. 1B and C) suggest that yAsf1 residues 188 to 231 may contribute to the yAsf1-H3/H4 interaction.
In vivo cross-linking reveals an interaction between Asf1 residue 210 and H3.
In order to determine whether the C-terminal tail of yAsf1 interacts directly with H3 or H4, we used an in vivo photo-cross-linking approach utilizing the unnatural photoreactive amino acid BPA (30, 32, 39, 48, 49). This method relies on the incorporation of BPA into yAsf1 by replacing specific codons with amber (UAG) stop codons. The stop codons are suppressed by supplying a CUA nonsense codon suppressor, tRNATyr, and a corresponding tyrosyl-tRNA synthetase. This results in the incorporation of BPA at the stop codon, leading to subsequent translational read-through and expression of full-length yAsf1 protein with a precisely placed cross-linkable amino acid. Cross-links are induced with UV light, and the resulting yAsf1 cross-linked proteins are isolated and analyzed by SDS-PAGE, followed by immunoblotting. An increase in the apparent molecular weight of Asf1 indicates the presence of a cross-linked protein. It has been suggested that the resolution limit of BPA is 10 Å (50), whereas common cross-linking reagents, such as dithiobis(succinimidylpropionate) (DSP), have a 12-Å spacer arm.
BPA incorporation and UV cross-linking were first validated in E. coli for the yAsf1-H3/H4 complex by introducing amber stop codons into the gene encoding yAsf1 at three positions (51, 140, and 149) near H3 or H4 in the crystal structure (7) (Fig. 2A). yAsf1-BPA-51 and yAsf1-BPA-140 cross-link to H3 in vitro, but yAsf1-BPA-149 did not (Fig. 2B), most likely because aa 149 is much closer to residues within Asf1 to which it can potentially cross-link more efficiently than to H3 or H4. The system was further validated in yeast using an amber stop codon at position 51 (Fig. 2C).
Fig 2.

Asf1 residue 210 with substitutions of BPA cross-links to histone H3. (A) Ribbon diagram of yAsf1 (aa 1 to 164) with H3 and H4 showing the proximity of yAsf1 residues Leu51, Leu140, and Phe149 (cyan) to H3. yAsf1 Trp153 is marked with an orange star. (B) Silver-stained gel (top) and Western blot analyses showing production of BPA-containing yAsf1 and cross-linking of Asf1-BPA-51 and Asf1-BPA-140 to histone H3 with UV treatment. E. coli transformed with a triple yAsf1-H3/H4 expression vector that harbors amber stop codons at position 51, 140, or 149 produced truncated yAsf1 in the absence of BPA. In the presence of BPA, full-length yAsf1 was observed. With UV treatment, cross-linked (xlinked) complexes that contain both yAsf1 and H3 appear. (C) yAsf1-Myc displays altered mobility (marked with an asterisk) only in the presence of photoactivatable artificial amino acid, BPA, the amber suppressor tRNA plasmid, and the Asf1-L51-UAG plasmid in yeast. Anti-myc immunoblot analysis of immunoprecipitated samples: in lanes 1 and 2, BKD094 (asf1Δ pRS314); in lanes 3 and 4, MCY001 (asf1Δ pRS314-Asf1-UAG-51); and in lanes 5 and 6, BKD0237 (asf1Δ pRS314-Asf1-UAG-51, pLH157-KAN). (D) Replacement of yAsf1 residues 51, 140, and 210 with BPA leads to cross-linked protein species. (Top) Anti-myc immunoblot analysis of extracts. (Bottom) Immunoprecipitated samples. The strains are BKD257 (None), BKD237 (51), BKD259 (140), BKD263 (149), BKD241 (210), BKD243 (231), BKD245 (246), BKD247 (249), BKD249 (257), BKD251 (260), BKD261 (267), and BKD255 (275). (E) yAsf1 residues 210 and 51, when replaced with BPA, cross-link to C-terminally FLAG-tagged histone H3. (Top) Immunoprecipitated yAsf1-Myc immunoblotted with anti-FLAG Ab. (Middle) Analysis of the same blot after being stripped and reprobed with anti-Myc Ab. (Bottom) Overlay of the anti-FLAG and anti-Myc Ab images. The blue/black bands indicate overlap between the Myc and FLAG signals. The HA and FLAG tags are not distinguishable by their electrophoretic mobilities. The identity of the Asf1-cross-linked central band, marked with a double asterisk, has not been established. Lanes 1 to 4, FLAG-H3 plus HA-H4; lanes 5 to 8, FLAG-H3 plus HA-H3. The strains are as follows: (lane 1) BKD382 (40), (lane 2) BKD378 (No UAG), (lane 3) BKD386 (210), (lane 4) BKD390 (asf1Δ), (lane 5) BKD379 (No UAG), (lane 6) BKD383 (51), (lane 7) BKD387 (210), and (lane 8) BKD391 (no Asf1). The yAsf1 signals seen in lanes 4 and 8 are due to interwell sample leakage.
Next, we determined whether substituting BPA for the stop codons in the C-terminal tail of full-length Asf1 (Fig. 1A) would allow in vivo cross-linking to other proteins when expressed in yeast. Consistent with the findings in E. coli (Fig. 2B), the mobility of yAsf1-Myc was decreased when BPA was substituted for yAsf1 core residues 51 and 140 but not 149 (Fig. 2D, lanes 2 to 4). Additionally, a more slowly migrating protein species was seen with the 210 substitution (Fig. 2D, lane 5) but not with other substitutions (Fig. 2D). However, the Asf1 cross-linked proteins were not reliably detected with antibodies to H3 or H4, likely due to weak antibody avidity.
To determine whether yAsf1-BPA-51 or yAsf1-BPA-210 cross-links to H3 or H4 in vivo, we epitope tagged both H3 and H4. HHT2 was C-terminally tagged with FLAG (H3-FLAG) in the genome in a yeast strain with ASF1 also deleted (BKD361; W1588 background). The strain was then transformed with either yAsf1-BPA-51 or yAsf1-BPA-210, the suppressor tRNA/tRNA synthetase plasmid, and a plasmid-borne copy of either N-terminally hemagglutinin (HA)-tagged HHT2 (HA-H3) or HHF2 (HA-H4).
Immunoprecipitation of Asf1 with anti-Myc Ab followed by immunoblotting and probing with anti-FLAG Ab indicates that H3-FLAG cross-links both to yAsf1-BPA-51, as expected, and to yAsf1-BPA-210 (Fig. 2E, top, lanes 1, 3, 6, and 7). We noted that the banding patterns of yAsf1-51-H3 (faster migration) and yAsf1-210-H3 (slower migration) were not identical to each other in either yeast or E. coli. This most likely results from the different positions (51 and 210) of BPA incorporation creating different cross-linked branched structures of Asf1 and histone H3, as has been suggested for the MOT1-TBP interaction probed by the same method (49). In yeast, we also noted other yAsf1-Myc cross-linked bands, in addition to the single bands representing the yAsf1-51-H3-FLAG and yAsf1-210-H3-FLAG cross-linked complexes. One of these is a faster-migrating species common to both Asf1-BPA-51 and Asf1-BPA-210 complexes. This band makes up the lower band of the Asf1-BPA-51 doublet and is the most prominent Asf1-BPA-210 cross-linked complex. The complex most likely arises from the endogenous HHT1 gene that encodes H3 and terminates with a native amber stop codon, especially as this species appears at low levels in strains where Asf1-Myc lacks the introduced UAG stop codon (Fig. 2E, middle, lanes 2 and 5). As noted above, yAsf1-210-H3 displays three distinct cross-linked species. The middle band represents a new species (Fig. 2E, middle, lane 7, marked with a double asterisk). To identify this protein, we tested yAsf1-BPA-210 for cross-linking to HA-H4 and other small Asf1 binding proteins (Sas5-FLAG, Rpc19-FLAG, and Spt15-FLAG) but failed to detect the other proteins (data not shown). Nonetheless, these data show that residue 210, when replaced with BPA, contacts histone H3, thus defining a novel interaction between the C terminus of Asf1 and H3.
The C terminus of Asf1 is important for its association with histones H3 and H4 in vitro.
C-terminal truncations of yAsf1 created by transposon insertion mutagenesis reduced the ability of yAsf1 to coimmunoprecipitate with histone H3 in yeast extracts (21). These results could be explained most simply by a decrease in the affinity of the histones for the truncated yAsf1 proteins. The C terminus of yAsf1 is highly acidic, which might suggest that the tail enhances histone interaction via charge-charge interactions. Alternatively, a specific region of the tail may contribute more significantly to histone binding. In order to distinguish between these possibilities, we compared the binding affinities of C-terminally truncated yAsf1 proteins for histones H3/H4. Previously, we found a tight association between full-length yAsf1 and the H3/H4 dimer using a fluorescence-quenching assay (36, 51). Using the same approach (Fig. 3A), we measured the binding affinities of yAsf11-155, yAsf11-169, yAsf11-185, yAsf11-210, yAsf11-246, and full-length yAsf11-279 for H3/H4 (Fig. 3B). Unlabeled H3/H4 dimers quenched the Alexa Fluor 532 signal of the labeled yAsf1 proteins. These fluorescence data were fitted with a ligand depletion binding model (equation 1) or single-site binding isotherm (equation 2), depending on the KD value. The resulting KD values vary from 1 nM to 228 nM for the full-length yAsf1 to yAsf1 lacking the C-terminal tail (Fig. 3C). Interestingly, yAsf11-210, yAsf11-246, and full-length yAsf11-279 bind with identical affinities to H3/H4, while yAsf11-185 binds with a 15-fold-weaker affinity. This demonstrates that the yAsf1 region between amino acids 186 and 210 contributes to histone binding and is consistent with the cross-link that we observed between Asf1 amino acid 210 and H3 in vivo (Fig. 3). The shorter constructs, yAsf11-169 and yAsf11-155, bind with increasingly weaker affinity and demonstrate that the C-terminal tail of Asf1 contributes directly to a more than 200-fold enhancement of histone H3/H4 heterodimer binding affinity in vitro.
Fig 3.
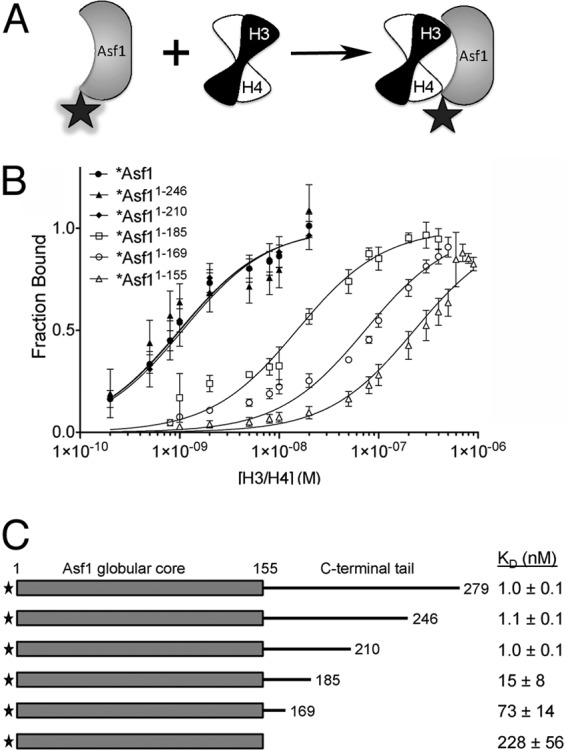
A specific region on the yAsf1 tail enhances binding affinity to H3/H4. (A) Asf1 proteins were fluorophore labeled at a position not directly involved in binding to H3/H4 dimers. The fluorophore (shaded star) fluoresces strongly in the free Asf1 protein. However, the binding of the H3/H4 dimer to Asf1 results in quenching of this fluorescence (plain star). (B) Analysis of fluorescence-quenching data for titration of histones H3/H4 into 1 nM Alexa Fluor 532-labeled full-length yAsf1 (1 to 279) and C-terminal truncations (1-246, 1-210, 1-185, 1-169, and 1-155). The data were fitted with a ligand-depleted binding model (equation 1) or single site-binding isotherm (equation 2), depending on the final KD values. The error bars indicate standard deviations from three independent experiments. (C) Table of KD values aligned with a diagram showing yAsf1 C-terminal tail truncations demonstrating that residues 156 to 209 in the C-terminal tail of yAsf1 contribute to the binding affinity of yAsf1 for H3/H4.
The C-terminal tails of hAsf1a and hAsf1b influence transcriptional silencing in yeast.
Deletion of the yAsf1 C terminus leads to a supersilencing phenotype, diminished yAsf1-histone interaction, and increased histone occupancy on the genome in the context of cac1 mutants (21, 22) (Fig. 1B). To indirectly determine whether the C-terminal tails of the human Asf1 proteins might also interact with histones, we asked whether they can suppress the supersilencing phenotype that is caused by lack of the C terminus of yAsf1. We fused the Myc-tagged hAsf1a tail (yAsf1-atail) or hAsf1b tail (yAsf1-btail) to the conserved N terminus of yAsf1 at the genomic yeast locus using the conserved W153 residue as the site of integration (Fig. 1A). After verifying equivalent protein expression (data not shown), these strains were assayed for silencing. In the sensitized cac1 background, the yAsf1-human Asf1b tail chimera (yAsf1-btail) displayed an increase in silencing over yAsf1 at the TELVIIL::URA3 reporter (Fig. 4A and B) that was only slightly less than that of the ASF1 CAC1 strain, indicating that the human Asf1b tail does not suppress the supersilencing phenotype that occurs upon deletion of the yAsf1 C-terminal tail (Fig. 1B, 188T). The yAsf1-human Asf1a tail chimera (yAsf1-atail) displayed only a subtle increase in silencing compared to yAsf1 in the cac1 background (Fig. 4A and B). Analysis of the silencing at HMRa::ADE2 showed that the C-terminal Myc tag itself appears to slightly increase silencing at HMRa::ADE2 (compare ASF1 cac1Δ to yAsf1 cac1Δ). Moreover, the fusion of the human Asf1a tail to the N terminus of yAsf1 (yAsf1-atail) suppressed the supersilencing phenotype observed for yAsf1 lacking its C-terminal tail (Fig. 4A and B). These results show that the tail of hAsf1a more closely resembles the tail of yAsf1 in its ability to suppress the supersilencing phenotype and likely also in mediating the additional interaction with H3/H4.
Fig 4.

Influence of hAsf1a and hAsf1b tails on the functions of the yAsf1 and hAsf1 cores. (A) Human tails enhance silencing at TELVIIL::URA3 in a cac1Δ background. Strains: ROY1172 (ASF1 CAC1), ROY1171 (ASF1 cac1Δ), BKD185 (yAsf1-myc cac1Δ), BKD198 (yAsf1-atail cac1Δ), BKD194 (yAsf1-btail cac1Δ), and ROY1169 (asf1Δ cac1Δ). (B) Influence of hAsf1a, hAsf1b, and hAsf1a/b and hAsf1b/a chimeras on silencing. (Top) ROY1172 (ASF1 CAC1), ROY1171 (ASF1 cac1Δ), BKD185 (yAsf1 cac1Δ), BKD200 (hAsf1a cac1Δ), BKD235 (hAsf1a-btail cac1Δ), BKD198 (yAsf1-atail cac1Δ), and ROY1169 (asf1Δ cac1Δ). (Bottom) ROY1172 (ASF1), ROY1171 (cac1Δ), BKD185 (yAsf cac1Δ), BKD202 (hAsf1b cac1Δ), BKD196 (hAsf1b-atail cac1Δ), BKD194 (yAsf1-btail cac1Δ), and ROY1169 (asf1Δ cac1Δ). (C) Sensitivity of hAsf1a, hAsf1b, and hAsf1a/b and hAsf11b/a chimeras to DNA-damaging and replicational-stress-inducing drugs (100 mM HU, 0.01% MMS, or 5 μg/ml zeocin). (Top) ROY1172 (ASF1), BKD187 (yAsf1), BKD188 (yAsf1), BKD135 (hAsf1a), BKD136 (hAsf1a), BKD130 (hAsf1b), BKD131 (hAsf1b), and ROY1170 (asf1Δ). (Bottom) ROY1172 (ASF1), BKD187 (yAsf1), BKD188 (yAsf1), BKD144 (hAsf1a-btail), BKD146 (hAsf1a-btail), BKD149 (hAsf1b-atail), BKD150 (hAsf1b-atail), and ROY1170 (asf1Δ).
To ask whether the identity of the human Asf1 tail can influence the function of the N-terminal domain of human Asf1a and Asf1b in yeast, we transplaced the Myc-tagged tail of hAsf1a onto the core of hAsf1b, and vice versa. We compared the functions of these chimeras to those of full-length Myc-tagged hAsf1a, hAsf1b, and yAsf1, all of which were integrated into the yeast genome. We found that the identity of the tail on the hAsf1 cores does not affect the growth rate or sensitivity to genotoxic stress (Fig. 4C, data not shown). Yeast cells expressing the hAsf1a core were highly sensitive to genotoxic stress, with growth defects on par with those of asf1 deletions (Fig. 4C). In contrast, yeast cells expressing the hAsf1b core were less sensitive to genotoxic stress, particularly to HU, than those expressing hAsf1a cores (Fig. 4C). The expression levels of yAsf1, hAsf1a, and hAsf1b were comparable (data not shown). These findings demonstrate that neither hAsf1a nor hAsf1b can fully compensate for yAsf1; that hAsf1a is a poor substitute for yAsf1; and, consistent with the conclusions of Tamburini et al. (22), that hAsf1b partially substitutes for yAsf1 in response to HU treatment, suggesting a functional role in protection against replicational stress. Notably, hAsf1b showed greater growth defects than were reported in a previous paper (22). This is likely because the hAsf1b strain (KDY006) previously used was a mixture of yAsf1- and hAsf1b-expressing yeast (data not shown), and although unfortunate, it is not surprising: deletion of a gene is accompanied by a duplication event nearly 8% of the time (52).
Yeast Asf1 is phosphorylated on threonine 270.
In yeast, the tail of yAsf1 has roles in both histone binding and transcriptional silencing. In human cells, the C terminus of Asf1 is phosphorylated by the Tousled-like kinases, which also phosphorylate Drosophila melanogaster, but not S. cerevisiae, Asf1 in vitro (14). However, a yAsf1-phospho-T270 peptide identified by mass spectrometry copurified with Rad53, a DNA damage checkpoint kinase (25), suggesting that the C terminus of Asf1 can be phosphorylated in yeast and that this phosphorylation might be important for interaction with Rad53. We found that yAsf1 is phosphorylated in yeast via multiple approaches. The acrylamide-pendant Phos-tag ligand binds to phosphate, causing an upward gel mobility shift (43) on SDS-PAGE. Immunoprecipitated yAsf1-Myc, expressed from the pRS314-Asf1-Myc plasmid and analyzed using Mn2+ Phos-tag SDS-PAGE, generated two distinct forms, one corresponding to yAsf1-Myc and a more slowly migrating species that was diminished upon lambda phosphatase treatment (Fig. 5A). Two-dimensional gel electrophoresis demonstrated a range of migration for yAsf1, which could result from multiple phosphorylations or other modifications (Fig. 5B). Liquid chromatography (LC)–MS-MS performed on immunoprecipitated samples of the wild type and T265A, T270A, and T265A/T270A mutants, all introduced onto the pRS314-Asf1-Myc plasmid, revealed that yAsf1 is phosphorylated on residue T270 (Fig. 5C and Table 3). Finally, an affinity-purified antibody raised to a yAsf1-T270 phosphopeptide recognized a protein of the expected molecular weight in wild-type but not yAsf1-T270A cells (Fig. 5D). These results demonstrate that full-length endogenous yAsf1 is phosphorylated at T270 in vivo.
Fig 5.

Asf1 is phosphorylated on T270, and mutations in the Asf1-histone binding surface prevent Asf1-Rad53 coimmunoprecipitation. (A) yAsf1-Myc extracts from strain HZY1161, expressing FLAG-tagged Rad53 and containing pRS314-Asf1-Myc, were treated with and without λ phosphatase and phosphatase inhibitors. The immunoprecipitates were resolved by Mn2+ Phos-tag SDS-PAGE and analyzed by immunoblotting with anti-Myc antibodies. (B) Asf1 might be multiply phosphorylated. Shown is a 2D gel analysis of yAsf1-Myc immunoprecipitated from strain HZY1161 containing pRS314-Asf1-Myc and blotted with anti-Myc antibodies. Putative yAsf1 phosphorylation states are circled. (C) LC–MS-MS analysis of a tryptic digest of Asf1-myc immunoprecipitated from strain JKT200 carrying pRS314-Asf1-Myc. The assigned fragment ions are shown on the peptide sequence. (D) An affinity-purified antibody raised to phosphorylated yAsf1 T270 shows a signal on an immunoblot of whole-cell extracts of JKT200 with pRS314-Asf1-Myc, but not with pRS314-Asf1T270A-Myc, at dilutions from 1:50 to 1:200. (E and F) pRS314-Asf1-Myc plasmids or an empty-vector control (pRS314) was transformed into HZY1161 expressing FLAG-tagged Rad53. JKT0200 lacks ASF1 and FLAG-tagged Rad53. yAsf1-Myc was coimmunoprecipitated with Rad53-FLAG, and immoprecipitated samples were analyzed by immunoblotting. (E) Coimmunoprecipitation of WT or mutated yAsf1-Myc with Rad53-FLAG shows that mutations of yAsf1 T265 and/or T270 did not disrupt its association with Rad53. A cross-reacting protein that migrates more quickly than yAsf1-Myc (crossreaction), is detected by the anti-Myc Ab and is present in the absence of Asf1 (JKT0200). A more slowly migrating cross-reacting species is occasionally seen with the anti-FLAG Ab, and is present in the absence of Rad53-FLAG (JKT0200). (F) yAsf1 mutations to prevent or mimic phosphorylation of T270 do not abolish or enhance the association of yAsf1 and Rad53. (G) Asf1's histone binding region is important for Rad53 association. Coimmunoprecipitation of WT or mutated yAsf1-Myc with Rad53-FLAG was analyzed by immunoblotting.
Table 3.
MS confirmation that Asf1 T270, not T265, is phosphorylated in vivo
| Straina | Peptideb | Scorec | Expect (P) value |
|---|---|---|---|
| Asf1 | KIEGGSTDIESTPK | 41.5 | <0.05 |
| KIEGGSTDIEStPK | 36.0 | ||
| KIEGGSTDIEsTPK | 34.2 | ||
| IEGGStDIESTPK | 20.9 | ||
| T265A | IEGGADIESTPK | 36.9 | <0.05 |
| IEGGADIEStPK | 23.2 | ||
| T270A | KIEGGSTDIESAPK | 45.3 | <0.05 |
| IEGGSTDIESAPK | 33.7 | ||
| T265A | KIEGGSADIESAPK | 43.0 | <0.05 |
| T270A | IEGGSADIESAPK | 34.5 |
Tandem MS performed on Asf1 immunoprecipitated from JKT200 strains possessing pAsf1-Myc or mutagenized pAsf1-Myc showed that only Asf1 from strains in which Asf1-T270A was present prevented the identification of phosphorylated peptides.
Residues in lowercase were identified as phosphorylated.
The scores are discriminate scores, a statistical measure of confidence in the identification of individual peptides generated by the Mascot or Protein Prospector analysis program.
Mutation of Asf1-T270 does not prevent Rad53 association.
Rad53 and yAsf1 interact with each other to the exclusion of histones (11), yet the regulation of the yAsf1-Rad53 interaction is not fully understood. Previously, Schwartz and others (13) demonstrated that the FHA1 domain of Rad53 could be used to purify yAsf1 from untreated yeast lysates but not from those treated with lambda phosphatase, suggesting that phosphorylation regulates the Asf1 interaction with Rad53. We tested whether phosphorylation of Asf1 at T270 was required for its interaction with full-length Rad53. We found that Rad53-FLAG coimmunoprecipitated with WT and mutant (T265A, T270A, and T265/T270) forms of yAsf1 (Fig. 5E), showing that phosphorylation of yAsf1 at T270 is not required for its interaction with Rad53. Similarly, when yAsf1 T270 was mutated to E to mimic constitutive phosphorylation, coimmunoprecipitation of yAsf1 and Rad53 was not enhanced (Fig. 5F). This agrees with the recent report of Jiao et al. (12), who found that although a peptide containing the Rad53 FHA1 domain associated with yAsf1 only when yAsf1 was phosphorylated in vitro on T270, the full-length yAsf1 and Rad53 yeast proteins coimmunoprecipitated independently of phosphorylation at T270. Therefore, other portions of Asf1 and Rad53 must contribute to the interaction of these two proteins.
Residue substitutions of Asf1 in the H3 histone binding interface disrupt Rad53 association.
yAsf1 can bind to either Rad53 or histones but not to both (11), suggesting that Rad53 and H3/H4 share the same binding surface on yAsf1. Amino acid substitutions in yAsf1 that disrupt histone binding (7) were tested by coimmunoprecipitation for their effects on the Asf1-Rad53 interaction (Fig. 5G). All mutations were introduced onto the plasmid pRS314-Asf1-Myc. The double mutants Y112A/T147E, T147E/S48R, Y112A/V146L, Y112A/R145E, and T147E/R145E, which abolish the Asf1-histone interaction, also abolish the yAsf1-Rad53 interaction (Fig. 5G). Single substitutions in residues important for the yAsf1-H3 interaction (V94R, R145E, and T147E) also disrupted the Rad53 interaction. However, substitutions in residues important for the yAsf1-H4 interaction gave intermediate results. V146L (Fig. 5G) and V109M (data not shown) had little effect on the yAsf1-Rad53 interaction, P144Y disrupted association, and L6M and Y162A had low to moderate levels of Rad53 association (Fig. 5G). Thus, Rad53 and Asf1 can interact through a common H3/H4 binding region of yAsf1.
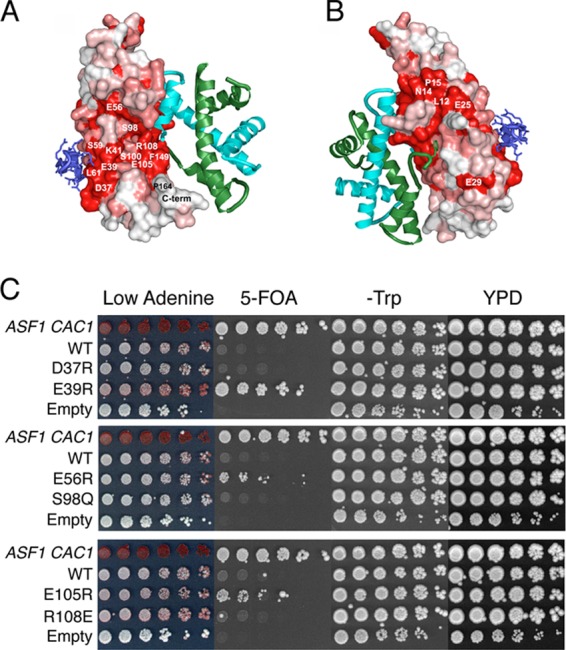
Surface residues on Asf1 near the site of histone interaction influence silencing.
The histone H3/H4 and CAF-1/HIRA histone chaperone binding sites contained within the globular core of yAsf1 are highly conserved. Two other conserved surfaces on Asf1 remain unoccluded even when histones H3/H4 and other chaperones interact with yAsf1. One surface lies between the histone and CAF-1/Hir binding sites (Fig. 6A) and includes a serine pocket (S98 and S100) surrounded by several charged residues, E105, R108, E39, K41, and E56. The other surface is on the opposite face of Asf1 and includes residues L12, N14, and E25 (Fig. 6B). To learn whether any of these residues are important for Asf1 function, we made S98Q, S100Q, D37R, E39R, K41E, E56E, E105R, and R108E amino acid substitutions in and around the serine pocket and L12A and -R, N14A and -R, E25R, and E29A and -R substitutions on the opposite face of Asf1.
Fig 6.
Surface mutations on Asf1's conserved core display various degrees of silencing. (A and B) The Asf1-H3/H4 complex is shown docked with the B-domain peptide of HIRA (blue). Asf1 colored according to sequence conservation, with absolutely conserved residues (red), highly similar residues (pink), and the least-conserved residues (white) mapped onto the surface. (C) Wild-type strain ROY1172 (ASF1 CAC1) carrying an empty vector marked with TRP1 or ROY1169 (asf1Δ cac1Δ) carrying plasmid-borne copies of either WT or mutated Asf1 (strains BKD094, -095, -103, -104, -106, -107, -109, and -110) were serially diluted 10-fold onto the indicated media.
Each mutation was introduced onto pRS314-Asf1-Myc and transformed into JKT200 (asf1Δ) and ROY1169 (asf1Δ cac1Δ). We examined the mutants' in vivo responses to hydroxyurea, MMS, and zeocin; their abilities to disassemble chromatin at the PHO5 promoter in a low-phosphate environment (6); and silencing at HMRa and TELVIIL. Except for the N14A mutation, which appeared to be poorly expressed and/or unstable (data not shown), none of these mutations impacted either growth in response to genotoxic stress (data not shown) or the induction of the PHO5 promoter (data not shown). However, the E39R, E56R, and E105R mutations all led to an increase in silencing over the WT control (Fig. 6C), with E39R showing the most striking effects. Taken together, these results show that the major functions of Asf1 are intact in these mutants but that some residues surrounding the serine pocket influence silencing, implicating the region as important for binding to one or more proteins that also affect silencing.
DISCUSSION
We have investigated the function of the Asf1 C terminus in yeast. Our studies reveal that Asf1 residue 210 cross-links to histone H3 in vivo and that loss of the Asf1 C-terminal tail reduces Asf1's affinity for H3/H4 nearly 200-fold in vitro. Functionally, the interaction between the C terminus of Asf1 and H3 appears to contribute to position-dependent transcriptional silencing. Additionally, we have shown that the tail of yAsf1 is phosphorylated at T270 but that this phosphorylation is not required for Asf1 and Rad53 to coimmunoprecipitate from yeast extracts. However, Asf1 surface residues that contribute to histone binding are required for a robust Asf1-Rad53 interaction. Finally, we show that other mutations on the surface of Asf1 that do not contribute to histone, Hir, or CAF-1 binding also influence Asf1's role in transcriptional silencing.
Previous structural analyses of the Asf1-H3/H4 complex revealed extensive interactions between the globular N-terminal core of Asf1 and both histones H3 and H4 (7, 8). The present study reveals an additional significant Asf1-H3 interaction between the C-terminal tail of Asf1 and H3. Exactly where on H3 the C terminus of Asf1 binds is not yet clear, but it is distinct from the H3-H3 dimerization surface that is occluded by binding to Asf1 (7). In addition, the interaction between the C-terminal tail of Asf1 and H3 also does not require histone posttranslational modification, as the proteins expressed in E. coli interact (Fig. 3). Given that acetylation of the N terminus of histone H3 and lysine 56 of H3 are required for efficient chromatin assembly following DNA synthesis (24, 53, 54), it will be interesting to determine whether these posttranslational modifications on free histones weaken the interaction between the tail of ASf1 and H3. If so, such a mechanism could promote the transfer of dimers of H3/H4 from Asf1 to the CAF-1 and Rtt106 histone chaperones to enable the formation of H3/H4 tetramers prior to chromatin assembly. Consistent with this idea, yeasts that lack the C-terminal tail of Asf1 have higher histone H3 occupancy on the DNA, perhaps due to the enhanced chromatin assembly that would result from weaker Asf1-H3 affinity (21).
The N terminus of Asf1 is highly conserved, but the C-terminal tail is not. In yeast, the tail is highly polyanionic, which might help stabilize the interaction between yAsf1 and its positively charged histone cargo. To begin to examine whether the tail displays specific or general interactions with histones, we tested several yAsf1 tail truncations for their functions in yeast. Truncations at residues 210 and 231 have small but reproducible increases in transcriptional silencing, and given that deletion of the tail at residue 188 also increases silencing (Fig. 1B and C) (21), this suggests that a region of the tail between residues 188 and 231 may contribute to histone binding. Using BPA substitutions, we determined that Asf1 C-terminal tail residue 210, as well as N-terminal core residues 51 and 140, cross-link to histone H3 in yeast, E. coli, or both (Fig. 2). Further, loss of the yAsf1 C terminus reduces Asf1's affinity for histones H3 and H4 in vitro, and the proximal 55 residues on the yAsf1 tail (155 to 210), but not the distal 67 residues (211 to 279), mediate this novel association (Fig. 3). Given that the C-terminal tail of Asf1 is highly acidic while histones are highly basic, it could be argued that there is a charge threshold that is required for the contribution of the Asf1 C-terminal tail to the histone interaction and that truncations beyond residue 185 progressively reduce the histone interaction due to the reduced remaining charge of the yAsf1 C-terminal tail. While this is a possibility, it is inconsistent with the finding that a small insertion mutation at Asf1 amino acid 153, with all of its acidic C-terminal residues still intact, has the supersilencing phenotype (Fig. 1B) that is diagnostic for loss of the interaction between the C terminus of Asf1 and H3/H4 (21). Furthermore, the C-terminal tail of human Asf1a, when fused to the N terminus of yAsf1, can largely suppress the supersilencing phenotype (Fig. 4B). Unlike yAsf1, the C-terminal tail of human Asf1a is not highly charged, arguing against a charge threshold requirement for the interaction between histones and the C terminus of yAsf1. Therefore, we propose that the region between residues 155 and 210 of the C terminus of yAsf1 mediates specific interactions with H3/H4.
In light of our previous findings that hAsf1a has an important role in DNA damage response whereas hAsf1b has an important role in DNA replicational-stress response (22), we sought to delineate the contribution of the human Asf1a and Asf1b tails to these functions. By making chimeric Asf1 proteins containing the core of one Asf1 protein (residues 1 to 153) and the tail of another, we found that the protein cores are most important for cellular function (Fig. 4B and C), which supports work showing that the yAsf1 core substitutes for full-length Asf1 in both S. cerevisiae (15) and S. pombe (18). However, both the hAsf1a and hAsf1b tail chimeras with the yeast core conferred an increase in silencing at TELVIIL::URA3 that was most striking for the hAsf1b tail (Fig. 4A and B). It has been suggested recently that the silencing assays we employed here may not directly measure transcription in silenced regions, but rather, may measure either an overall upregulation of poorly expressed genes or changes in purine/pyrimidine metabolism (47). Therefore, it is possible that the phenotypic variations we see in these assays are due to the influence of Asf1 on base metabolism rather than silencing per se. Nonetheless, these results indicate a functional role for the C-terminal tail of Asf1.
The HIRA complex is important for heterochromatic gene silencing in mammals (55), and the related Hir complex is important for position-dependent transcriptional silencing in yeast (56, 57). The fact that the hAsf1b tail had a stronger influence on silencing than did the hAsf1a tail seems opposed to the finding that the hAsf1b tail interfered with HIRA binding whereas the hAsf1a tail enhanced HIRA binding (58), even though both parts of the core (aa 1 to 30) and the tail (aa 155-end) of hAsf1a and hAsf1b differentially influenced their abilities to interact with the B domain of HIRA. Alternatively, because the hAsf1a tail behaves more like the endogenous yeast tail, it may be that the hAsf1a tail is a better substitute, in yeast, for transcriptional silencing than the hAsf1b tail. This result is interesting, given that hAsf1a can immunoprecipitate with both HIRA and CAF-1, but hAsf1b can do so only with CAF-1 (58). Thus, while the globular cores of the two human Asf1 isoforms may be capable of interaction with HIRA and CAF-1, the respective tails may enhance or weaken those interactions.
Interestingly, the differentiation of hAsf1a and hAsf1b tail functions in yeast was lost in the context of the human cores: the hAsf1a-btail chimera behaved like full-length hAsf1a, and the hAsf1b-atail chimera behaved like full-length hAsf1b (Fig. 4). Therefore, it is likely that core differences, rather than tail differences, primarily dictate hAsf1a and hAsf1b functions in yeast. Notably, neither human Asf1a nor Asf1b could fully substitute for yAsf1, but in logarithmically growing cells, hAsf1b more fully compensated for yAsf1 (Fig. 4 and data not shown) (22). This agrees with previous demonstrations that hAsf1a could not substitute for either S. pombe Asf1 (18) or S. cerevisiae Asf1 (16).
In human cells, phosphorylation of hAsf1a by the Tousled-like kinases (14) protects it from degradation via a proteasome-dependent mechanism (23). We have shown that full-length yAsf1 is phosphorylated at T270 (Fig. 5) and that yAsf1 may be multiply phosphorylated (Fig. 5B). If phospho-T270 were to influence yAsf1 stability, one might expect a decrease in yAsf1 protein with T270 mutants, but no obvious decreases in yAsf1 protein were seen (Fig. 5E and F), nor was there an obvious loss-of function phenotype, as might be expected for a subfunctional amount of protein, associated with mutations at T270, T265, or T265 and T270 (data not shown). Given that yAsf1 is potentially multiply phosphorylated, it is still possible that phosphorylation stabilizes yAsf1 but that loss of phosphorylation at T270 was not sufficient to see an effect. Alternatively, if yAsf1 functions more like hAsf1b, as our assays indicate, then the phosphorylation state of yAsf1 might not impact its stability or function, as phosphorylation of hAsf1b, unlike hAsf1a, does not correlate with stability or function (23). Future studies will reveal whether the phosphorylation of yAsf1 changes through the cell cycle or influences other protein interactions.
The finding that lambda phosphatase treatment of yeast extracts inhibited the interaction between yAsf1 and GST-Rad53-FHA1 provided evidence that the interaction between yAsf1 and Rad53 might depend on phosphorylation (13). Indeed, phosphorylation at yAsf1 T270 is required for the binding of the Rad53 FHA1 domain (12), but disruption of this phosphorylation site is not sufficient to prevent coimmunoprecipitation of yAsf1 with Rad53 (Fig. 5E and F) (12), suggesting that yAsf1 and Rad53 must have other sites of interaction. In fact, mutations influencing histone binding to Asf1 (7), particularly those important for H3 binding, decreased the ability of Rad53 to coimmunoprecipitate Asf1 (Fig. 5G). Others have shown, using different Asf1 mutations, that both the H3/H4 histone interaction surface and the HIRA/CAF-1 binding surface on yAsf1 contribute to Rad53 association (12). Further investigation will be required to reveal the mechanism and functional consequences of the exchange of Asf1 and Rad53 as histone binding partners.
Although it is increasingly apparent that Rad53, histones H3 and H4, and CAF-1/HIRA all compete for binding to yAsf1, the region on the surface of yAsf1 near the Rad53/histone and CAF-1/HIRA binding sites has not been thoroughly explored. Mutations in conserved residues near these regions revealed that E39R, E56R, and E105R influence the activity of yAsf1 in transcriptional- silencing assays. The E39R mutation, which had the most profound influence (Fig. 6C) when combined with D37R, influences silencing without altering histone H3 binding (16), decreases the Asf1-Rad53 interaction (12), and contributes to HIRA/CAF-1 binding (58). Mutations in D37 disrupt the Asf1-Hip1B interaction, and the E39 residue may interact with HIRA/CAF-1 (59). The E56 residue has not been shown to interact with chaperones, histones, or Rad53, but the nearby residue D54 undergoes a chemical shift upon H3 binding and, when mutated, weakens the H3-Asf1 interaction (16). Additionally, D58 may help support Asf1 binding to Hip1B and Cac2 (59). Likewise, mutations at R108 have been shown to reduce the Asf1-histone interaction accompanied by an increase in silencing at HMRa::URA3 (16). Taken together, these results suggest it is likely that both E39R and E56R impact silencing via HIRA, but a reason for the effect of the E105R mutation is less clear. Future studies will determine exactly how the Asf1-Rad53 and Asf1-histone interactions are regulated, as well as whether and how this binding might regulate the association of Asf1 with CAF-1 and Hir, as Asf1 cannot associate with these complexes when bound by Rad53 (12). It will also be important to determine the roles of cell cycle progression, DNA replication, and DNA damage in regulating the binding partners and activity of Asf1. This is especially intriguing as newly synthesized histones H3 and H4 interact with yAsf1 (5) while Rad53 has a role in the degradation of excess free histones in yeast (60). Further, the association of Asf1 and Rad53 is disrupted in the presence of HU, suggesting that binding partners change in response to DNA damage and/or replicational stress. The fact that the same mutations on yAsf1 can influence both H3/H4 and Rad53 binding might also suggest that the previously identified loss of yAsf1 function phenotypes (7) is due not only to abrogation of histone binding, but also to alterations in Rad53 binding. If possible, it will be interesting to identify Asf1 mutants that do not interact with histones but that are still competent to immunoprecipitate Rad53, and vice versa, in order to more carefully define and distinguish the functions of these two important proteins.
ACKNOWLEDGMENTS
We are grateful to David Jones for the use of the Horiba Fluorolog-3 spectrometer, Robert Hodges for the use of the Rayonet photochemical reactor RPR-100, Michael McAndrew for help with PCR screening of the FLAG-tagged genes, Stephanie Williams for performing PHO5 induction assays, and Elizabeth Doggett for the construction of EDY002. We thank Jean Scorgie for help with the fluorescence assay, Myrriah Chavez for help with some of the early studies, Yumeng Hao for help with plasmid construction, Steve Hahn for the gift of pLH157, and Richard Baker for the gift of pRB294 containing a NotI-HA cassette.
We acknowledge support from NIH GM 064475 and CPRIT to J.K.T. and NIH GM GM079154 to M.E.A.C.
Footnotes
Published ahead of print 26 November 2012
REFERENCES
- 1. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251– 260 [DOI] [PubMed] [Google Scholar]
- 2. Ransom M, Dennehey BK, Tyler JK. 2010. Chaperoning histones during DNA replication and repair. Cell 140: 183– 195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Le S, Davis C, Konopka JB, Sternglanz R. 1997. Two new S-phase-specific genes from Saccharomyces cerevisiae. Yeast 13: 1029– 1042 [DOI] [PubMed] [Google Scholar]
- 4. Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. 1998. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics 150: 613– 632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tyler JK, Adams CR, Chen SR, Kobayashi R, Kamakaka RT, Kadonaga JT. 1999. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402: 555– 560 [DOI] [PubMed] [Google Scholar]
- 6. Adkins MW, Howar SR, Tyler JK. 2004. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol. Cell 14: 657– 666 [DOI] [PubMed] [Google Scholar]
- 7. English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. 2006. Structural basis for the histone chaperone activity of Asf1. Cell 127: 495– 508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Natsume R, Eitoku M, Akai Y, Sano N, Horikoshi M, Senda T. 2007. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature 446: 338– 341 [DOI] [PubMed] [Google Scholar]
- 9. Tyler JK, Collins KA, Prasad-Sinha J, Amiott E, Bulger M, Harte PJ, Kobayashi R, Kadonaga JT. 2001. Interaction between the Drosophila CAF-1 and ASF1 chromatin assembly factors. Mol. Cell Biol. 21: 6574– 6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Green EM, Antczak AJ, Bailey AO, Franco AA, Wu KJ, Yates JRIII, Kaufman PD. 2005. Replication-independent histone deposition by the HIR complex and Asf1. Curr. Biol. 15: 2044– 2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emili A, Schieltz DM, Yates JRIII, Hartwell LH. 2001. Dynamic interaction of DNA damage checkpoint protein Rad53 with chromatin assembly factor Asf1. Mol. Cell 7: 13– 20 [DOI] [PubMed] [Google Scholar]
- 12. Jiao Y, Seeger K, Lautrette A, Gaubert A, Mousson F, Guerois R, Mann C, Ochsenbein F. 2012. Surprising complexity of the Asf1 histone chaperone-Rad53 kinase interaction. Proc. Natl. Acad. Sci. U. S. A. 109: 2866– 2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwartz MF, Lee SJ, Duong JK, Eminaga S, Stern DF. 2003. FHA domain-mediated DNA checkpoint regulation of Rad53. Cell Cycle 2: 384– 396 [PubMed] [Google Scholar]
- 14. Sillje HH, Nigg EA. 2001. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr. Biol. 11: 1068– 1073 [DOI] [PubMed] [Google Scholar]
- 15. Daganzo SM, Erzberger JP, Lam WM, Skordalakes E, Zhang R, Franco AA, Brill SJ, Adams PD, Berger JM, Kaufman PD. 2003. Structure and function of the conserved core of histone deposition protein Asf1. Curr. Biol. 13: 2148– 2158 [DOI] [PubMed] [Google Scholar]
- 16. Mousson F, Lautrette A, Thuret JY, Agez M, Courbeyrette R, Amigues B, Becker E, Neumann JM, Guerois R, Mann C, Ochsenbein F. 2005. Structural basis for the interaction of Asf1 with histone H3 and its functional implications. Proc. Natl. Acad. Sci. U. S. A. 102: 5975– 5980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Padmanabhan B, Kataoka K, Adachi N, Horikoshi M. 2002. Purification, crystallization and preliminary X-ray diffraction analysis of yeast nucleosome-assembly factor Cia1p. Acta Crystallogr. D 58: 1876– 1878 [DOI] [PubMed] [Google Scholar]
- 18. Umehara T, Chimura T, Ichikawa N, Horikoshi M. 2002. Polyanionic stretch-deleted histone chaperone cia1/Asf1p is functional both in vivo and in vitro. Genes Cells 7: 59– 73 [DOI] [PubMed] [Google Scholar]
- 19. Franco AA, Lam WM, Burgers PM, Kaufman PD. 2005. Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 19: 1365– 1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minard LV, Williams JS, Walker AC, Schultz MC. 2011. Transcriptional regulation by Asf1: new mechanistic insights from studies of the DNA damage response to replication stress. J. Biol. Chem. 286: 7082– 7092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tamburini BA, Carson JJ, Linger JG, Tyler JK. 2006. Dominant mutants of the Saccharomyces cerevisiae ASF1 histone chaperone bypass the need for CAF-1 in transcriptional silencing by altering histone and Sir protein recruitment. Genetics 173: 599– 610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tamburini BA, Carson JJ, Adkins MW, Tyler JK. 2005. Functional conservation and specialization among eukaryotic anti-silencing function 1 histone chaperones. Eukaryot. Cell 4: 1583– 1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pilyugin M, Demmers J, Verrijzer CP, Karch F, Moshkin YM. 2009. Phosphorylation-mediated control of histone chaperone ASF1 levels by Tousled-like kinases. PLoS One 4: e8328 doi:10.1371/journal.pone.0008328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. 2008. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134:231– 243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smolka MB, Albuquerque CP, Chen S-H, Schmidt KH, Wei XX, Kolodner RD, Zhou H. 2005. Dynamic changes in protein-protein interaction and protein phosphorylation probed with amine-reactive isotope tag. Mol. Cell. Proteomics 4: 1358– 1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Longtine MS, McKenzie AIII, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953– 961 [DOI] [PubMed] [Google Scholar]
- 27. Funakoshi M, Hochstrasser M. 2009. Small epitope-linker modules for PCR-based C-terminal tagging in Saccharomyces cerevisiae. Yeast 26: 185– 192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19– 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hyland EM, Cosgrove MS, Molina H, Wang D, Pandey A, Cottee RJ, Boeke JD. 2005. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol. Cell. Biol. 25: 10060– 10070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryu Y, Schultz PG. 2006. Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods 3: 263– 265 [DOI] [PubMed] [Google Scholar]
- 31. English CM, Maluf NK, Tripet B, Churchill ME, Tyler JK. 2005. ASF1 binds to a heterodimer of histones H3 and H4: a two-step mechanism for the assembly of the H3-H4 heterotetramer on DNA. Biochemistry 44: 13673– 13682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen H-T, Warfield L, Hahn S. 2007. The positions of TFIIF and TFIIE in the RNA polymerase II transcription preinitiation complex. Nat. Struct. Mol. Biol. 14: 696– 703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mann RK, Grunstein M. 1992. Histone H3 N-terminal mutations allow hyperactivation of the yeast GAL1 gene in vivo. EMBO J. 11: 3297– 3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guthrie C, Fink GR. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194: 1– 863 [PubMed] [Google Scholar]
- 35. Chavez MS, Scorgie JK, Dennehey BK, Noone SM, Tyler JK, Churchill ME. 2012. The conformational flexibility of the C-terminus of histone H4 promotes histone octamer and nucleosome stability and yeast viability. Epigenetics Chromatin 5: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Donham DCII, Scorgie JK, Churchill MEA. 2011. The activity of the histone chaperone yeast Asf1 in the assembly and disassembly of histone H3/H4-DNA complexes. Nucleic Acids Res. 39:5449– 5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luger K, Rechsteiner TJ, Richmond TJ. 1999. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 119: 1– 16 [DOI] [PubMed] [Google Scholar]
- 38. Chalkley RJ, Baker PR, Huang L, Hansen KC, Allen NP, Rexach M, Burlingame AL. 2005. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer. II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol. Cell. Proteomics 4: 1194– 1204 [DOI] [PubMed] [Google Scholar]
- 39. Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. 2003. An expanded eukaryotic genetic code. Science 301: 964– 967 [DOI] [PubMed] [Google Scholar]
- 40. Tong AHY, Boone C. 2006. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol. Biol. 313: 171– 192 [DOI] [PubMed] [Google Scholar]
- 41. Pont-Kingdon G. 1994. Construction of chimeric molecules by a two-step recombinant PCR method. Biotechniques 16: 1010– 1011 [PubMed] [Google Scholar]
- 42. Sarkar G, Sommer SS. 1990. The “megaprimer” method of site-directed mutagenesis. Biotechniques 8: 404– 407 [PubMed] [Google Scholar]
- 43. Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5: 749– 757 [DOI] [PubMed] [Google Scholar]
- 44. Hellman U, Wernstedt C, Gonez J, Heldin CH. 1995. Improvement of an “in-gel” digestion procedure for the micropreparation of internal protein fragments for amino acid sequencing. Anal. Biochem. 224: 451– 455 [DOI] [PubMed] [Google Scholar]
- 45. Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. 1992. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem. 203: 173– 179 [DOI] [PubMed] [Google Scholar]
- 46. Chalkley RJ, Baker PR, Hansen KC, Medzihradszky KF, Allen NP, Rexach M, Burlingame AL. 2005. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer. I. How much of the data is theoretically interpretable by search engines? Mol. Cell. Proteomics 4: 1189– 1193 [DOI] [PubMed] [Google Scholar]
- 47. Rossmann MP, Luo W, Tsaponina O, Chabes A, Stillman B. 2011. A common telomeric gene silencing assay is affected by nucleotide metabolism. Mol. Cell 42: 127– 136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang L-Y, Umanah G, Hauser M, Son C, Arshava B, Naider F, Becker JM. 2008. Unnatural amino acid replacement in a yeast G protein-coupled receptor in its native environment. Biochemistry 47: 5638– 5648 [DOI] [PubMed] [Google Scholar]
- 49. Mohibullah N, Hahn S. 2008. Site-specific cross-linking of TBP in vivo and in vitro reveals a direct functional interaction with the SAGA subunit Spt3. Genes Dev. 22: 2994– 3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wittelsberger A, Mierke DF, Rosenblatt M. 2008. Mapping ligand-receptor interfaces: approaching the resolution limit of benzophenone-based photoaffinity scanning. Chem. Biol. Drug Des. 71: 380– 383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scorgie JK, Donham DCIII, Churchill ME. 2012. Analysis of histone chaperone antisilencing function 1 interactions. Methods Enzymol. 512: 223– 241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johnston M, Riles L, Hegemann JH. 2002. Gene disruption. Methods Enzymol. 350: 290– 315 [DOI] [PubMed] [Google Scholar]
- 53. Burgess RJ, Zhou H, Han J, Zhang Z. 2010. A role for Gcn5 in replication-coupled nucleosome assembly. Mol. Cell 37: 469– 480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. 2008. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 134: 244– 255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD. 2005. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 8: 19– 30 [DOI] [PubMed] [Google Scholar]
- 56. Kaufman PD, Cohen JL, Osley MA. 1998. Hir proteins are required for position-dependent gene silencing in Saccharomyces cerevisiae in the absence of chromatin assembly factor I. Mol. Cell. Biol. 18: 4793– 4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kaufman PD, Kobayashi R, Stillman B. 1997. Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 11: 345– 357 [DOI] [PubMed] [Google Scholar]
- 58. Tang Y, Poustovoitov MV, Zhao K, Garfinkel M, Canutescu A, Dunbrack R, Adams PD, Marmorstein R. 2006. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat. Struct. Mol. Biol. 13: 921– 929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Malay AD, Umehara T, Matsubara-Malay K, Padmanabhan B, Yokoyama S. 2008. Crystal structures of fission yeast histone chaperone Asf1 complexed with the Hip1 B-domain or the Cac2 C terminus. J. Biol. Chem. 283: 14022– 14031 [DOI] [PubMed] [Google Scholar]
- 60. Singh RK, Kabbaj M-HM, Paik J, Gunjan A. 2009. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat. Cell Biol. 11: 925– 933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gupta SK, Kececioglu JD, Schaffer AA. 1995. Improving the practical space and time efficiency of the shortest-paths approach to sum-of-pairs multiple sequence alignment. J. Comput. Biol. 2: 459– 472 [DOI] [PubMed] [Google Scholar]