

Abstract
The coordinated activity of Protein Tyrosine Phosphatases (PTP) is crucial to initiate, modulate and terminate diverse cellular processes. The catalytic activity of this protein depends on a nucleophilic cysteine at the active site that mediates the hydrolysis of the incoming phosphotyrosine substrate. While the role of conserved residues in the catalytic mechanism of PTPs has been extensively examined, the diversity in the mechanisms of substrate recognition and modulation of catalytic activity suggest that other, less conserved sequence and structural features could contribute to this process. Here we describe the crystal structures of Drosophila melanogaster PTP10D in the apo form as well as in complex with a substrate peptide and an inhibitor. These studies reveal the role of aromatic ring stacking interactions at the boundary of the active site of PTPs in mediating substrate recruitment. We note that Phenylalanine 76, of the so-called KNRY loop, is crucial for orienting the phosphotyrosine residue towards the nucleophilic cysteine. Mutation of Phenylalanine 76 to Leucine results in a sixty-fold decrease in the catalytic efficiency of the enzyme. Fluorescence measurements with a competitive inhibitor, p-nitrocatechol sulphate, suggest that Phe76 also influences the formation of the enzyme-substrate intermediate. The structural and biochemical data on PTP10D thus highlight the role of relatively less conserved residues in PTP domains in both substrate recruitment and modulation of reaction kinetics.
Protein tyrosine phosphatases (PTPs) are key regulators of various signalling processes and are crucial for the balance of phosphorylated tyrosines to maintain cellular homeostasis (1). Forming the reverse switch for tyrosine kinase mediated signalling, the role of PTPs is now recognized in initiating, sustaining and terminating cellular signalling (2-3). A comparison of PTP domains across diverse organisms suggests extensive sequence conservation and a conserved catalytic mechanism (4). The involvement of PTP homologues in similar biological processes in both vertebrates and invertebrates emphasises the evolutionary relationship of the PTP domains (5). PTPs are broadly divided into two classes; ones anchored to the membrane by trans-membrane helices, known as Receptor Protein Tyrosine Phosphatases (RPTPs), and cytosolic PTPs. RPTPs are further divided into sub-classes based on their extracellular domain arrangement (4).
Drosophila melanogaster has about half the number of PTPs than either humans or the worm. These observations suggest that the fruit-fly uses an optimal set of PTPs in consortia, in related pathways (6). Genetic studies reveal that five RPTPs of Drosophila melanogaster are expressed exclusively in the central nervous system (CNS) of the embryo. While four of these RPTPs (PTP10D, DLAR, PTP69D and PTP99A) are expressed on CNS axons, PTP52F is present on both the CNS axons and cell-bodies (7-9). The crucial role of PTP10D and PTP69D in regulating the repulsion of growth cones from the embryonic midline has been extensively studied (10). More recently, behavioural and genetic experiments on memory formation in Drosophila melanogaster reveal that PTP10D deletion mutants are chronically impaired in long-term memory (11).
The dephosphorylation catalyzed by PTPs primarily depends on their active site cysteine residue located in the phosphate binding loop (P loop). The backbone dipoles of the P loop and the side chain dipole of the conserved serine in the P loop contribute to lower the pKa of the active site cysteine (12). This cysteine is thus retained in its thiolate (deprotonated) form and functions as a nucleophile to attack the phosphate of the incoming phosphotyrosine residue. Subsequently, the general acid aspartate of the WPD loop protonates the leaving group following substrate binding, resulting in the formation of a cysteinyl-phosphate enzyme substrate intermediate. The intermediate is then hydrolyzed by the addition of a water molecule to release the active enzyme and the inorganic phosphate. This addition of water is facilitated by two glutamine residues of the Q loop (13). While conserved residues of the P loop, WPD loop and Q loop have been extensively studied (14), the role of residues in the substrate recognition loop that form the boundary of the PTP active site has received much less attention.
A ca 9 Å deep cleft at the active site distinguishes a PTP from a dual specificity phosphatase (ca 6 Å deep cleft). While the overall mechanism of dephosphorylation is the same in both PTP and dual specificity phosphatases, a much deeper active site selects for a phosphotyrosine as opposed to a phosphothreonine or phosphoserine residue (14). The boundary of this cleft is marked by an aromatic residue of the substrate recognition loop. The crystal structure of the PTP domain of Drosophila melanogaster PTP10D in both native and the phosphopeptide substrate (GP4) bound forms provides an insight into the role of this residue in substrate recruitment. Two other complexes, with the inhibitors vanadate and p-nitrocatechol sulphate (PNC) provide a conformational basis for PTP activity. These structures provide the first evidence for the role of an aromatic residue in the substrate recognition loop in localizing the phosphotyrosine residue. In PTP10D, the aromatic ring of Phe76 stacks with the incoming substrate, thus facilitating substrate recruitment. This forms the first step for the insertion of a phosphotyrosine residue into the active site cleft of a PTP domain to interact with the nucleophilic cysteine at its base. We note that mutation of Phe76 to Leu results in a ca 60 fold decrease in the catalytic efficiency of the enzyme. The mean life time of the phosphatase reaction is doubled while the half time of the enzyme-substrate/enzyme-inhibitor complex formation is increased by three fold. Put together, the crystal structures, enzymatic assays and fluorescence measurements on inhibitor binding provide a conformational rationale for the role of the substrate recognition loop in mediating enzyme-substrate complex formation in this class of enzymes.
Experimental Procedures
Cloning, expression and purification of the PTP domain of PTP10D
The cDNA cosmids to PCR amplify PTP10D were obtained from Prof. Kai Zinn (Caltech). The DNA fragment encoding the catalytic domain of PTP10D (Arg1226 – Asn1533) was cloned between the NheI and NotI sites of the E. coli expression vector pET28a. 5′-ATATGCTAGCCGCCCCATTCTGATCAAGAACTTTGCC-3′ and 5′-AATAATGCGGCCGCCTAGTTCTCCTTCCCCTCGAGCACCGC-3′ were used as the forward and reverse primers respectively. A stop codon was incorporated in the reverse primer so that the recombinant protein harboured only the N-terminal poly-histidine tag. The plasmid containing the recombinant PTP domain was transformed into BL21(DE3) cells (Novagen, Inc.). The cells were grown to an optical density of 0.6 at 37°C, where upon the cells were induced with 0.3mM isopropyl-β-D-galactopyranoside (final concentration). Following this, the temperature for growth was lowered to 15 °C and cells were grown for 10-12 hrs before they were spun down. The cells were resuspended in lysis buffer (Buffer L) containing 50mM Tris-HCl pH 8.0, 300mM NaCl, 5% Glycerol and 1mM β-mercaptoethanol (βME). After sonication on ice, the cell debris was separated from the crude cell lysate by centrifugation for 30min at 15000rev.min−1 in a centrifuge. The cell-free lysate was incubated with Ni-NTA affinity resin (approximately 4ml of resin was used for cell-free lysate from 10g cell paste) at 4°C for 2 hours in an end-to-end rotor. The resin was then packed into a XK16 column (Amersham Biosciences) and washed with ca three bed volumes of a buffer containing 50mM Tris-HCl pH 8.0, 7mM imidazole, 300mM NaCl, 5% Glycerol and 1mM βME. The protein was eluted by a gradient of imidazole (7-200mM) prepared in Buffer L. The pure fractions were pooled and concentrated using a centricon (Millipore) and loaded onto a S200 size exclusion column (Amersham). The protein was eluted in 30mM Tris-HCl pH 7.5, 300mM NaCl and 5% Glycerol. The peak fractions were pooled, concentrated and used for crystallization and biochemical studies.
Site directed mutagenesis
The catalytic domain of the PTP10D was modified to obtain single (F76L, D210A, C242S) and double mutant (D210A/Q286A; C242S/Q286A) proteins. While the mutations D210A, C242S and Q286A were at the active site of the PTP domain, F76L was in the phosphotyrosine recognition loop. A single primer approach was used for site-directed mutagenesis, with an overlap of 10 nucleotides on either side of the mutation. Unique restrictions site(s) were incorporated in the primer to aid screening of mutants. After PCR (Phusion Polymerase: Finnzymes), the parent template was digested with DpnI at 37 °C and the mutated template was transformed in DH5α cells. All mutants were confirmed by sequencing (Macrogen Inc.)
Substrates and inhibitors for structural and biochemical analysis
Peptide sequences were designed corresponding to the SH2 interacting regions of the glycoprotein GP150 which was previously shown to be a substrate for PTP10D in Drosophila S2 cells (15). The sequence of GP150 was obtained from the non-redundant protein database at the NCBI. Four peptides were designed incorporating one phosphotyrosine residue each from the four immunoreceptor family tyrosine-based activation motifs (ITAM) in the cytosolic tail of GP150. The peptide sequences used were GP1: KRGERpYRMALL, GP2: LMRVIpYWLRKR, GP3: PKVHRpYAPINQ and GP4: KNSFVpYQKLSE. All four peptides in their phosphorylated form were obtained commercially at >99% purity from GL Bioscience, China. Physicochemical properties of the peptides were estimated from the peptide calculator at the Novagen custom peptide website (http://www.innovagen.se/custom-peptide-synthesis/peptide-property-calculator/). The small molecule substrate p-nitrophenyl phosphate (pNPP) and the inhibitors p-nitrocatechol sulfate (PNC) and sodium vanadate were obtained from Sigma Aldrich, Inc.
Crystallization and structure determination
Purified PTP10D at 10 mg/ml concentration was screened for crystallization conditions using commercially available screens. Crystals were observed in several conditions containing small chain alcohols and PEG 3350. Optimal crystals were obtained in crystallization drops containing 3μl of ca 7mg/ml protein and 3μl of a solution containing 120mM Citrate, 15% PEG4000, 10% iso-Propanol, 10% n-Butanol and 10% 1,4-Butanediol. These crystals appeared within 10days in microbatch trials using 2:1 ratio of Paraffin to Silicon oil (Hampton Research). Crystals were flash frozen in cryo-protectant containing 5% PEG 400 in the crystallization condition. Inclusion of 5mM tris 2-carboxyethyl phosphine (TCEP) in the cryoprotectant was crucial to obtain an isotropic diffraction pattern. The diffraction datasets corresponding to the substrate and inhibitor complexes were obtained on crystals soaked in 0.8mM Phosphopeptides GP1-4 (substrate peptide) and 5mM PNC or 5mM sodium vanadate for the inhibitor complexes.
X-ray diffraction data was collected at the beamline BM14 of the European Synchrotron Radiation Facility, Grenoble. Diffraction images were processed using MOSFLM (16) and scaled using SCALA (17). Phase information was obtained by molecular replacement, performed using PHASER (18-19) with the crystal structure of the catalytic domain of Human Tyrosine Receptor Phosphatase β (PDB ID: 2AHS, 55% identity) as the search model. The molecular replacement utility CHAINSAW (20) was used to modify the model structure by pruning non-conserved residues. The structures of the complexes were solved by using native PTP10D as the search model. Structures were refined using REFMAC5 (21) and the fit of the model to the electron density was evaluated using COOT(22). The topology and parameter files for various ligands were obtained from the PRODRG server (23). Models were validated using MolProbity (24) and the WHATIF server (25). Inter-atomic distances were calculated using CONTACT from the CCP4 suite(26).
In-silico mutation of Phe76 to Leu was performed on the WHATIF server (25). Neuronal network combinations at the HotPatch server (27) were used to identify residues lining the active site of the catalytic domain of PTP10D. All protein structural representations were made using PyMOL (DeLano Scientific). The sequences of individual PTP domains were obtained from the PTP database (4). Multiple sequence alignment and percentage occurrence of the amino acid was calculated by the Jalview software (28).
Circular Dichroism and steady state fluorescence measurements
Secondary structure and tertiary structure changes upon mutagenesis in PTP10D were ascertained by Circular Dichroism (CD) and intrinsic tryptophan fluorescence measurements. CD measurements were recorded at a protein concentration of 1μM on a Jasco-J715A spectropolarimeter using a 0.1cm path-length cuvette. Spectra were collected with a slit width of 1nm and a response time of 4sec at a scan rate of 50nm/min. Fluorescence emission spectra were collected on a Jasco SPEX Fluoromax-3 instrument using a 1cm cuvette. Samples were excited at 280nm and spectra were recorded from 300–400nm. Each data point was an average of 3 scans. The excitation and emission slit widths were maintained at 3.5 nm and 5nm respectively. For all spectral measurements, the buffer used was 25mM HEPES pH 7.0, 150mM NaCl with 2mM DTT.
Phosphatase assays and time course measurements
Phosphatase assays using pNPP as the substrate were performed as described earlier (29) in Buffer A (50 mM Citrate, Glycine and HEPES (CGH), pH 6.5, 100mM NaCl and 2mM DTT). Phosphatase activity with the phosphotyrosine containing peptides was ascertained by the detection of inorganic phosphate released by the malachite green reaction (30). Phosphopeptide stocks were prepared in 50mM citrate pH 6.5 and 100mM NaCl. Peptide concentration was estimated by measuring the absorbance at 268nm. Increasing concentrations of peptides were incubated with 0.125μM PTP10D (0.5μM of F76L mutant) in buffer B (25mM Citrate buffer pH 6.5, 2% glycerol, 150mM NaCl and 2mM DTT) at 25°C for 15min. The reaction was stopped by the addition of five times the reaction volume of Biomol Green reagent (Enzo Life Sciences). The inorganic phosphate released was estimated by measuring the absorbance at 650nm. For all assays, kinetic constants for the steady state catalysis were obtained by fitting the reaction curves to the Michaelis-Menten equation using the nonlinear regression module of the Sigma Plot software (Systat Software, Inc.).
For time course measurements, dephosphorylation was initiated by addition of 0.5μg of PTP10D to 35mM pNPP in buffer A. Formation of para-NitroPhenol (pNP) was monitored at 405nm in continuous mode (every 0.5sec) for 25min. For time course measurements with peptides, 0.5μg of protein was incubated with peptides at concentrations 5 times of their Km values (Table 2) in reaction buffer B. An aliquot was taken from the main reaction mixture and the reaction stopped by the addition of Biomol green reagent every 2min for 25mins. As the time course measurements were performed at saturating concentrations of the substrate, the rate of product formation was fitted to a pseudo-first order reaction (dependent only on the concentration of the [ES] complex) using the equation
| (1) |
where v is the reaction velocity (nmol of product/min), t is time (minutes), α is the maximum amount of product formed at the end of the time course, λ is the rate of increase of the reaction velocity to maximum and is related to the t1/2 of the reaction as
| (2) |
The mean lifetime of the reaction is defined as
| (3) |
Table 2. Enzyme kinetics parameters for PTP10D and the F76L mutant determined using different substrates.
| PTP10D | ||||
|---|---|---|---|---|
| Vmax (μmole/min/mg) |
Km (μM) |
Kcat (sec−1) |
Kcat / Km (sec−1 M−1) × 103 |
|
| pNPP | 13.74 ± 1.40 | 5312.80 ± 58.42 | 8.24 ± 0.89 | 1.55 ± 0.16 |
| GP1 | 10.45 ± 0.48 | 138.05 ± 6.43 | 6.65 ± 0.32 | 48.18 ± 2.39 |
| GP2 | 6.24 ± 0.55 | 508.11 ± 45.21 | 3.92 ± 0.34 | 7.71 ± 0.67 |
| GP3 | 10.33 ± 0.41 | 129.67 ± 5.07 | 6.47 ± 0.26 | 50.15 ± 2.11 |
| GP4 | 6.82 ± 0.24 | 69.88 ± 2.24 | 4.27 ± 0.15 | 61.11 ± 2.14 |
| PTP10D F76L mutant | ||||
|
Vmax (μmole/min/mg) |
Km (μM) |
Kcat (sec−1) |
Kcat / Km (sec−1 M−1) × 103 |
|
| pNPP | 8.01 ± 0.84 | 24891.9 ± 387.09 | 4.80 ± 0.50 | 0.19 ± 0.02 |
| GP1 | 3.33 ± 0.39 | 1081.24 ± 133.17 | 2.00 ± 0.24 | 1.85 ± 0.22 |
| GP2 | 0.83 ± 0.11 | 2564.83 ± 355.26 | 0.52 ± 0.07 | 0.21 ± 0.03 |
| GP3 | 3.54 ± 0.46 | 1009.29 ± 137.87 | 2.12 ± 0.27 | 2.11 ± 0.27 |
| GP4 | 1.49 ± 0.13 | 542.95 ± 48.67 | 0.89 ± 0.08 | 1.64 ± 0.15 |
Inhibitor binding studies
The inhibition constant (Ki) was estimated using protocols reported earlier (31). Sodium vanadate (1mM) was prepared in water and concentrations of subsequent dilutions were estimated by absorbance at 260 nm (εM = 3550 M−1cm−1). Steady state Michaelis-Menten kinetics for pNPP was monitored in the presence of 0.2, 0.5, 0.75, 1.0 and 2.5 μM sodium vanadate in reaction buffer A at 25°C. The data was simultaneously fit to non-linear regression equations for competitive inhibitor binding by the Graph Pad Prism software (Graph Pad). A Dixon plot was plotted for increasing concentrations of vanadate and reciprocal of reaction velocities to obtain a set of intersecting lines that provide an independent estimate of the inhibition constant Ki.
PNC binding was monitored using the intrinsic tryptophan quenching of the WPD loop of the PTP domain upon inhibitor binding (32). 5.0 μM of PTP10D and its single and double mutants were incubated with increasing concentrations of PNC (from 0 to 1.2mM) in buffer B for 15min at 25°C. Fluorescence measurements were performed on a Jasco SPEX Fluoromax-3 instrument using a 1cm cuvette. Excitation and emission wavelengths were fixed at 280nm and 360nm respectively. The slit-width for the excitation and emission monochromators was fixed at 3.5nm and 5.0nm respectively. All measurements were an average of five readings with an integration time of 0.2sec.
PNC binding kinetics was monitored on a Jasco FP-6300 spectro-fluorometer in a 1cm cuvette to minimize the dead-time of the reaction. The reaction was initiated by adding 600μM of PNC to 1.5μM of protein. Fluorescence quenching was monitored in real time at 360nm upon excitation at 280nm for 300sec (response time 0.1sec). A slit width of 3.0nm was used for both the excitation and emission monochromators.
The fraction of protein bound to PNC was estimated using
| (4) |
where Y(x) is the fluorescence signal of the protein at (x) concentration of PNC, Yunbound is the fluorescence signal of the protein in the absence of PNC and Ybound is the fluorescence signal of the protein at saturating concentration of PNC. The graph was then fitted to a conventional ligand binding non-linear regression equation using the sigma plot software.
The equation used to fit the kinetic data was
| (5) |
where V is the rate of PNC binding, vo is the unbound baseline correction, α is the maximum difference in the fluorescence signal of the bound and unbound protein, β describes the slope of the curve and t1/2 is half time of PNC binding defined by the first derivative maximum of the sigmoid function. to was used to define the lag phase of the curve. All equations were fit to experimental data using the non-linear regression module of Sigma Plot software.
Results
The structure of the catalytic domain of PTP10D and substrate complexes
The catalytic domain of PTP10D and its active site mutants were purified to homogeneity. Circular Dichroism (CD) and fluorescence spectra of the recombinant proteins suggest that the mutations do not alter the overall structure of PTP10D. A modification of the recombinant protein construct incorporating an additional 47 residues to the N-terminus of the classical PTP domain was crucial to obtain protein samples at the concentration required for crystallization. As reported earlier, despite poor sequence similarity in the N-terminal segment (preceding the M1 PTP motif), this polypeptide stretch adopts an α helical conformation and contributes to the stability of the PTP domain, a feature seen both in PTP10D as well as in few other structures where this segment has been included in the PTP domain construct (For example, RPTPα; PDB: 1YFO) (29). The native PTP10D, as well as the PTP10D active site mutant proteins, are monomers in solution (Supplementary Figure S1).
This catalytic domain of PTP10D crystallized in the space group P3121 with two molecules in the asymmetric unit. Molecular Replacement using the catalytic domain of PTP-β yielded a single solution with a Log Likelihood Gain (LLG) of 2886. The data, refinement and model statistics are compiled in Table 1. For ease in comparison with other characterized PTP domains, residues in the catalytic domain of PTP10D were numbered from 1-305 (Figure 1; Supplementary Figure S1). PTP10D adopts the classical PTP fold with a central twisted β-sheet composed of 8 β-strands (Figure 1). This β-sheet is flanked by eight α-helices, two of which form the N-terminal helix-turn-helix motif crucial for protein solubility and stability (29). The P-loop located between β8 and α4 contains the nucleophilic cysteine (Cys242 in PTP10D) and forms the base of the active site. This cysteine is flanked by the general acid/base Aspartate (Asp210) from the WPD loop (located between β7 and α3) and the Glutamines (residues 286 and 290) of the Q loop (between α5 and α6). A structure based sequence analysis revealed that the ten PTP motifs (reviewed in 4) are conserved in PTP10D (Supplementary Table 1; Figures S3 and S4). The structure of PTP10D with the phosphopeptide substrate GP4 and the inhibitors vanadate and PNC showed clear electron density for the ligand at the active site of one monomer (Chain B) of the two in the asymmetric unit. This observation is consistent with crystal packing that reveals that the active site of Chain B is more accessible to the solvent (Supplementary Figure S2). An analysis of the PTP10D structure and the ligand complexes using Molprobity (24) revealed that a few residues in these structures were outliers in the Ramachandran plot. A prominent outlier is the active site Cysteine (Cys242). Other outliers include residues in the loop adjacent to the active site. This loop segment changes conformation upon ligand binding. An inspection of the fit of the model to the electron density as well as high real space correlation coefficients (ca 0.8–0.9) suggests that these residues have been correctly modeled in these crystal structures.
Table 1. Data collection, refinement and model statistics for apo and ligand bound PTP10D structures.
| PDB ID | PTP10D 3S3E |
PTP10D with GP4 3S3H |
PTP10D with Vanadate 3S3F |
PTP10D with PNC 3S3K |
|---|---|---|---|---|
| I. Data Collection | ||||
| Wavelength | 0.954 Å | 1 Å | 1 Å | 1 Å |
| Space Group | P 3121 | P 3121 | P 3121 | P 3121 |
| Cell Dimensions | a = 102.45 Å b = 102.45 Å c = 171.81 Å |
a = 102.70 Å b = 102.70 Å c = 173.22 Å |
a = 103.07 Å b = 103.07 Å c = 173.40 Å |
a = 102.51 Å b = 102.51 Å c = 172.80 Å |
| Resolution Limits (Å) |
44.0 – 2.40 (2.53 – 2.40) |
45.0 – 2.80 (2.95 – 2.80) |
45.0 – 2.70 (2.85 – 2.7) |
45.0 – 3.20 (3.37 – 3.20) |
| aRmerge (%) | 11.9 (48.4) | 11.4 (45.6) | 11.3 (43.5) | 16.4 (42.6) |
| (I) / σ (I) | 15.9 (5.2) | 10.6 (3.1) | 11.9 (3.7) | 4.7 (2.4) |
| Number of Unique Reflections |
41551 (5989) | 26708 (3832) | 29943 (4297) | 17564 (2523) |
| Completeness | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 98.7 (99.0) |
| Multiplicity | 11.2 (11.3) | 5.3 (5.4) | 5.8 (5.8) | 3.1 (3.1) |
| Average Mosaicity | 0.41 | 0.66 | 0.57 | 0.67 |
| II. Refinement | ||||
| bRcryst(%) | 20.45 | 25.01 | 23.81 | 25.29 |
| Rfree(%) | 22.72 | 28.92 | 27.18 | 30.86 |
| III. Model | ||||
| Ramachandran Plot |
||||
| Preferred | 538 (94.72 %) | 532 (93.66 %) | 540 (95.07 %) | 514 (90.49 %) |
| Allowed | 26 (4.58 %) | 31 (5.46 %) | 24 (4.23 %) | 47 (8.27 %) |
| Outliers | 4 (0.70 %) | 5 (0.88 %) | 4 (0.70 %) | 7 (1.23 %) |
| RMSD bond Length (Å) |
0.007 | 0.011 | 0.009 | 0.008 |
| RMSD bond Angle (°) |
1.178 | 1.401 | 1.261 | 1.194 |
| Wilson B (Å2) | 38.12 | 54.12 | 44.99 | 56.63 |
| Mean B (Å2) | 27.75 | 49.60 | 38.07 | 54.07 |
Values in parenthesis are for outer resolution shell.
Rmerge = Σj |<I>-Ij|/Σ<I> where Ij is the intensity of the jth reflection and <I> is the average intensity.
Rcryst = Σ|Fo - Fc|/Σ|Fo|; Rfree : the same as Rcryst but on 5 % of the data excluded from the refinement calculation.
Figure 1. Sequence and structural features of the catalytic domain of PTP10D.

(a) The domain organization of D. melanogaster PTP10D. The sequence of the catalytic domain has been renumbered from 1-305 to facilitate comparisons with other PTP domains. (b) The catalytic domain of PTP10D. (c) The PTP sequence motifs mapped onto the structure of PTP10D.
Conformational features of the active site
Superposition of the ligand bound and the apo structure of PTP10D with the ‘closed’ conformation of PTP1B (PDB ID: 1SUG) reveal that all PTP10D structures adopt the ‘open’ conformation of the active-site loop (Figure 2; Supplementary Figure S5). This superposition suggests that the hinge Prolines (Pro209 and Pro214 in PTP10D) retain the WPD loop in the open conformation. The general acid, Asp210, in PTP10D is 13.8 Å away from the active site Cys242 as opposed to the equivalent Asp181 in PTP1B which is 8.8 Å away from Cys215. The difference in the orientation of the side chain of Phe211 is the most distinct between the bound and free PTP10D structures.
Figure 2. The crystal structure of PTP10D offers mechanistic insights into distinct steps of the phosphatase reaction.

I. The de-protonated active site cysteine prior to substrate entry into the active site. Three conserved water molecules hydrate the active site of PTP10D (PTP10D open structure). II. Substrate entry into the active site is mediated by Phe76 of the KNRY loop (based on the structure of PTP10D with the GP4 peptide). The incoming substrate displaces water W3. III. The substrate approaches the active site cysteine by displacing water W1. Aryl stacking interactions with Phe76 maintains the optimum conformation of the incoming substrate (based on the crystal structure of PTP10D with PNC). IV. The WPD loop closes for the conserved active site Phenylalanine (Phe182) to make stacking interactions with the substrate. Conformation of the substrate is optimized to aid catalysis. This orientation is perpendicular to that noted in step II. (based on the PTP1B-phosphotyrosine complex). V. Optimized orientation of the substrate ensures catalysis. The cysteinyl-phosphate intermediate as seen at the active of PTP1B. VI. The cysteinyl–phosphate intermediate is hydrolyzed by addition of water by Glutamines of the Q loop. The active site cysteine reverts to its free, de-protonated state with three waters at the active site (based on the PTP1B closed form structure).
The water molecule network at the active site of the PTPs has been described earlier. Generally, two ordered and one disordered water molecule are seen in the active site of PTPs (33). In PTP10D, all three water molecules could be unambiguously modelled with B factors of 12.64 Å2, 18.97 Å2 and 21.05 Å2 for waters W1, W2 and W3 respectively (Figure 3a, Supplementary Figures S6, S7). While W1 is coordinated by Cys242 and Gln286, W2 is bound by Cys242 and Lys154. W3 is loosely associated with Cys242 and Gln286 and represents the disordered water at the PTP active site. Ligand binding at the PTP active site requires sequential removal of these water molecules as noted from the structures of the enzyme-substrate and enzyme-inhibitor complexes of PTP10D.
Figure 3. The apo and the substrate/inhibitor bound active site of PTP10D.
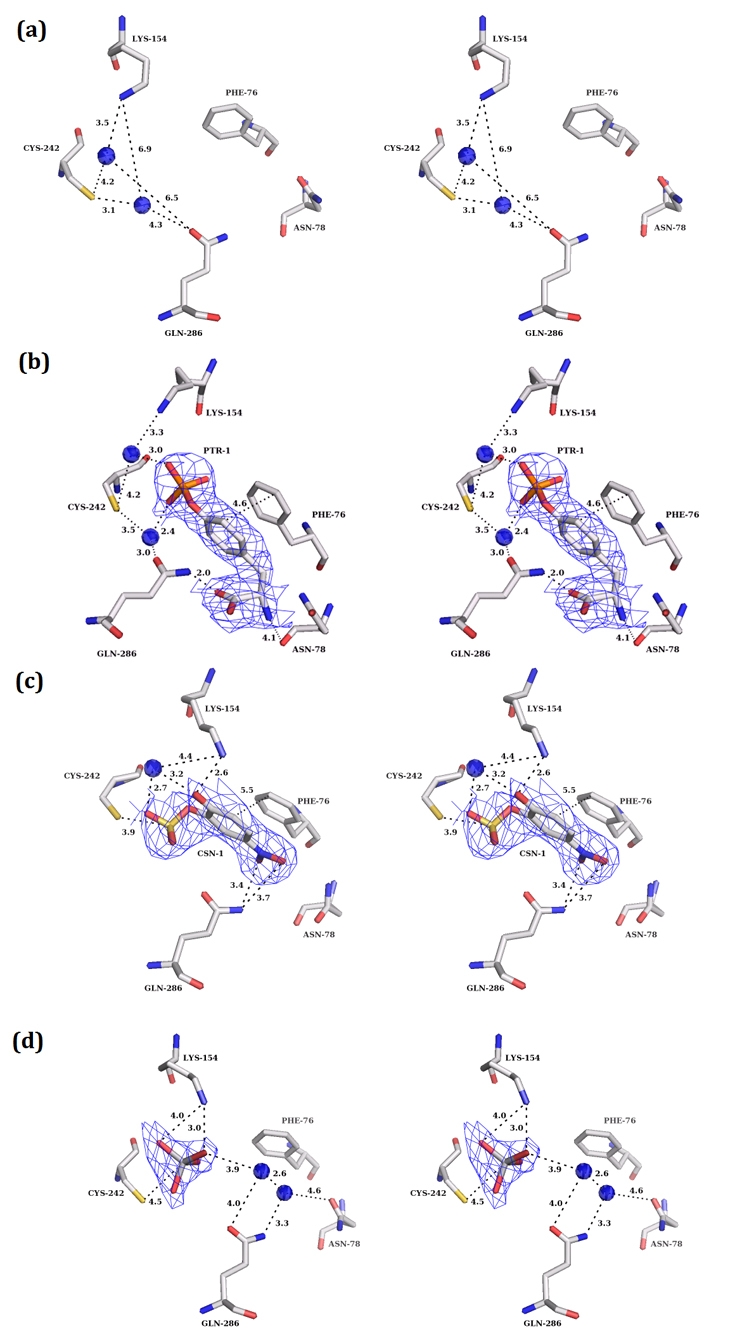
(mFo-DFc) electron density maps (at 3.0σ level) for the apo and substrate/inhibitor bound states of PTP10D. a. The active site of apo PTP10D (PDB: 3S3E; resolution 2.4Å). b. The phosphotyrosine of the GP4 peptide at the active site of PTP10D (PDB ID: 3S3H; resolution 2.8Å). c. PNC bound at the active site of PTP10D (PDB ID: 3S3K; resolution 3.2Å). d. Vanadate bound at the active site of PTP10D (PDB ID: 3S3F; 2.7Å).
PTP10D crystals incubated with the phosphopeptide GP4 showed unambiguous electron density at the active site which could be attributed to a phosphotyrosine residue. The electron density for the rest of the bound peptide was poor; the flanking segments were thus not modelled in this crystal structure (2.8Å resolution). The bound phosphotyrosine, however, reveals several facets of substrate recognition. The phenyl ring of this residue makes planar stacking interactions with Phe76 (aromatic ring stacking distance of 4.5 Å) such that the phosphate of the phosphotyrosine is aligned with the nucleophilic Cys242. The phosphotyrosine also displaces one of the water molecules (W3) which is most loosely associated in the active site. Waters W1 and W2 could be modelled at the active site with the side chain of Lys154 coming closer to W2 by 0.3 Å. The carboxyl and amino terminal of the phosphotyrosine residue make hydrogen bonds with Gln286 and Asn78 that regulate access to the PTP10D active site (Figure 3b).
para-Nitrocatechol Sulphate (PNC), a competitive inhibitor of PTPs, mimics phosphotyrosine (34). In the structure of the PTP10D-PNC complex, one PNC molecule could be modelled into the active site. Similar to the substrate peptide complex, Phe76 makes planar stacking interactions with the PNC molecule. However, this molecule is located deeper in the active site of PTP10D when compared to the phosphotyrosine of the peptide substrate (Figure 3c). We also note that the sulphate of the PNC molecule displaces water W1. This position of the sulphate is the same as that of the phosphate of the phosphotyrosine seen in PTP-substrate structures reported previously (35-36). In this study, sodium vanadate was also used as an inhibitor as it adopts an inorganic phosphate-like geometry as well as a five coordinate trigonal bipyramidal structure that resembles the transition state of the phosphotranfer reaction (37). Crystals soaked in vanadate showed a tetrahedral electron density in Chain B which could be attributed to vanadate (Figure 3d). We note that vanadate binding displaces the waters at the active site.
Phosphatase activity and time course measurements for PTP10D and the F76L mutant
The phosphatase activity measured using either pNPP or phosphopeptide substrates reveal PTP10D to be a highly efficient enzyme. PTP10D could dephosphorylate all the four GP150 immuno-receptor family tyrosine-based activation motif (ITAM) peptide substrates. While the catalytic efficiency was highest in the case of peptides harbouring charged groups around the central phosphotyrosine (GP1 and GP3), affinity of PTP10D was the maximum for a peptide having small chain residues adjacent to the phosphotyrosine (GP4). The dephosphorylation catalyzed by PTP10D was least efficient for GP2 which harboured non-polar and aromatic residues around the phosphotyrosine. This suggested that substrate recognition is largely influenced by charges on the surface and the size of the side chains (Table 2 and Figure 4). Mutation of Phe76 to Leu led to a seven-fold decrease in the Vmax for all substrates of PTP10D and an eight-fold increase in the Km values. While this mutation decreased the overall efficiency of PTP10D, it did not alter substrate preference characteristics. Time course measurements reveal that the mean lifetime (τ) as well as the t1/2 of the phosphatase reaction increased by more than two-fold in the F76L mutant as compared to native PTP10D (Table 3 and Figure 4). The amount of product formed at the end of the time course for the F76L is lower by a factor of ca 0.45 when compared to the native protein. This observation is consistent with the decrease in the Vmax of the phosphatase reaction and is also reflected in the decrease in the overall rate of the pseudo-first order reaction (λ).
Figure 4. Phosphatase assays and time course measurements on PTP10D.

a. Michealis-Menten kinetics for the phosphatase activity of PTP10D and the F76L mutant with various substrates. b. Time course measurements to ascertain the life time of the phosphatase reaction as catalyzed by PTP10D and the F76L mutant.
Table 3. Parameters for the phosphatase reaction catalyzed by PTP10D and the F76L mutant.
| PTP10D | PTP10D F76L | |||||||
|---|---|---|---|---|---|---|---|---|
| α (nmol) | λ(min−1) × 10−2 |
τ (min) | t1/2 (min) |
μ (nmol) | λ(min−1) × 10−2 |
τ (min) | t1/2 (min) | |
| pNPP | 55.77 ± 1.12 |
16.43 ± 0.32 |
6.08 ± 0.12 |
4.22 ± 0.08 |
52.80 ± 0.79 |
6.12 ± 0.92 | 16.34 ± 0.25 |
11.32 ± 0.17 |
| GP1 | 9.96 ± 0.49 |
20.99 ± 1.06 |
4.76 ± 0.24 |
3.30 ± 0.17 |
5.36 ± 0.56 |
8.51 ± 0.84 | 11.74 ± 1.15 |
8.13 ± 0.79 |
| GP2 | 2.21 ± 0.14 |
11.07 ± 0.72 |
9.03 ± 0.59 |
6.26 ± 0.41 |
1.61 ± 0.17 |
3.89 ± 0.41 | 25.70 ± 2.69 |
17.81 ± 1.87 |
| GP3 | 14.58 ± 0.37 |
22.26 ± 0.56 |
4.50 ± 0.11 |
3.11 ± 0.08 |
10.81 ± 0.65 |
8.39 ± 0.41 | 11.91 ± 0.59 |
8.26 ± 0.49 |
| GP4 | 5.12 ± 0.32 |
15.76 ± 0.95 |
6.35 ± 0.41 |
4.39 ± 0.28 |
2.54 ± 0.24 |
7.18 ± 0.68 | 13.92 ± 1.32 |
9.65 ± 0.93 |
Addition of vanadate to the reaction mixture increased the Km of the reaction but did not affect the Vmax of the reaction. The enzyme inhibition constant (Ki) was found to be 1.53 ± 0.14 μM. The Michaelis-Menten and Dixon plots are shown in Supplementary Figure S8. This Ki value is three times higher when compared to the vanadate inhibition of PTP1B activity (38). Steady state measurements to ascertain the dissociation constant of PNC binding (KD) showed that mutations at the active site increase the KD for PNC binding (Figure 5). As expected, a mutation of the active site Cys242 to serine caused a three-fold decrease in the affinity for PNC, while that of the general acid/base Asp210 was less disruptive. Double mutations that serve as substrate trap mutants (39) also show lower PNC binding to PTP10D. The F76L mutation was seen to influence PNC binding as much as the C242S/Q286A mutant protein (Figure 5).
Figure 5. para-nitrocatechol sulphate (PNC) binding to PTP10D and mutant enzymes.

a. Intrinsic Tryptophan fluorescence quenching of PTP10D and its mutants upon PNC binding. (b and d) Dissociation constants for PNC binding to PTP10D and its mutants. c. Fraction of PTP10D bound to PNC to ascertain the ligand saturation. (e and f) Kinetics of PNC association with PTP10D.
Kinetic measurements reveal that the lag time (to) for PNC binding is the longest (160sec) for the F76L mutant (Figure 5 and Table 4) when compared to either wild type PTP10D or the active site mutants. While mutations at the active site decreased the lag time for PNC binding (likely by altering the water network at the active site), the F76L mutation drastically increased the lag time for PNC binding to PTP10D. An increase of ca 130 sec in the t1/2 of PNC binding thus supports the role of Phe76 in mediating ligand entry at the PTP active site.
Table 4. Kinetic parameters, rate constants and the half life of PNC binding obtained for PTP10D and mutant proteins.
| PTP10D | t0 (sec) | α (A.U.) | β (A.U./sec) × 10−2 |
t1/2 (sec) |
|---|---|---|---|---|
| Wild Type | 50.0 | 0.88 ± 0.01 | 35.97 ± 0.04 | 62.89 ± 0.04 |
| F76L | 160.0 | 0.87 ± 0.03 | 4.57 ± 0.01 | 197.22 ± 0.25 |
| C242S | 35.0 | 0.90 ±0.01 | 12.51 ± 0.07 | 52.14 ± 0.08 |
| D210A | 30.0 | 0.87 ± 0.02 | 42.01 ± 0.05 | 37.73 ± 0.05 |
| C242S/Q286A | 40.0 | 1.04 ± 0.02 | 2.49 ± 0.03 | 111.07 ± 0.14 |
| D210A/Q286A | 16.0 | 0.85 ± 0.02 | 44.24 ± 0.04 | 22.9 ± 0.05 |
Discussion
The structure of the PTP domain of a Drosophila melanogaster RPTP (PTP10D), a protein implicated in memory and nervous system development, was determined in both apo and substrate bound forms. An analysis of the surface of PTP10D by the HotPatch server suggests that the active site cleft is demarcated by residues Asn78, Phe76, Lys154, Gly247, Arg248 and Gln286 (Supplementary Figure S5). The entry of a substrate into the active site cleft is governed by residues Asn78 and Gln286. The nucleophilic Cys242 lies at the bottom of this cleft and is hydrated by three conserved water molecules. A close inspection of the native, phosphotyrosine and PNC bound structures shows the protein backbone superposes well with an RMSD of less than 0.25 Å. Variations in the EKGERK, AEQR and DDD loops in the ligand-bound and unbound forms of PTP10D provide an insight into the conformational changes at the active site upon ligand binding (Supplementary Figures S9 and S10). Interactions between the active site and the WPD loop are mediated by the EKGERK loop. While Glutamate (Glu149) makes bi-dentate salt bridges with Arg248 (connected to the AEQR loop) at the base of the active site, Lys150 makes a salt bridge with the Asp210 of the WPD loop.
A comparison of the substrate peptide and PNC bound structures of PTP10D with other PTP complexes (34-35) provides further insights into the mode of phosphotyrosine binding at the active site (Figure 2). The cavity of the active site of the PTP domain is constrained by Asn78 (Asp48 in PTP1B; Asp69 in YopH) and Gln286 (Gln262 in PTP1B; Gln284 in YopH) so as to specifically allow a phosphorylated tyrosine residue. Phe76 (equivalent to Tyr46 in PTP1B; Phe67 in YopH) forms the boundary of the active site and makes hydrophobic interactions with the phenyl ring of the incoming substrate. The structure reveals that Phe76 assists the substrate in adopting a conformation required to access the active site. The entry of the phosphotyrosine at the active site displaces the water W3 which is loosely bound, thereby contacting the nucleophilic cysteine by water mediated hydrogen bonds (Figures 2 and 3). The PTP10D-GP4 structure further suggests that the active site caves in to trap the substrate. This involves movement of the WPD loop to bring the acid Asp210 (Asp181 in PTP1B; Asp194 in YopH) closer to Cys242 (Cys215 in PTP1B; Cys241 in YopH). The incoming phosphotyrosine thus approaches the nucleophilic cysteine, displacing waters W1 and W2 coordinated by Gln286 (Gln262 of PTP1B; Gln284 of YopH) for the second step of catalysis. This observation is also supported by the PTP10D-PNC and the PTP10D-vanadate complexes. Consistent with other PTP complexes, the closure of the WPD loop facilitates the conserved Phenylalanine (Phe211 in PTP10D; Phe182 in PTP1B) to make ring stacking interactions with the bound phosphotyrosine. The phosphotyrosine thus adopts a position at the base of the active site in an orientation perpendicular to that corresponding to the initial stage of entry (Stage II in Figure 2). This orientation of the phosphotyrosine is consistent with the substrate trap mutant structures of PTP1B and YopH (RCSB: 1G1G and 1PA9). This re-orientation of the phosphotyrosine ring is important for the coordinated movement of Asp210 allowing Cys242 to complete the formation of the cyteinyl phosphate intermediate. The movement of the product (dephosphorylated tyrosine) perhaps triggers the waters trapped by the Gln286 (Q loop) to be used for the hydrolysis of the cysteinyl-phosphate intermediate.
A comparison of PTP10D with PTP sequences from Homo sapiens, Drosophila melanogaster and Caenorhabditis elegans suggests Phe76 to be a part of the phosphotyrosine recognition (KNRY) loop (Supplementary Table 1 and Figures S3, S4). This comparison across 75 PTP sequences (Supplementary Figure S4) shows the last position of the KNRY loop is either a Tyrosine or Phenylalanine in 67% of the sequences. The sequences that do not harbour aromatic residues at this position belong to either the membrane distal domains of double domain RPTPs or pseudo-phosphatases, both of which are inactive. The absence of this aromatic residue along with mutations in the Asp of the WPD loop is speculated to be the reason of for the lack of activity in these domains (4, 13). While the role of Asp as the general acid has been well studied, the role of the aromatic residue of the KNRY loop in mediating substrate entry at the PTP active site and facilitating enzyme-substrate complex formation has received peripheral attention. The fact that the aromatic residue corresponding to Phe76 is not strictly conserved across PTP homologues was perhaps seen to reflect a less important role for this residue. To biochemically elucidate the role of Phe76 in the dephosphorylation mechanism and kinetics of PTP10D, phosphopeptide and PNC binding experiments were performed with both the native and the F76L mutant of PTP10D. A decrease in the Vmax with a corresponding increase in the Km for the dephosphorylation of the phosphotyrosine containing peptides without a concomitant change in their peptide sequence preference suggests that Phe76 does not function in substrate selection but affects the reaction rates of phosphatase activity. It thus appears likely that substrate selection occurs at the surface of the active site cavity whereas Phe76 is involved in the subsequent steps after peptide binding at the active site. The kinetic time-course experiments allow an estimate of the role of Phe76 in the reaction mechanism of PTP10D. A PTP catalyzed reaction can be represented by the following equations:
| (6) |
| (7) |
The rate of the formation of the cysteinyl phosphate enzyme intermediate (equation 6) is defined by a forward rate constant k1 and a reverse rate constant k−1. The rate of the equation (7) is defined by a forward rate constant k2. The overall rate is thus
| (8) |
The rate of the reverse reaction of the dissociation of the ES complex (E—P(OR)) is negligible at saturating concentrations of the substrate in the time course experiments. The forward reaction, under these experimental conditions, thus depends only on the rates k1 and k2. While the rate of ES complex dissociation to release the free enzyme and the product depends on the catalytic residues (Cys, Asp and Gln), the rate of ES complex formation (k1) depends on residues that mediate intermediate formation. A decrease in the Vmax for all substrates in the case of the F76L mutant suggests that Phe76 affects k2 of the PTP10D catalyzed reaction. An overall decrease in the rate of the time course (λ) in the F76L mutant for the five substrates suggests that the F76L mutation also affects the rate constant k1. Phe76 thus serves as a mediating residue that facilitates ES formation (Tables 3 and 4).
Binding of PNC at the PTP10D active site depends on two features-electrostatic interactions mediated by the nucleophilic cysteine (34) and the water network at the PTP active site (25). Mutations in the conserved Cys242 lead to a decrease in the rate of PNC binding at the active site as is seen by the lower rates (β) for the C242S and C242S/Q286A mutants (Table 4). A decreased lag time and t1/2 noted in the case of the D210A/Q286A mutant can probably be rationalized based on the disruption of the water network without any change in the electrostatic environment due to the active site Cysteine. Mutation of Phe76, which neither disrupts the nucleophile nor the water network, shows a lag of 160 sec and an increased t1/2 of PNC binding by 130 seconds. This suggests that Phe76 plays a role in helping the aryl ring of the substrate progress towards the PTP active site, displacing the water molecules and eventually docking at the nucleophilic cysteine. In the absence of a navigating phenyl group, the substrate ring is likely to adopt multiple conformations, most of which would hinder proper entry of the phosphotyrosine into the active site. The effect on PNC binding kinetics seen in the case of F76L with an increase in the lag time and t1/2 is consistent with the role of Phe76 at the step of ES complex formation. These observations are consistent with biochemical studies and molecular dynamics simulations on two membrane associated receptor PTPs, PTP99A and DLAR, reported recently. These proteins have two PTP domains D1 and D2. The membrane distal D2 domain is inactive (40). This observation can be correlated with a mutation of the aromatic residue in the KNRY motif of the D2 domain (Supplementary Table 1).
To conclude, the structural and biochemical analysis of PTP10D reveals a hitherto unexpected observation on substrate recognition in this class of enzymes. These studies suggest specific roles for relatively less conserved residues that lie at the periphery of the active site in substrate recruitment and modulation of catalytic activity. It thus appears likely that a nuanced regulation of catalytic activity in PTPs occurs at the stage of substrate recruitment, in addition to the other characterized mechanisms such as redox sensitivity or oligomerization.
Supplementary Material
Table 1 summarizing details of the PTP sequence motifs and Figures S1-S10 with additional biochemical data, representative sections of electron density and sequence and structural analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations
- PTP
Protein Tyrosine Phosphatase
- IPTG
isopropyl β-D-thiogalactoside
- LB
Luria-Bertani
- pNPP
p-nitrophenyl phosphate
- PNC
p-nitrocatechol sulphate
Footnotes
This work was supported by a grant from the Department of Science and Technology, Government of India.
REFERENCES
- 1.Hunter T. Signaling--2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 2.Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 3.Fischer EH. Cell signaling by protein tyrosine phosphorylation. Adv Enzyme Regul. 1999;39:359–369. doi: 10.1016/s0065-2571(98)00014-4. [DOI] [PubMed] [Google Scholar]
- 4.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desai CJ, Sun Q, Zinn K. Tyrosine phosphorylation and axon guidance: of mice and flies. Curr Opin Neurobiol. 1997;7:70–74. doi: 10.1016/s0959-4388(97)80122-5. [DOI] [PubMed] [Google Scholar]
- 6.Morrison DK, Murakami MS, Cleghon V. Protein kinases and phosphatases in the Drosophila genome. J Cell Biol. 2000;150:F57–62. doi: 10.1083/jcb.150.2.f57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schindelholz B, Knirr M, Warrior R, Zinn K. Regulation of CNS and motor axon guidance in Drosophila by the receptor tyrosine phosphatase DPTP52F. Development. 2001;128:4371–4382. doi: 10.1242/dev.128.21.4371. [DOI] [PubMed] [Google Scholar]
- 8.Tian SS, Tsoulfas P, Zinn K. Three receptor-linked protein-tyrosine phosphatases are selectively expressed on central nervous system axons in the Drosophila embryo. Cell. 1991;67:675–685. doi: 10.1016/0092-8674(91)90063-5. [DOI] [PubMed] [Google Scholar]
- 9.Yang XH, Seow KT, Bahri SM, Oon SH, Chia W. Two Drosophila receptor-like tyrosine phosphatase genes are expressed in a subset of developing axons and pioneer neurons in the embryonic CNS. Cell. 1991;67:661–673. doi: 10.1016/0092-8674(91)90062-4. [DOI] [PubMed] [Google Scholar]
- 10.Sun Q, Bahri S, Schmid A, Chia W, Zinn K. Receptor tyrosine phosphatases regulate axon guidance across the midline of the Drosophila embryo. Development. 2000;127:801–812. doi: 10.1242/dev.127.4.801. [DOI] [PubMed] [Google Scholar]
- 11.Qian M, Pan G, Sun L, Feng C, Xie Z, Tully T, Zhong Y. Receptor-like tyrosine phosphatase PTP10D is required for long-term memory in Drosophila. J Neurosci. 2007;27:4396–4402. doi: 10.1523/JNEUROSCI.4054-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dillet V, Van Etten RL, Bashford D. Stabilization of Charges and Protonation States in the Active Site of the Protein Tyrosine Phosphatases: A Computational Study†. The Journal of Physical Chemistry B. 2000;104:11321–11333. [Google Scholar]
- 13.Zhang ZY. Protein-tyrosine phosphatases: biological function, structural characteristics, and mechanism of catalysis. Crit Rev Biochem Mol Biol. 1998;33:1–52. doi: 10.1080/10409239891204161. [DOI] [PubMed] [Google Scholar]
- 14.Zhang ZY. Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 15.Fashena SJ, Zinn K. Transmembrane glycoprotein gp150 is a substrate for receptor tyrosine phosphatase DPTP10D in Drosophila cells. Mol Cell Biol. 1997;17:6859–6867. doi: 10.1128/mcb.17.12.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell HR. The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr D Biol Crystallogr. 1999;55:1690–1695. doi: 10.1107/s0907444999009506. [DOI] [PubMed] [Google Scholar]
- 17.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 18.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 19.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 20.Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. Journal of Applied Crystallography. 2008;41:641–643. [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 24.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 26.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.Pettit FK, Bare E, Tsai A, Bowie JU. HotPatch: a statistical approach to finding biologically relevant features on protein surfaces. J Mol Biol. 2007;369:863–879. doi: 10.1016/j.jmb.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madan LL, Gopal B. Addition of a polypeptide stretch at the N-terminus improves the expression, stability and solubility of recombinant protein tyrosine phosphatases from Drosophila melanogaster. Protein Expr Purif. 2008;57:234–243. doi: 10.1016/j.pep.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. A malachite green colorimetric assay for protein phosphatase activity. Anal Biochem. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- 31.Kakkar T, Boxenbaum H, Mayersohn M. Estimation of Ki in a competitive enzyme-inhibition model: comparisons among three methods of data analysis. Drug Metab Dispos. 1999;27:756–762. [PubMed] [Google Scholar]
- 32.Khajehpour M, Wu L, Liu S, Zhadin N, Zhang ZY, Callender R. Loop dynamics and ligand binding kinetics in the reaction catalyzed by the Yersinia protein tyrosine phosphatase. Biochemistry. 2007;46:4370–4378. doi: 10.1021/bi602335x. [DOI] [PubMed] [Google Scholar]
- 33.Pedersen AK, Peters GG, Moller KB, Iversen LF, Kastrup JS. Water-molecule network and active-site flexibility of apo protein tyrosine phosphatase 1B. Acta Crystallogr D Biol Crystallogr. 2004;60:1527–1534. doi: 10.1107/S0907444904015094. [DOI] [PubMed] [Google Scholar]
- 34.Sun JP, Wu L, Fedorov AA, Almo SC, Zhang ZY. Crystal structure of the Yersinia protein-tyrosine phosphatase YopH complexed with a specific small molecule inhibitor. J Biol Chem. 2003;278:33392–33399. doi: 10.1074/jbc.M304693200. [DOI] [PubMed] [Google Scholar]
- 35.Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 36.Jia Z, Barford D, Flint A, Tonks N. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 37.Tsiani E, Bogdanovic E, Sorisky A, Nagy L, Fantus IG. Tyrosine phosphatase inhibitors, vanadate and pervanadate, stimulate glucose transport and GLUT translocation in muscle cells by a mechanism independent of phosphatidylinositol 3-kinase and protein kinase C. Diabetes. 1998;47:1676–1686. doi: 10.2337/diabetes.47.11.1676. [DOI] [PubMed] [Google Scholar]
- 38.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 39.Xie L, Zhang YL, Zhang ZY. Design and characterization of an improved protein tyrosine phosphatase substrate-trapping mutant. Biochemistry. 2002;41:4032–4039. doi: 10.1021/bi015904r. [DOI] [PubMed] [Google Scholar]
- 40.Madan LL, Veeranna S, Shameer K, Reddy CCS, Sowdhamini R, Gopal B. Modulation of Catalytic Activity in Multi-Domain Protein Tyrosine Phosphatases. PLoS ONE. 2011;6:e24766. doi: 10.1371/journal.pone.0024766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1 summarizing details of the PTP sequence motifs and Figures S1-S10 with additional biochemical data, representative sections of electron density and sequence and structural analysis. This material is available free of charge via the Internet at http://pubs.acs.org.