

Abstract
Previous work has established the existence of dystrophin–nitric oxide (NO) signaling to histone deacetylases (HDACs) that is deregulated in dystrophic muscles. As such, pharmacological interventions that target HDACs (that is, HDAC inhibitors) are of potential therapeutic interest for the treatment of muscular dystrophies. In this study, we explored the effectiveness of long-term treatment with different doses of the HDAC inhibitor givinostat in mdx mice—the mouse model of Duchenne muscular dystrophy (DMD). This study identified an efficacy for recovering functional and histological parameters within a window between 5 and 10 mg/kg/d of givinostat, with evident reduction of the beneficial effects with 1 mg/kg/d dosage. The long-term (3.5 months) exposure of 1.5-month-old mdx mice to optimal concentrations of givinostat promoted the formation of muscles with increased cross-sectional area and reduced fibrotic scars and fatty infiltration, leading to an overall improvement of endurance performance in treadmill tests and increased membrane stability. Interestingly, a reduced inflammatory infiltrate was observed in muscles of mdx mice exposed to 5 and 10 mg/kg/d of givinostat. A parallel pharmacokinetic/pharmacodynamic analysis confirmed the relationship between the effective doses of givinostat and the drug distribution in muscles and blood of treated mice. These findings provide the preclinical basis for an immediate translation of givinostat into clinical studies with DMD patients.
INTRODUCTION
The most common muscular dystrophy (MD) is Duchenne muscular dystrophy (DMD), a severe recessive X-linked disease that affects 1 in 3500 males and is characterized by rapid progression of muscle degeneration, eventually leading to loss of ambulation and death within the second decade of life (1,2). This disorder is caused by mutations in the dystrophin gene that result in the complete absence or, very infrequently, in the expression of a truncated, nonfunctional protein. There is currently no available therapy for children with DMD, and current treatment is based on steroids, which only marginally affect the natural history of the disease (3,4). Pharmacological strategies for the treatment of muscular dystrophies are normally designed to counter the disease progression by targeting events downstream of the genetic mutation, such as inflammation, fibrosis, fat deposition and calcium homeostasis, or by promoting endogenous regeneration (5). Because of the hurdles that still prevent the application to dystrophic patients of gene- and cell-mediated therapies, pharmacological strategies provide a unique, immediate and suitable resource for the treatment of the current generation of dystrophic patients.
We have previously demonstrated the effectiveness of histone deacetylase inhibitors (HDACi) in the treatment of muscular dystrophies, using mdx mice as models (6,7). The mdx mice are the mouse model of human DMD and therefore provide the most amenable and approachable disease model for exploratory and preclinical evaluation of experimental interventions in muscular dystrophies. We have shown that exposure to HDACi counters the disease progression in mdx mice (6). HDACi produced functional and morphological beneficial effects, such as increased cross-sectional area (CSA) of myofibers, restoration of muscle force, decreased inflammatory infiltrate and prevention of fibrotic scars, which contribute to counter the muscle loss and the functional decline that are typically observed in mdx mice (6). Interestingly, the extent to which HDACi ameliorate the mdx phenotype varies significantly among these compounds, with trichostatin A (TSA) being the most effective drug at defined concentrations (TSA 0.6 mg/kg, delivered by daily intraperitoneal injection). An interesting insight into the specific role of individual HDACs in the pathogenesis of muscular dystrophy is suggested by the comparable efficacy of MS275, which selectively inhibits class I HDACs, and pan HDACi, which inhibit both class I and II HDACs (6,7). This suggests that inhibition of class I HDACs is sufficient to exert most of the beneficial effects observed in HDACi-treated mdx mice, once again emphasizing the key contribution to DMD pathogenesis by class I HDACs.
Current studies are seeking to define the relative ability to counter DMD progression with a number of different HDACi that have long been used in clinical practice (valproic acid [VPA] and phenylbutyrate) or have recently been approved for treatment of cancer and other diseases (6,8,9). Among them, suberoylanilide hydroxamic acid (SAHA) was also effective in ameliorating the dystrophic phenotype of mdx mice. A dose-finding study was performed by Colussi et al. with SAHA, using escalating doses ranging from 0.3 to 100 mg/kg/d delivered to mdx mice for 3 months (10). This study identified efficacy in recovering functional and histological parameters within a window of doses between 0.6 and 5 mg of SAHA, with evident reduction of the beneficial effects with doses lower than 0.6 mg and higher than 5 mg. Although the reason for such a dose-dependent response of mdx mice to SAHA is still unclear, this evidence indicates that dose-finding studies should be extended to all HDACi used for the experimental treatment of muscular dystrophies. The same study revealed an interesting correlation between mdx exposure to SAHA (5 mg/kg/d) and restoration of the profile of specific plasma proteins that were differentially expressed in mdx mice versus their normal counterparts (10).
One of the hurdles that complicates the translation into clinical trials of experimental drugs that have shown a therapeutic potential in animal models of muscular dystrophy is the absence of information on critical pharmacological parameters in pediatric populations, such as DMD boys. A notable exception is represented by ITF2357 (givinostat), which is currently being tested in a phase I safety study in children affected by systemic onset juvenile arthritis (11,12). This has inspired preclinical studies addressing the effectiveness of givinostat in preventing disease progression in mdx mice after prolonged exposure.
MATERIALS AND METHODS
Animals and In Vivo Treatments
C57Bl6J mdx mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Animals were used at the specified age and treated for the indicated periods with daily intraperitoneal injections of TSA (0.6 mg/kg/d) (Sigma-Aldrich, St. Louis, MO, USA) dissolved in saline solution, with gavage administration of givinostat (ITF2357) (1 mg/kg/d, 5 mg/kg/d and 10 mg/kg/d dissolved in methylcellulose 0.01%) or methylcellulose (Sigma-Aldrich) alone as control (CTR). Mice were maintained according to the standard animal facility procedures, and all experimental protocols were approved by the internal Animal Research Ethics Committee according to the Italian Ministry of Health and complied with the NIH-used Guide for the Care and Use of Laboratory Animals(13).
Exhaustion Treadmill Test
The analyses were carried out using a six-lane motorized treadmill (Exer 3/6 Treadmill; Columbus Instruments, Columbus, OH, USA) supplied with shocker plates. The first trial was performed at low intensity and for a short duration to accustom the mice to the exercise. In the protocol used, the treadmill was run at an inclination of 0° at 8 cm/s for 5 min, after which the speed was increased by 2 cm/s every 2 min up to a speed of 9 m/min. The test was stopped when the mouse remained on the shocker plate for more than 10 s without attempting to reengage the treadmill. The time to exhaustion was determined from the beginning of the test. Four tests were performed on the same animal, monthly.
Evans Blue Staining
Mice were intraperitoneally injected with 1% Evans blue dye (Sigma-Aldrich) (w/v) in phosphate-buffered saline (PBS) (pH 7.5) sterilized by passage through a Millex®-GP 0.22-μm filter (Millipore, Bedford, MA, USA). After injection, animals were returned to their cages and allowed food and water ad libitum. After 24 h the mice were killed and diaphragms were frozen (methyl butanol and N2 liquid).
Histology and Immunofluorescence
Tibialis anterior muscles were prefixed in paraformaldehyde (PFA) 0.5% and incubated at 4°C for 2 h. After fixation, to maintain good tissue morphology, the tissues were incubated in a 20% sucrose solution overnight at 4°C. The day after, the muscles were snap frozen in liquid nitrogen–cooled isopentane and then cut transversally into cryosections (10 μm) using a Leica CM 3050 S cryostat (Leica, Wetzlar, Germany).
For hematoxylin and eosin staining, cryosections were fixed in 4% PFA, washed in 1× PBS and then stained in hematoxylin for 4 min and eosin for 6 min. Then the cryosections were dried in ethanol and fixed in xylene and mounted with Eukitt mounting (O. Kindler GmbH, Freiburg, Germany).
To stain lipids, 10% formalin-fixed cells and tissues were rinsed with water and then with 60% isopropanol, stained with oil red O (Sigma-Aldrich) in 60% isopropanol and rinsed with water.
To stain fibrotic tissue Masson trichrome analysis was used. Muscle cryosections were stained in working Weigert’s iron hematoxylin solution (Sigma-Aldrich) for 5 min, washed in running tap water for 5 min and stained in Biebrich scarlet-acid fuchsin (Sigma-Aldrich) for 5 min. The cryosections were then rinsed in deionized water, placed in working phosphotungstic/phosphomolybdic acid solution for 5 min and stained in aniline blue solution for 5 min and in acid acetic 1% for 2 min. The slides were mounted with Eukitt mounting.
To stain inflammatory infiltration and in particular neutrophils and monocytes, the anti-human myeloperoxidase (MPO) monoclonal antibody (R&D Systems, Minneapolis, MN, USA) was used on tibialis anterior paraformaldehyde-fixed cryosections. The antibody was used at a concentration of 8 ug/mL after methanol (−20°C) permeabilization.
Images were acquired with a Leica confocal microscope and edited using the Photoshop software (Adobe, Seattle, WA, USA). Fields reported in the figures are representative of all examined fields.
Quantitative Analysis
The CSA was calculated using ImageJ software downloaded from http://rsb.info.nih.gov/ij. Fibrotic areas were measured by selecting four representative and nonadjacent sections and photographing up to three microscopic fields. The total fibrosis was calculated from sections by evaluating image analysis algorithms for color deconvolution. ImageJ was used for image processing. The original image was segmented with three clusters, and the plugin assumes images generated by color subtraction (white represents background, blue represents collagen and magenta represents noncollagen regions). Oil red O areas were measured by using ImageJ and calculating the area of red pixels (pixel2) per field. Inflammation was quantified by counting the number of MPO-positive cells per field. Each measurement was performed by selecting four representative and non-adjacent sections and photographing up to three microscopic fields. Statistical significance was determined with the Student t test.
Pharmacokinetic/Pharmacodynamic Analysis
The Emax model (also named the Hill equation model) was applied for linking the fibrosis and MPO effects with the individual areas under the curves (AUCs). The general equation of the model is defined by four parameters: the maximum effect Emax, the shape factor m, the baseline effect E0 and the AUC50, which measures the exposure for achieving 50% of the maximum effect. On the basis of different biological and model-building criteria, different parameterizations were tested. Finally, Emax was fixed to the median value in untreated mice (Emax = 52,347 pixels for fibrosis and Emax = 17.5 for MPO), m was set to 1 and E0 = 0, which means absence of fibrosis and/or MPO as the maximum therapeutic effect. Therefore, a one-parameter Hill equation model was applied for fitting pharmacodynamic (PD) data, and the AUC50 was estimated for each PD endpoint (fibrosis and MPO). Errors (for example, differences between observed and model-predicted values), considered homoscedastic and serially uncorrelated, were described using an additive model.
The cumulative frequency function was computed for each CSA distribution curve and the 50% cumulative frequencies (MTF50) were identified and linked to the individual blood concentration AUCs.
A categorical population model was applied to describe a PD effect in terms of the running time values on treadmill.
Score values of 1 and 0 were assigned to running time values showing a time increase higher or lower than 30%, respectively, in comparison to the median running time in controls (mice treated with vehicle). Each endpoint was considered to pertain to a binomial distribution, with 1 meaning the appearance of the desired event and 0 the absence of the event. On this basis, the dependence of the probability of a successful therapeutic effect from the blood concentration AUC was defined using a two-parameter logistic function:
where π is the probability of success, b0 yields the probability in the absence of drug treatment and b1 adjusts for how quickly the probability p(1) changes, changing the AUC a single unit. Considering the nature of the data and the high variability (in particular in controls, which ranged between 705 and 1935 s) data were preprocessed for outlier identification. In particular, individual running times were compared with the median values computed considering animals treated with a similar dosing regimen. A difference greater than 20% in respect to the median running time was considered to pertain to an outlier. The blood concentration AUC for obtaining an 80% probability of the desired effect was indicated (AUCp80).
RESULTS
In preliminary studies, we initially tested the effects of defined concentrations of givinostat (ITF2357) in vitro on cultured primary human skeletal myoblasts. Concentrations ranging from 80 to 200 nmol/L promoted the formation of myosin–heavy chain (MyHC)-positive multinucleated myotubes with a size larger than those formed by control-treated myoblasts—an effect that was equivalent to that observed with previously tested HDACi, such as TSA (Figure 1) (14). The potential availability of givinostat in clinical practice, and the known pharmacological profile of givinostat in pediatric patients (15), prompted an interest in testing givinostat in preclinical studies for the treatment of DMD. To this end, we performed a dose-finding study using escalating daily doses ranging from 1 to 10 mg/kg/d delivered to 1.5-month-old mdx mice (n = 8 for each experimental group) for 3.5 months (105 d). The effect of these doses was compared with that of vehicle alone (orally delivered) or TSA (0.6 mg/kg, delivered by daily intraperitoneal injection). The effect of these treatments was assessed by monitoring the weight of single muscles at the end of the treatment and by measuring morphohistological and functional parameters. In mdx mice there was progressive muscle degeneration starting by 3 wks of age. Muscle atrophy, inflammation and fibrosis are detected in the mdx mice at 8 wks of age in coincidence with a decrease in strength of the forelimb (16). Thus, this analysis in mdx mice between 1.5 and 3.5 months should capture the effect of the treatment with HDACi on the morphohistological and functional parameters of disease progression.
Figure 1.

Givinostat treatment of human skeletal myoblasts increases myotube size. The myogenic potential of human skeletal myoblasts was assessed after 24 h of TSA (50 nmol/L) and givinostat (150 nmol/L) in vitro treatment. Representative images of MyHC (red) staining are shown. Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich) (blue).
Although we observed a slight increase in total body weight of mdx mice only when they were exposed to 5 mg/kg/d of givinostat (Figure 2A), the exposure at each of the doses of givinostat tested (1, 5 and 10 mg/kg/d) equally increased the weight of a single muscle analyzed, with a statistically significant increase observed in the quadriceps of mdx mice exposed to 5 and 10 mg/kg/d of givinostat (Figure 2B). The increased weight of muscles from mdx mice exposed to givinostat was paralleled by a consensual increase in the CSA detected in muscles isolated from mdx mice treated with givinostat at 5 and 10 mg/kg/d (Figures 3A, B). The dose-related increased muscle mass and CSA in mdx mice exposed to givinostat correlated well with the preservation of the muscle architecture, which was otherwise subverted in control-treated mdx mice (Figure 3A). Importantly, the givinostat-dependent increase in muscle size was not due to infiltration of adipocytes or fibrosis, as oil red O and Masson trichome staining showed dose-dependent reductions of fat and collagen deposition, respectively, in muscles of all mdx mice exposed to givinostat (Figures 4 and 5). In both cases, muscles of mdx mice treated with 5 and 10 mg/kg/d doses of givinostat showed a dramatic reduction of fibrosis (Figure 4) and fatty infiltration (Figure 5), whereas the reduction of fibroadipogenic degeneration observed in muscles of mdx mice exposed to 1 mg/kg/d givinostat, versus control-treated mice, was less pronounced (fibrosis, Figure 4B) or statistically not significant (fatty infiltration, Figure 5B).
Figure 2.

Givinostat increases the weight of mdx muscles. (A) Whole naked bodies of mdx control-untreated (CTR), TSA-treated (TSA) or givinostat-treated mice (givinostat 1 mg/kg/d = ITF1; givinostat 5mg/kg/d = ITF5; givinostat 10 mg/kg/kg = ITF10) after 105 d of treatment. (B) Graph representing the body weight of mdx mice measured as grams (g) before the treatments (time 1) and every week along the course of the treatments. Data are presented as the average ± standard error of the mean (SEM); n = 8. (C) Hind limb legs isolated from CTR-, TSA- or givinostat-treated mdx after 105 d of treatment. (D) Graph representing the quadriceps weight measured as milligrams (mg) at the end of CTR, TSA or givinostat treatment. Data are presented as the average ± SEM; n = 6. *p < 0.05; **p < 0.01.
Figure 3.

Givinostat increases the size of mdx muscles. (A) Representative images of hematoxylin and eosin staining (H&E) of tibialis anterior (TA) transverse sections of CTR-, TSA- or givinostat-treated mdx mice (givinostat 1 mg/kg/d = ITF1; givinostat 5mg/kg/d = ITF5; givinostat 10 mg/kg/kg = ITF10) at the end of the treatment. Original magnification, 10×. (B) Graph of the analysis of myofiber CSA of muscles represented in (A). Data are represented as average ± SEM; n = 6. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 4.

Givinostat reduces fibrosis in mdx muscles. (A) Representative images of Masson trichrome staining of TA transverse sections of CTR-, TSA- or givinostat-treated mdx mice (givinostat 1 mg/kg/d = ITF1; givinostat 5 mg/kg/d = ITF5; givinostat 10 mg/kg/kg = ITF10) at the end of the treatment. Original magnification, 10×. (B) Graph representing the fibrotic index (quantification of collagen deposition) of muscles shown in (A) measured as blue area (reported as pixel2) per field. Data are presented as the average ± SEM; n = 6. ***p < 0.001.
Figure 5.

Givinostat reduces fat deposition in mdx muscles. (A) Representative images of oil red O staining of TA transverse sections of CTR-, TSA-, or givinostat-treated mdx mice (givinostat 1 mg/kg/d = ITF1; givinostat 5 mg/kg/d = ITF5; givinostat 10 mg/kg/kg = ITF10) at the end of the treatment. Original magnification, 10×. (B) Graph representing fat accumulation in muscles shown in (A) measured as red area (reported as pixel2) per field. Data are presented as the average ± SEM; n = 6. **p < 0.01.
Fibrosis is typically considered the most deleterious consequence of disease progression in DMD children, and it results from a complex functional interplay between resident cell types and the inflammatory infiltrate (17). We therefore measured the MPO activity in muscles of mdx mice killed at the end of the treatment with different doses of givinostat, compared with control vehicle-treated mdx mice. MPO is an enzyme produced by neutrophils, monocytes and macrophages, and MPO activity can be used to quantitate the magnitude of inflammation associated with muscle degeneration in muscular dystrophies (18,19). Once again, a significant reduction of MPO was observed in muscles from mdx mice exposed to 5 and 10 mg/kg/d doses of givinostat (Figure 6). No significant reduction of MPO was observed with TSA and the lowest dose of givinostat (1 mg/kg/d). This might be a direct consequence of the most pronounced effect of high doses of givinostat on muscle mass increase and on reduction of fibroadipogenic degeneration, compared with TSA and lower doses of givinostat. Moreover, an antiinflammatory effect of givinostat has been previously reported at specific concentrations (20) and might account for the reduction of the inflammatory infiltrate in mdx muscles. However, we could not detect significant reductions in circulating inflammatory cytokines (data not shown), suggesting that the effect of givinostat is local (intramuscular) rather than systemic. Further studies will clarify this issue.
Figure 6.

Givinostat reduces inflammation (neutrophil granulocytes) in mdx mice. Representative images of MPO staining on TA transverse sections of CTR-, TSA-, or givinostat-treated mdx mice (givinostat 1 mg/kg/d = ITF1; givinostat 5 mg/kg/d = ITF5; givinostat 10 mg/kg/kg = ITF10). Original magnification, 10×. (B) Graph representing MPO evaluation scored as the number of MPO-positive cells per field. Data are presented as the average ± SEM; n = 6. **p < 0.01.
We next evaluated the effect of givinostat on functional parameters of skeletal muscle activity by monitoring the performance of mdx mice on a treadmill test, at defined time points during the treatment. The mdx mice exposed to 1 mg/kg/d doses of givinostat displayed a trend toward an increased performance starting from the very beginning and extending for the entire duration of the treatment (Figure 7), whereas exposure to 5 mg/kg/d led to a significantly better performance at months 1 and 2. By contrast, the beneficial effects of 10 mg/kg/d doses of givinostat, if any, were observed only at the end of treatment (Figure 7). The maintenance of myofiber integrity after exercise, which reflects a protective effect of the treatment on the major consequence of dystrophin deficiency—that is, degeneration of dystrophic muscles at each contraction—was determined by measuring the Evans blue uptake in muscles of mdx mice killed at the end of the treadmill test (21). Evans blue dye was delivered to the mice by intraperitoneal injection, and an abundant uptake was detected in degenerating muscles of control mdx mice, as expected. Mice treated with givinostat showed a reduction of Evans blue uptake at all doses tested (Figure 8). However, the effect was statistically significant only at the 10 mg/kg/d dose, and TSA did not affect this parameter (Figure 8B).
Figure 7.

Givinostat ameliorates fatigue resistance of mdx mice. Treadmill test on CTR-, TSA-, or givinostat-treated mdx mice (givinostat 1 mg/kg/d = ITF1; givinostat 5 mg/kg/d = ITF5; givinostat 10 mg/kg/kg = ITF10) at four time points during the treatment from d 0 to d 105 (1 = d 0; 2 = 1 month; 3 = 2 months; 4 = 3.5 months). Data are presented as the average ± SEM; n = 8. *p < 0.05; **p < 0.01.
Figure 8.

Givinostat reduces Evans blue uptake in diaphragms of mdx mice. (A) Representative images of Evans blue uptake in diaphragm transverse sections of CTR-, TSA-, or givinostat-treated mdx mice (givinostat 1 mg/kg/d = ITF1; givinostat 5mg/kg/d = ITF5; givinostat 10 mg/kg/kg = ITF10). Diaphragms were harvested after a treadmill test at the end of the treatment. Original magnification, 2×. (B) Graph representing Evans blue uptake scored as red area (reported as pixel2) per field. Data are presented as the average ± SEM; n = 2. **p < 0.01.
To further elucidate the relationship between givinostat exposure and effects on muscle tissue and on performance, we conducted a pharmacokinetic (PK)/PD analysis. We developed a population PK approach (Advan 1 model using Nonmem, version VI) aimed at determining the exposure parameters (Cmax and daily AUC) in blood after repeated oral daily administration of givinostat at 1, 5 and 10 mg/kg/d in mdx male mice. Individual daily blood AUCs were linked with the PD effects in terms of fibrosis, MPO, CSA and treadmill running time. In Figure 9A, the observed fibrosis values and the model-derived profile are displayed. The blood AUC of 512 h/nmol/L (coefficient of variation [CV%] of 38.1) was estimated to be necessary to reduce fibrosis by 50%.
Figure 9.

A) Observed fibrosis as a function of blood concentration AUC (h/nmol/L) and predicted profile using a one-parameter Hill equation (AUC50 = 512 h/nmol/L, CV% = 38.1). (B) Observed MPO as a function of blood concentration AUC (h/nmol/L) and predicted profile using a one-parameter Hill equation (AUC50 = 632 h/nmol/L, CV% = 35.7).
Observed and predicted MPO endpoints are displayed in Figure 9B. Considering the MPO effect, an AUC50 value of 632 h/nmol/L (CV% = 35.7) was obtained in good agreement with the value previously estimated for the fibrosis endpoint. Givinostat was shown to shift the muscle fiber distribution toward fibers with larger diameters. Such effects were expressed by the parameter muscle fiber thickness 50 (MFT50), that is, the muscle fiber thickness matching a 50% cumulative frequency. Figure 10 shows that, in this case, MTF50 was also correlated with givinostat exposure, that is, increasing exposures determined an increase in MTF50, namely a shift toward thicker muscle fibers.
Figure 10.

Individual 50% cumulative frequencies, MFT50 as a function of blood concentration AUCs.
Finally, we estimated the givinostat exposures expected to improve by at least 30% muscular function, measured by the running time on the treadmill test, with a probability of 80%. Categorical score values of 1 and 0 were assigned to running time values showing a time increase higher or lower than 30%, respectively, in comparison to the median running time in controls. Three outliers were identified and excluded from the analysis. The population logistic model allowed us to estimate an AUCp80 of about 610 h/nmol/L, in good agreement with the AUC50 values (512 and 632 h/nmol/L) obtained for fibrosis and MPO endpoints (Figure 11).
Figure 11.
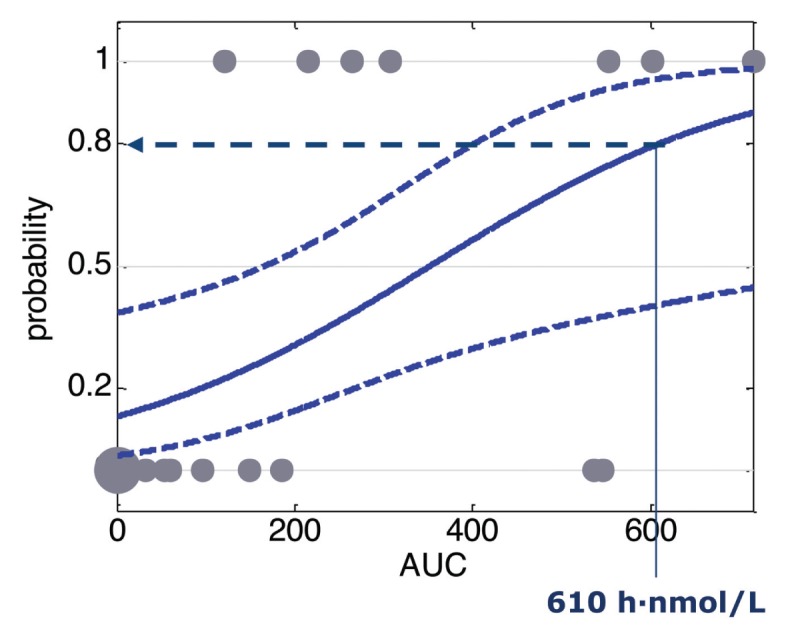
Observed running times computed as binary variable and predicted probability of successful effect defined using a logistic regression function versus blood concentration AUC. The 80% of probability to have a positive outcome is indicated (AUC80 = 610 h/nmol/L).
DISCUSSION
The results of these preclinical studies demonstrate that givinostat exerts beneficial effects in the mouse model of DMD (mdx mice) that are similar or even better than those previously reported with TSA (6). The dose-response study indicates that doses higher than 1 mg/kg/d are required for a therapeutic effect of givinostat and suggests an optimal range of doses between 5–10 mg/kg/d in the mdx model. Furthermore the PK/PD analysis identified exposures of the drug expected to be beneficial. Interestingly, an AUC of approximately 600 h/nmol/L was consistently associated with a 50% effect on histological parameters, and such exposure was needed for a meaningful improvement in performance of the mdx mice on the treadmill test.
The actual reason accounting for the beneficial effect observed in mdx mice treated with doses of givinostat between 5–10 mg/kg/d is currently unknown. While these doses of givinostat could ameliorate most of the functional and histological parameters, we observed a better effect of 10 mg/kg/d in promoting increased CSA (Figure 3) and preventing fibrosis (Figure 4) and adipose infiltration (Figure 5), while treatment with 5 mg/kg/d appeared to have the best effect in restoring muscle endurance, as shown by the treadmill test (Figure 6). It is interesting to note that despite the better treadmill performance of mdx mice treated with 5 mg/kg/d of givinostat, these mice showed increased postexercise membrane permeability of diaphragms (Figure 8) as well as other muscles of the posterior legs (not shown), compared with 10 mg/kg/d concentration. This might depend on the differential impact of these doses on distinct parameters, such as muscle strength and membrane stabilization of dystrophic muscles. For instance, 5 mg/kg/d might exert maximal activity on muscle strength and endurance but have limited efficacy in stabilizing the membrane of dystrophin-deficient muscles, as suggested by the reduced effects on histological parameters in comparison with 10 mg/kg/d. The overall outcome of such effects would be paradoxical degeneration of muscles that have undergone more strenuous exercise, as shown in Figures 7 and 8.
An additional effect observed with both 5- and 10-mg/kg/d doses of givinostat was the reduction of the inflammatory infiltrate in mdx muscles (Figure 6). The relationship between reactive inflammation of dystrophic muscles and the role of local concentration of inflammatory cytokines in promoting compensatory regeneration or fibroadipogenic degeneration is still controversial (22). Although the role of chronic inflammation in the pathogenesis of DMD has been firmly established (23) and is well supported by the beneficial effects observed with steroid treatment in DMD patients (4) and antiinflammatory interventions in mdx mice (24), the inflammatory infiltrate also provides a source of environmental cues that can positively affect the activity of the cellular network that promotes muscle regeneration (22). However, over time these beneficial effects can turn into negative ones, with an exhaustion of the regeneration potential of dystrophic muscles and the compensatory repair by fibrosis and fat deposition. Because inflammation typically occurs in response to the degeneration after contraction in dystrophic muscles, the reduction of the inflammatory infiltrate observed in muscles of mdx mice treated with 5 and 10 mg/kg/d of givinostat can be the consequence of their primary beneficial effects—for example, stimulation of regeneration and inhibition of fibrosis and adipogenesis. Alternatively, the antiinflammatory action of givinostat can directly contribute to the long-term ability to reduce fibrosis and improve functional parameters.
CONCLUSION
Overall, these data demonstrate the efficacy of givinostat in a preclinical model of DMD and support the progression of givinostat into clinical studies in children affected by DMD. Importantly, the current study predicts exposures to doses that could be necessary to exert a beneficial effect, and this will help in defining the dose to be used in the clinical studies. Because pharmacological interventions with HDACi do not restore dystrophin expression (6) but instead target downstream effectors of dystrophin-NO signaling (5), they require a prolonged schedule of treatment or even life-long exposure to achieve a persistent effect in dystrophic muscles. Thus, it will be important to define the effectiveness and the potential of long-term treatment with givinostat in DMD patients in clinical trials. In this regard, an important goal of future studies will be to better understand the interactions of givinostat with other pharmacological interventions, including steroids, to optimize synergistic schedules of drug administration.
ACKNOWLEDGMENTS
PL Puri is an Associate Investigator of Sanford Children’s Health Research Center. This work has been supported by the following grants to PL Puri: R01AR052779 and P30 AR061303 from the National Institute of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), MDA, AFM, FILAS and EPIGEN. This work has benefited from research funding from the European Community’s Seventh Framework Programme in the project FP7-Health 2009 ENDOSTEM 241440 (Activation of vasculature associated stem cells and muscle stem cells for the repair and maintenance of muscle tissue). C Mozzetta was supported by an AFM fellowship. We thank A Sandri (Plaisant) for the excellent assistant in treating and monitoring in vivo functions of mdx mice.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Dalkilic I, Kunkel LM. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. 2003;13:231–8. doi: 10.1016/s0959-437x(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 3.Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol. 2006;9:427–43. doi: 10.2350/06-07-0127.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muntoni F, Fisher I, Morgan JE, Abraham D. Steroids in Duchenne muscular dystrophy: from clinical trials to genomic research. Neuromuscul. Disord. 2002;12(Suppl 1):S162–5. doi: 10.1016/s0960-8966(02)00101-3. [DOI] [PubMed] [Google Scholar]
- 5.Consalvi S, et al. Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases. Mol Med. 2011;17:457–65. doi: 10.2119/molmed.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minetti GC, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–50. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 7.Colussi C, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A. 2008;105:19183–7. doi: 10.1073/pnas.0805514105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iezzi S, et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6:673–84. doi: 10.1016/s1534-5807(04)00107-8. [DOI] [PubMed] [Google Scholar]
- 9.Dinarello CA, Fossati G, Mascagni P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol Med. 2011;17:333–52. doi: 10.2119/molmed.2011.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colussi C, et al. Proteomic profile of differentially expressed plasma proteins from dystrophic mice and following suberoylanilide hydroxamic acid treatment. Proteomics Clin Appl. 2010;4:71–83. doi: 10.1002/prca.200900116. [DOI] [PubMed] [Google Scholar]
- 11.Vojinovic J, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:1452–8. doi: 10.1002/art.30238. [DOI] [PubMed] [Google Scholar]
- 12.Vojinovic J, Damjanov N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol Med. 2011;17:397–403. doi: 10.2119/molmed.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Studies, National Research Council of the National Academies. Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press; 2011. [cited 2013 Apr 23]. Available from: http://oacu.od.nih.gov/regs/ [Google Scholar]
- 14.Iezzi S, Cossu G, Nervi C, Sartorelli V, Puri PL. Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc Natl Acad Sci U S A. 2002;99:7757–62. doi: 10.1073/pnas.112218599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furlan A, et al. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat) Mol Med. 2011;17:353–62. doi: 10.2119/molmed.2011.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grounds MD, et al. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol Dis. 2008;31:1–19. doi: 10.1016/j.nbd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vidal B, et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22:1747–52. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barone FC, et al. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–45. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- 19.Zhang ZG, Chopp M. Measurement of myeloperoxidase immunoreactive cells in ischemic brain after transient middle cerebral artery occlusion in the rat. Neurosci Res Commun. 1997;20:85–91. [Google Scholar]
- 20.Leoni F, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11:1–15. doi: 10.2119/2006-00005.Dinarello. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamer PW, McGeachie JM, Davies MJ, Grounds MD. Evans Blue dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J Anat. 2002;200:69–79. doi: 10.1046/j.0021-8782.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mozzetta C, Minetti G, Puri PL. Regenerative pharmacology in the treatment of genetic diseases: the paradigm of muscular dystrophy. Int J Biochem Cell Biol. 2009;41:701–10. doi: 10.1016/j.biocel.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J, Grange RW. Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in Duchenne muscular dystrophy. Am J Phys Med Rehabil. 2009;88:502–22. doi: 10.1097/PHM.0b013e3181a5a24f. [DOI] [PubMed] [Google Scholar]
- 24.Brunelli S, et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci U S A. 2007;104:264–9. doi: 10.1073/pnas.0608277104. [DOI] [PMC free article] [PubMed] [Google Scholar]