

Abstract
Background:
An increased or normal serum TSH concentration, despite elevated thyroid hormone levels, is observed in resistance to thyroid hormone (RTH) and TSH-secreting adenomas (TSHomas). When coexistent with a differentiated thyroid cancer (DTC), maintenance of a suppression of TSH is challenging.
Objectives:
The aim of the study was to discuss the pitfalls arising from the failure to suppress TSH secretion in DTC and the strategies for proper treatment of DTC in association with RTH and TSHoma.
Methods:
Four unusual cases of DTC associated with TSHoma (2 cases), RTH (1 case), and an elevated TSH of unknown etiology (1 case) are presented, and the literature is reviewed.
Results:
Although a persistent mild TSH elevation may not be a risk factor for the development of DTC, it represents an important problem during the treatment of DTC. Aggressive treatment options should be applied in the proper order to prevent tumor recurrence and persistence in the absence of ideal TSH suppression.
Conclusions:
Although there is no agreed consensus regarding the management of DTC in the presence of persistent hyperthyrotropinemia, complete tumor removal followed by radioablation and attempts to reduce the serum TSH to the lowest tolerable level are recommended. The outcomes in the reported cases have not been unfavorable, despite the persistence of nonsuppressed TSH.
The state of an increased or normal TSH level despite elevated levels of thyroid hormones is termed as inappropriate TSH secretion (1). This condition occurs in TSH-secreting adenomas (TSHomas). However, it is being used generically for various abnormalities such as resistance to thyroid hormone (RTH), the presence of antibodies to T4, familial dysalbuminemic hyperthyroxinemia, increased circulating transport proteins during the neonatal period, systemic or acute psychiatric illnesses, and with some pharmaceuticals (amiodarone, contrast agents, amphetamine, levothyroxine [l-T4]) (2). Physiologically, serum TSH levels that are seen in RTH are appropriate for the reduced sensitivity of thyroid hormones resulting in maintain a near euthyroid state (3). Therefore, instead of “inappropriate TSH secretion,” this TSH state can be termed as “compensative TSH secretion.” A detailed history, measurement of free thyroid hormone levels, and application of specific laboratory tests may be sufficient to distinguish most of these etiologies. In addition, the differential diagnosis of RTH and TSHoma is essential and needs further investigation (4).
Thyroid cancers account for more than 90% of all endocrine malignancies, most of which are papillary thyroid carcinomas (PTCs) (5). In differentiated thyroid cancers (DTCs), l-T4 is used to suppress TSH release in order to prevent the relapse or progression of the thyroid cancer (6). The rationale for the suppressive l-T4 treatment was well demonstrated in multiple experimental and clinical studies (6–15). Therefore, TSH suppression has been considered as the key step of DTC treatment after total thyroidectomy and radioactive iodine therapy. However, when a patient with inappropriate TSH secretion or compensative high TSH secretion is present, TSH suppression may not be obtained despite increasing doses of l-T4. Yet there is no consensus on how to overcome the persistently high TSH concentration barrier in association with DTC.
To the best of our knowledge, there are only a few cases reporting the coexistence of DTC with RTH or TSHoma (16–25). Herein we report a total of 4 cases and their follow-up results, 2 of which show the coexistence of TSHoma with PTC, one with the presence of RTH in association with PTC, as well as a final case of hyperthyrotropinemia of unknown etiology in association with PTC. To discuss the pitfalls arising from the failure to inhibit TSH secretion and to focus on the strategy for proper treatment, a review of the literature was also undertaken (Tables 1 and 2).
Table 1.
Literature Review of DTCs with TSHoma
| First Author, Year (Ref.) | Age (y)/ Gender | Order of Diagnosis (Diagnosis Time) | Type of DTC (Diameter) | IHC Exam or Biochemical Diagnosisa | Diameter of Pituitary Mass (Expansion) | Applied Treatment (Respectively) | Follow-up for DTCb |
|---|---|---|---|---|---|---|---|
| Calle-Pascual, 1991 (16) | 55/M | Concurrent | Follicular (50 mm) | TSHa | ? (suprasellar invasion) | 1. Total thyroidectomy and radioiodine | 1 y/Remission |
| 2. Pituitary adenomectomy | |||||||
| Gasparoni, 1998 (17) | 37/F | 1. PTC | Papillary (20 mm) | TSHa | 10 mm (intrasellar) | 1. Total thyroidectomy and radioiodine | NR/remission |
| 2. TSHoma (just after first diagnosis) | 2. ? | ||||||
| Kishida, 2000 (18) | 27/F | 1. TSHoma | Papillary (30 mm) | TSH β-subunit | 10 mm (intrasellar) | 1. Somatostatin analog and iodine (preoperative only) | 3 mo/NR |
| 2. PTC (5 mo after first diagnosis) | 2. Pituitary adenomectomy | ||||||
| 3. Hemi- thyroidectomy | |||||||
| Ohta, 2001 (19) | 45/F | Concurrent | Papillary (20 mm) | TSH and GH | 15 mm (frontal base invasion) | 1. Somatostatin analog (preoperative only) | 4 mo/NR |
| 2. Total thyroidectomy | |||||||
| 3. Pituitary adenomectomy | |||||||
| Gessl, 2006 (20) | 47/F | 1. FTC | Follicular (8 mm) | TSH and prolactin | 4 mm (intrasellar) | 1. Total thyroidectomy and radioiodine | 13 y/NR |
| 2. TSHoma (13 y after first diagnosis) | 2. Pituitary adenomectomy | ||||||
| Poggi, 2009 (21) | 50/M | Concurrent | Follicular (17 mm) | TSHa | 3 mm (intrasellar) | 1. Total thyroidectomy | 12 mo/remission |
| 2. Patient refused pituitary surgery | |||||||
| 3. Watchful waiting for pituitary adenoma | |||||||
| Nguyen, 2010 (22) | 57/F | Concurrent | Papillary (multifocal and largest one 8 mm) | TSH and GHa | 26 mm (left cavernous sinus invasion) | 1. Somatostatin analog | 12 mo/remission |
| 2. Patient refused pituitary surgery | |||||||
| 3. Somatostatin analog | |||||||
| 4. Total thyroidectomy and radioiodine | |||||||
| 5. Somatostatin analog | |||||||
| Present case 1 | 38/F | 1. TSHoma | Papillary (multifocal and largest one 40 mm) | TSH and FSH | 22 mm (indentation to suprasellar cistern) | 1. Patient refused pituitary surgery | 7 y/remission |
| 2. PTC (9 y after first diagnosis) | 2. Somatostatin analog | ||||||
| 3. Total thyroidectomy and radioiodine | |||||||
| 4. Pituitary adenomectomy | |||||||
| 5. Irradiation by γ knife | |||||||
| Present case 2 | 27/F | Concurrent | Papillary (multifocal and largest one 10 mm) | TSH | 28 mm replaced the infundibulum to left side | 1. Pituitary adenomectomy | 6 mo/remission |
| 2. Total thyroidectomy and radioiodine | |||||||
| 3. Somatostatin analog |
Abbreviations: FTC, follicular thyroid carcinoma; F, female; M, male; NR, not reported.
IHC exam: immunohistochemical examination of pituitary mass.
Follow-up for DTC: the duration of the follow-up after the initial treatment of thyroid cancer/result.
Table 2.
Literature Review of DTCs with RTH
| Author, Year (Ref.) | Age (y)/ Gender | Order of Diagnosis (Diagnosis Time) | Type of DTC | Diameter | Mutation Type | Applied Treatment (Respectively) | Follow-up for DTCa |
|---|---|---|---|---|---|---|---|
| Taniyama, 2001 (23) | 46/F | 1. TMNG | Follicular variant of papillary | 5 mm | R429Q | 1. Methimazole | NR/NR |
| 2. PTC | 2. Subtotal thyroidectomy | ||||||
| 3. RTH | |||||||
| Kim, 2010 (24) | 38/F | 1. RTH | Papillary | 4 mm (multifocal) | M310T | 1. Total thyroidectomy | NR/NR |
| 2. PTC | 2. l-T4 | ||||||
| Paragliola, 2011 (25) | 48/M | 1. RTH | Papillary | 24 mm | No mutation found | 1. Total thyroidectomy and radioiodine | 9.5 y/remission |
| 2. MNG | 2. l-T4 | ||||||
| 3. PTC | |||||||
| 63/M | 1. MNG | Papillary | 6 mm | P453T | 1. Total thyroidectomy | 5 y/remission | |
| 2. RTH | 2. l-T4 | ||||||
| Present case 3 | 29/F | 1. TDG | Papillary | 8 mm | T334C | 1. Subtotal thyroidectomy | 21 y/remission |
| 2. PTC | 2. Completion thyroidectomy and radioiodine | ||||||
| 3. RTH | 3. l-T4 and bromocriptine | ||||||
| 4. l-T4 | |||||||
| Present case 4 | 33/M | 1. MNG | Papillary | 12 mm | No mutation found | 1. Total thyroidectomy and radioiodine | 1 y/remission |
| 2. PTC | 2. l-T4 | ||||||
| 3. Hyperthyrotropinemia of unknown etiology |
Abbreviations: TMNG, toxic multinodular goiter; MNG, multinodular goiter; FTC, follicular thyroid carcinoma; F, female; M, male; NR, not reported.
Follow-up for DTC: the duration of the follow-up after the initial treatment of thyroid cancer/result.
Patients and Methods
Case 1
A 38-year-old woman presented with a palpable goiter and nervousness. She had no history of irradiation or family history of thyroid cancer. Laboratory investigations revealed elevated serum TSH of 10.1 mU/L (normal range [N]; 0.30–4.0), free T4 (fT4) of 24.4 pmol/L (N; 10–22), and free T3 (fT3) of 7.1 pmol/L (N; 2.8–7), as well as elevated thyroglobulin (Tg) and thyroperoxidase (TPO) antibodies. Her thyroid ultrasound (US) showed marked heterogeneity of the parenchyma with pseudonodules. Thyroidal 131I uptake was 97%. Fine-needle aspiration cytology (FNAC) of the hypoactive area on scan revealed regressive changes. Her pituitary magnetic resonance imaging (MRI) revealed an adenoma 7 × 14 mm in diameter. The baseline TSH level of 10.5 mU/L was 10.1 mU/L 7 d after the administration of 100 μg/d l-T3 and increased from a baseline of 12.3 mU/L to only 13.1 mU/L 20 min after the administration of TRH. Antibodies to T3 and T4 were negative and the α-subunit/TSH molar ratio was 4.2. Octreotide scan indicated a pathological uptake in the pituitary. Collectively, these findings were compatible with TSHoma. The response to 3-month treatment with 100 μg/d octreotide was minimal. The patient refused surgical resection and was lost to follow-up. Nine years later, she presented with headache and a palpable goiter. Her pituitary adenoma was now 15 × 22 mm in diameter, invading the suprasellar cistern. Her thyroid US revealed multiple nodules, and FNAC of the largest nodule was compatible with PTC. Total thyroidectomy was performed, and a 4-cm oncocytic variant of PTC with extrathyroidal invasion was observed. Subsequently, she received 150 mCi of 131I. One year later, the patient consented to undergo transsphenoidal pituitary adenomectomy. The resected tissue was an adenoma, 100% TSH and 40% FSH on immunohistochemical staining. The presence of a residual pituitary tumor on MRI and a decrease of serum TSH levels from 15.6 to 3.7 mU/L nonsuppressible with 175 μg l-T4 led to treatment with 10 mg octreotide LAR. Octreotide LAR dose was gradually increased during the course of treatment. Because neither TSH reduction (values ranging from 4.3 to 9.9 mU/L while on 30 mg) nor reduction of residual tumor size was observed, stereotactic radiosurgery (Gamma Knife, Elekta Instruments, Stockholm, Sweden) was performed. After 3 years, the patient discontinued octreotide LAR injections. Six years after radiosurgery, her serum TSH levels were 3.4 and 3.7 mU/L, residual pituitary tumor remained stable, and her PTC was in remission.
Case 2
A 27-year-old woman presented with a 2-year history of treatment with propylthiouracil, allegedly for thyrotoxicosis, which the patient had discontinued 3 weeks earlier. Physical examination revealed a 1-cm thyroid nodule. She had no history of irradiation or family history of thyroid cancer. Laboratory investigations revealed elevated serum TSH of 11.29 mU/L, fT4 of 26 pmol/L (N; 7–16), and fT3 of 9.13 pmol/L (N; 3–6.8). Tg and TPO antibodies were not detectable. Thyroid US showed 2 hypoechogenic nodules measuring 8 × 4 × 6 mm in diameter and 12 × 8 × 10 mm in each lobe. Radioiodide distribution was nonhomogeneous, and uptake at 24 hours was 66.3%. FNAC of the nodules was suspicious of PTC. Pituitary MRI revealed a 28 × 22-mm adenoma that was near the optic chiasm. The patient's α-subunit/TSH molar ratio was 6.1, compatible with TSHoma. Treatment with 20 mg/d methimazole was given prior to transsphenoidal pituitary adenomectomy. Histopathological examination confirmed the diagnosis of pituitary adenoma (ie, 100% immunohistochemical positivity for TSH). Three months later, her serum TSH level was still elevated at 17.6 mU/L. The patient underwent total thyroidectomy with central lymph node dissection. Histopathological examination revealed multifocal PTC, being 1 cm in the longest diameter, and 2 other lesions of 2 mm residing in the left thyroid lobe with reactive central lymph nodes. The patient received 150 mCi of 131I. Six months later, although on 200 μg/d l-T4, her serum TSH levels were still elevated at 10 mU/L with fT4 of 17.7 pmol/L. Serum Tg and Tg antibodies were undetectable. Her cervical US examination showed no pathologically appearing lymph node. Due to the persistence of a residual pituitary mass (9 × 5 mm in diameter) and high serum TSH levels that could not be suppressed with 200 μg/d l-T4, 60 mg/month, lanreotide acetate was added to her treatment regimen. In her last follow-up visit 9 months later, her serum TSH level had decreased to 4.7 mU/L.
Case 3
A 29-year-old woman presented with a previous diagnosis of toxic diffuse goiter and 5-year symptoms of palpitation and nervousness. She had no exposure to irradiation or family history of thyroid cancer. She had no ophthalmopathy. Thyroid function tests demonstrated a normal serum TSH of 1.1 mU/L but elevated fT3 of 8.2 pmol/L and fT4 of 35.6 pmol/L. Tg and TPO antibodies were undetectable. Failure of response to antithyroid drugs led to subtotal thyroidectomy. Histopathological examination revealed an 8-mm PTC. Thyroidectomy was subsequently completed, followed by 100 mCi of 131I. Treatment with 300 μg/d of l-T4 did not suppress her TSH, which was 4.6 mU/L with fT4 of 35.8 pmol/L and fT3 of 7.6 pmol/L. Her anterior pituitary hormones as well as SHBG levels were within normal ranges. TSH increased from 11.9 to 91.2 mU/L at 20 min after the administration of TRH. After 100 μg/d T3 for 10 d, her serum TSH level of 10.9 mU/L at baseline decreased to only 7.4 mU/L. Pituitary MRI showed a partially empty sella. Screening of family members revealed 2 siblings with elevated serum fT3 and fT4 levels with normal serum TSH concentrations but no thyrotoxic manifestations. Sequencing of the thyroid hormone receptor β (TRβ) gene revealed a single nucleotide substitution (T>C) in codon 334, resulting in the replacement of the normal methionine with a threonine (M334T; Figure 1). This missense mutation has been previously reported (26). A short period of treatment with 2.5 mg/d of bromocriptine and 150 μg/d of l-T4 decreased her TSH level from 8.8 to 4.1 mU/L. However, due to the severe gastrointestinal intolerance, bromocriptine was discontinued and treatment with l-T4 was maintained. The l-T4 treatment dose could not be further increased to suppress TSH due to the appearance of tachycardia. Nevertheless, after 20 years, there is no evidence of PTC recurrence. Bone mineral density is normal. Because of sinus tachycardia, 50 mg/d metoprolol was added to her treatment.
Figure 1.

Pedigree of case 3 (II-1), including the thyroid function tests and thyroid antibody levels as well as the TRβ gene mutation. Abnormal values are represented in bold, high values are underlined, and low values are in italics. AITD, autoimmune thyroid disease; TT4, total T4; TT3, total T3; TrT3, total rT3.
Case 4
A 33-year-old man presented with a multinodular goiter. His physical examination revealed a slightly enlarged thyroid gland. Thyroid function tests demonstrated elevated levels of fT4 of 18.4 pmol/L (N; 7–16) and fT3 of 7.6 pmol/L (N; 3.8–6) with a normal TSH of 1.85 mU/L. Antibodies to Tg, TPO, T4, and T3 were absent, and SHBG was 43.7 nmol/L (N; 15.5–48.4). MRI showed a normal pituitary gland. Thyroid US demonstrated a hypoechoic nodule measuring 8 × 10 × 8 mm. FNAC of the nodule was compatible with PTC. Histopathological examination after total thyroidectomy with central lymph node dissection revealed a PTC focus of 12 mm in the longest diameter at the nodule location, showing capsular invasion, and reactive central lymph nodes. The patient received 150 mCi 131I. Six months later, his serum TSH level was 16 mU/L while on 200 μg l-T4 daily, with fT4 within the normal range of 13.5 pmol/L. Both TRβ and albumin genes were sequenced to determine whether the patient had RTH or familial dysalbuminemic hyperthyroxinemia. However, no mutations were found. The TSH receptor (TSHR) gene was also sequenced to determine whether the patient had partial insensitivity to TSH. A novel nucleotide substitution (A>T) in 1 allele of the TSHR gene resulting in the replacement of the normal isoleucine, located in the extracellular domain of the TSHR, with a phenylalanine (I364F) was demonstrated. The patient and 4 of 7 family members had the same nucleotide substitution (Figure 2). However, the phenotype did not cosegregate with the genotype.
Figure 2.
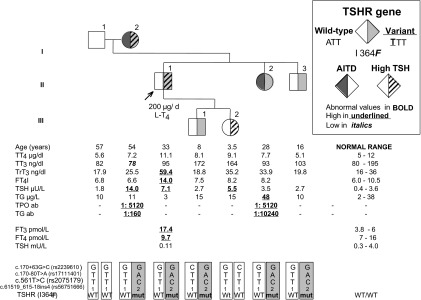
Pedigree of case 4 (II-1), including the thyroid function tests and thyroid antibody levels as well as the TSHR mutation profiles. Abnormal values are represented in bold, high values are underlined, and low values are in italics. AITD, autoimmune thyroid disease; WT, wild type; mut, mutant; TT4, total T4; TT3, total T3; TrT3, total rT3.
l-T4 treatment was gradually increased to 250 μg/d. After 12 months, his serum TSH level was 3.4 mU/L, whereas the fT4 level was slightly increased at 20.17 pmol/L, with a fT3 level of 5.21 pmol/L and within normal range. Tg antibodies remained undetectable, and after stimulation with recombinant human TSH, Tg level was 0.29 ng/ml. Cervical US examination revealed no pathological lymph nodes.
Studies were approved by the Institutional Review Board, and written consents were obtained from all subjects studied and guardians of minors.
Thyroid function tests
The quantitative determination of serum Tg, Tg antibodies, fT3, fT4, TSH, and TPO antibodies was performed by chemiluminescent immunometric assays, employing the Unicell DxI-800 Access Immunoassay Systems (Beckman Coulter GmBH, Krefeld, Germany), according to the manufacturer's instructions. The analytical sensitivities of TSH and Tg measurements were 0.003 mU/L and 0.1 ng/mL, respectively.
In Chicago, total T4, total T3, and TSH were measured by chemiluminescence immunometric assays using the Elecsys Automated System (Roche Molecular Biochemicals GmbH and Hitachi, Ltd, Indianapolis, Indiana). Total rT3 was measured by RIA (Adaltis Italia S.p.A, Bologna, Italy), and Tg was measured by an in-house RIA. Serum fT4 and fT3 indexes were calculated as the product of their total serum concentrations of each iodothyronine and the normalized resin T4 uptake ratio. Antibodies to Tg and TPO were measured by passive hemagglutination (Fujirebio, Inc, Tokyo, Japan).
DNA isolation and TRβ sequencing
DNA was isolated from peripheral blood leukocytes using QIAamp DNA Mini Kit (QIAGEN, Valencia, California) following the manufacturer's protocol. Exons and adjacent intronic TRß gene sequences were amplified by the PCR and sequenced using automated fluorescence-based sequencing (3730XL 96-capillary; Applied Biosystems, Carlsbad, California). The primers used for amplification and sequencing are available upon request.
Discussion
Herein, we describe 4 unusual cases of PTC in coexistence with TSHoma, RTH, and hyperthyrotropinemia of unknown etiology. To our knowledge, there are only a few reports of TSHomas or RTH along with DTC (reviewed in Tables 1 and 2).
Case 1 was exposed to high TSH levels for at least 10 years after the diagnosis of TSHoma because she refused treatment, which could have been the cause of the PTC development. Previous case reports have also mentioned the contribution of TSHoma or high TSH levels to the development of DTC (16–18). However, none of the previously reported cases had such a long delay between the initial diagnosis of TSHoma and the diagnosis of DTC (Table 1). Case 2 is another example of long-term exposure to high TSH, with TSHoma undiagnosed for at least 2 years. A recent dose-response meta-analysis of 22 studies also demonstrated a positive association between TSH levels and thyroid cancer diagnosis (27).
Although there are several treatment options for TSHoma, surgical resection is the recommended first-line therapy (4). Most TSHomas are macroadenomas, probably due to delayed diagnosis, and total removal of a macro-TSHoma is difficult in most cases (2, 4, 28, 29). Thus, in the case of a TSHoma, the thyroid parenchyma is often exposed to high levels of TSH, which could contribute to thyroid oncogenesis. In addition, because of the low rate of TSHoma eradication, the control of DTC with a coexisting TSHoma by TSH suppression could be rather difficult. Thus, in DTC patients with coexisting TSHoma, all treatment options targeting hypophysis, such as pituitary surgery, the administration of somatostatin analogs, and radiotherapy, are recommended in order to maintain serum TSH levels as low as possible (17). We also suggest that a patient with TSHoma should be periodically screened for thyroid nodules, and if DTC is detected by FNAC, a total thyroidectomy and RAI ablation should be performed. Preoperative imaging or inspection during surgery is not helpful to detect central lymph node metastasis in many cases, and central lymph node dissection can be applied with low morbidity in experienced hands (30). Therefore, central lymph node dissection (ipsilateral or bilateral) should be added to surgical treatment of DTC associated with an incurable TSHoma case. This treatment may reduce the risks for locoregional recurrence and mortality (30, 31), although the long-term prognosis of this rare association is not well known (16–22). In case of a small (<10 mm), noninvasive, apparently node-negative tumor, total thyroidectomy with close intraoperative inspection of the central compartment would be a safer surgical approach in less experienced surgical hands, although this approach may increase the risk of future locoregional recurrence (30).
Another challenging issue in patients with DTC and TSHoma is to determine the optimal order of the surgical tumor treatment (16–22). If total thyroidectomy or other ablative treatment strategies are performed first, the growth rate of the TSHoma might, at least theoretically, be affected due to diminished negative feedback effect of thyroid hormones, and resection of TSHoma might become more complicated (4, 32). Thus, pituitary adenomectomy could be planned and performed first, when possible. This strategy could also help to reduce thyroid vascularization and control thyrotoxicosis for an ideal thyroid surgery. If central hypothyroidism develops after pituitary surgery, recombinant human TSH can be used effectively to increase radioactive iodine uptake after total thyroidectomy (33). The review of the literature reveals that in these cases, thyroidectomy was commonly selected as the first treatment of choice because of refusal of pituitary surgery or the delayed diagnosis of the pituitary tumor (Table 1). After application of all treatment options targeting hypophysis in incurable cases of TSHoma, when multinodular goiter with an uncontrolled thyrotoxicosis is present, a total thyroidectomy may be considered and discussed with the patient, instead of radioiodine ablation (34), due to the increased risk of developing thyroid cancer.
Patients in cases 3 and 4 were also exposed to high levels of TSH due to thyroid hormone receptor mutation and an unknown cause, respectively. In case 4, a novel nucleotide substitution in 1 allele of the TSHR gene, which affected 5 of 7 family members, was demonstrated. The resulting substitution of an isoleucine to a phenylalanine was found to be located in the “inserted” sequence of the hinge region, showing no homology with either LH or FSH receptors (35). Moreover, this inserted segment could be deleted from the TSHR with little or no functional consequences, and it is not very well conserved even in mammals. This incidental finding did not correlate with high serum TSH or any of the thyroid test abnormalities in the subject.
Suzuki et al (36) reported a mouse model with mutant TRß gene that resulted in high levels of circulating TSH. In this model, spontaneous thyroid cancers occurred only in homozygotes but not in heterozygotes, a rare occurrence in humans (37). The TSH levels in the common, heterozygous form of RTH are mildly elevated and compensative due to the reduced sensitivity of thyroid hormones (3). In fact, only a few of the approximately 3000 known cases with RTH were found to have DTC. Thus, the likelihood of a precancerous role of high TSH in RTH is less likely. In fact, the 2 children of case 3, carrying the same TRß gene mutation, had no thyroid nodules on US. Furthermore, the diameters of thyroid tumors in patients with TSHoma were larger than those of patients with RTH (Table 1 and 2).
The suppression of TSH is also challenging when DTC coexists with RTH. Increasing the dose of l-T4 can result in thyrotoxicosis without TSH suppression. Therefore, the lowest possible TSH level that does not lead to excessive thyroid hormone effect should be targeted by watching for tachycardia. Alternatively, the TSH levels in these patients could be kept at levels lower than the mean TSH level of the affected family members (38). Another approach to treatment could be to give more thyroid hormone together with a β-blocker to reduce the tachycardia. However, β-blockers do not prevent accelerated osteoporosis, and therefore follow-up with bone mineral density studies and treatment with calcium and vitamin D should be added to this recommendation. There are also data about the usage of thyroid hormone analogs, such as 3,3,5 triiodothyroacetic acid and dextrothyroxine to suppress TSH in RTH (37, 39). It was demonstrated that 3,3,5 triiodothyroacetic acid exerts thyromimetic effects predominantly on pituitary and on hepatic tissue (39–41). Thus, these alternatives may also be tried to suppress TSH, without causing as much peripheral tissue effect in RTH with DTC.
In conclusion, the outcomes in the reported cases were favorable despite the persistence of nonsuppressed TSH levels. Nevertheless, persistently high TSH secretion might be 1 of the risk factors for the development of DTC, especially in cases with TSHoma. Although an agreed consensus regarding the management of DTC in the presence of persistent hyperthyrotropinemia is not yet present, effective treatment options such as the complete removal of the tumor followed by radioablation and attempts to reduce serum TSH to the lowest tolerable level are recommended to prevent the tumor recurrence elicited by persistently high TSH.
Acknowledgments
The authors thank Dr Sacit Altuğ Kesikli for his assistance.
This work was supported in part by Grants DK15070 and RR04999 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health.
Disclosure Summary: S.R. is an academic associate for Quest Diagnostics. All other authors have no conflicts of interest.
Footnotes
- DTC
- differentiated thyroid cancer
- FNAC
- fine-needle aspiration cytology
- fT3
- free T3
- fT4
- free T4
- l-T4
- levothyroxine
- MRI
- magnetic resonance imaging
- N
- normal range
- PTC
- papillary thyroid carcinoma
- RTH
- resistance to thyroid hormone
- Tg
- thyroglobulin
- TPO
- thyroperoxidase
- TRβ
- thyroid hormone receptor β
- TSHoma
- TSH-secreting adenoma
- TSHR
- TSH receptor
- US
- ultrasound.
References
- 1. Faglia G, Beck-Peccoz P, Piscitelli G, Medri G. Inappropriate secretion of thyrotropin by the pituitary. Horm Res. 1987;26:79–99 [DOI] [PubMed] [Google Scholar]
- 2. Beck-Peccoz P, Brucker-Davis F, Persani L, Smallridge RC, Weintraub BD. Thyrotropin-secreting pituitary tumors. Endocr Rev. 1996;17:610–638 [DOI] [PubMed] [Google Scholar]
- 3. Dumitrescu AM, Refetoff S. The syndromes of reduced sensitivity to thyroid hormone [published online August 16, 2012]. Biochim Biophys Acta. doi.org/10.1016/j.bbagen.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beck-Peccoz P, Persani L. Thyrotropinomas. Endocrinol Metab Clin North Am. 2008;37:123–134, viii-ix [DOI] [PubMed] [Google Scholar]
- 5. Tuttle RM, Leboeuf R, Martorella AJ. Papillary thyroid cancer: monitoring and therapy. Endocrinol Metab Clin North Am. 2007;36:753–778, vii [DOI] [PubMed] [Google Scholar]
- 6. Hovens GC, Stokkel MP, Kievit J, et al. Associations of serum thyrotropin concentrations with recurrence and death in differentiated thyroid cancer. J Clin Endocrinol Metab. 2007;92:2610–2615 [DOI] [PubMed] [Google Scholar]
- 7. Crile G., Jr Endocrine dependency of papillary carcinomas of the thyroid. JAMA. 1966;195:721–724 [PubMed] [Google Scholar]
- 8. Ichikawa Y, Saito E, Abe Y, Homma M, Muraki T. Presence of TSH receptor in thyroid neoplasms. J Clin Endocrinol Metab. 1976;42:395–398 [DOI] [PubMed] [Google Scholar]
- 9. Goretzki PE, Frilling A, Simon D, Roeher HD. Growth regulation of normal thyroids and thyroid tumors in man. Recent Results Cancer Res. 1990;118:48–63 [DOI] [PubMed] [Google Scholar]
- 10. Mazzaferri EL, Jhiang SM. Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am J Med. 1994;97:418–428 [DOI] [PubMed] [Google Scholar]
- 11. Pujol P, Daures JP, Nsakala N, Baldet L, Bringer J, Jaffiol C. Degree of thyrotropin suppression as a prognostic determinant in differentiated thyroid cancer. J Clin Endocrinol Metab. 1996;81:4318–4323 [DOI] [PubMed] [Google Scholar]
- 12. Cooper DS, Specker B, Ho M, et al. Thyrotropin suppression and disease progression in patients with differentiated thyroid cancer: results from the National Thyroid Cancer Treatment Cooperative Registry. Thyroid. 1998;8:737–744 [DOI] [PubMed] [Google Scholar]
- 13. Haugen BR, Pacini F, Reiners C, et al. A comparison of recombinant human thyrotropin and thyroid hormone withdrawal for the detection of thyroid remnant or cancer. J Clin Endocrinol Metab. 1999;84:3877–3885 [DOI] [PubMed] [Google Scholar]
- 14. Biondi B, Filetti S, Schlumberger M. Thyroid-hormone therapy and thyroid cancer: a reassessment. Nat Clin Pract Endocrinol Metab. 2005;1:32–40 [DOI] [PubMed] [Google Scholar]
- 15. Jonklaas J, Sarlis NJ, Litofsky D, et al. Outcomes of patients with differentiated thyroid carcinoma following initial therapy. Thyroid. 2006;16:1229–1242 [DOI] [PubMed] [Google Scholar]
- 16. Calle-Pascual AL, Yuste E, Martin P, et al. Association of a thyrotropin-secreting pituitary adenoma and a thyroid follicular carcinoma. J Endocrinol Invest. 1991;14:499–502 [DOI] [PubMed] [Google Scholar]
- 17. Gasparoni P, Rubello D, Persani L, Beck-Peccoz P. Unusual association between a thyrotropin-secreting pituitary adenoma and a papillary thyroid carcinoma. Thyroid. 1998;8:181–183 [DOI] [PubMed] [Google Scholar]
- 18. Kishida M, Otsuka F, Kataoka H, et al. Hyperthyroidism in a patient with TSH-producing pituitary adenoma coexisting with thyroid papillary adenocarcinoma. Endocr J. 2000;47:731–738 [DOI] [PubMed] [Google Scholar]
- 19. Ohta S, Nishizawa S, Oki Y, Namba H. Coexistence of thyrotropin-producing pituitary adenoma with papillary adenocarcinoma of the thyroid—a case report and surgical strategy. Pituitary. 2001;4:271–274 [DOI] [PubMed] [Google Scholar]
- 20. Gessl A, Vierhapper H, Feichtinger H. Non-suppressible TSH in a patient thyroidectomized due to follicular thyroid carcinoma. Exp Clin Endocrinol Diabetes. 2006;114:389–392 [DOI] [PubMed] [Google Scholar]
- 21. Poggi M, Monti S, Pascucci C, Toscano V. A rare case of follicular thyroid carcinoma in a patient with thyrotropin-secreting pituitary adenoma. Am J Med Sci. 2009;337:462–465 [DOI] [PubMed] [Google Scholar]
- 22. Nguyen HD, Galitz MS, Mai VQ, Clyde PW, Glister BC, Shakir MK. Management of coexisting thyrotropin/growth-hormone-secreting pituitary adenoma and papillary thyroid carcinoma: a therapeutic challenge. Thyroid. 2010;20:99–103 [DOI] [PubMed] [Google Scholar]
- 23. Taniyama M, Ishikawa N, Momotani N, Ito K, Ban Y. Toxic multinodular goitre in a patient with generalized resistance to thyroid hormone who harbours the R429Q mutation in the thyroid hormone receptor β gene. Clin Endocrinol (Oxf). 2001;54:121–124 [DOI] [PubMed] [Google Scholar]
- 24. Kim HK, Kim D, Yoo EH, et al. A case of resistance to thyroid hormone with thyroid cancer. J Korean Med Sci. 2010;25:1368–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paragliola RM, Lovicu RM, Locantore P, et al. Differentiated thyroid cancer in two patients with resistance to thyroid hormone. Thyroid. 2011;21:793–797 [DOI] [PubMed] [Google Scholar]
- 26. Mannavola D, Vannucchi G, Fugazzola L, Cerutti N, Persani L, Beck-Peccoz P. Genetic analyses and evaluation of peripheral parameters of thyroid hormone action for the differential diagnosis of RTH. A novel heterozygous missense mutation (M334T) discovered. J Endocrinol Invest. 2002;25:RC4–RC6 [DOI] [PubMed] [Google Scholar]
- 27. McLeod DS, Watters KF, Carpenter AD, Ladenson PW, Cooper DS, Ding EL. Thyrotropin and thyroid cancer diagnosis: a systematic review and dose-response meta-analysis. J Clin Endocrinol Metab. 2012;97:2682–2692 [DOI] [PubMed] [Google Scholar]
- 28. Brucker-Davis F, Oldfield EH, Skarulis MC, Doppman JL, Weintraub BD. Thyrotropin-secreting pituitary tumors: diagnostic criteria, thyroid hormone sensitivity, and treatment outcome in 25 patients followed at the National Institutes of Health. J Clin Endocrinol Metab. 1999;84:476–486 [DOI] [PubMed] [Google Scholar]
- 29. Sanno N, Teramoto A, Osamura RY. Long-term surgical outcome in 16 patients with thyrotropin pituitary adenoma. J Neurosurg. 2000;93:194–200 [DOI] [PubMed] [Google Scholar]
- 30. Cooper DS, Doherty GM, Haugen BR, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19:1167–1214 [DOI] [PubMed] [Google Scholar]
- 31. Mazzaferri EL, Kloos RT. Clinical review 128: current approaches to primary therapy for papillary and follicular thyroid cancer. J Clin Endocrinol Metab. 2001;86:1447–1463 [DOI] [PubMed] [Google Scholar]
- 32. Beck-Peccoz P, Persani L. Medical management of thyrotropin-secreting pituitary adenomas. Pituitary. 2002;5:83–88 [DOI] [PubMed] [Google Scholar]
- 33. Pacini F, Castagna MG. Diagnostic and therapeutic use of recombinant human TSH (rhTSH) in differentiated thyroid cancer. Best Pract Res Clin Endocrinol Metab. 2008;22:1009–1021 [DOI] [PubMed] [Google Scholar]
- 34. Daousi C, Foy PM, MacFarlane IA. Ablative thyroid treatment for thyrotoxicosis due to thyrotropin-producing pituitary tumours. J Neurol Neurosurg Psychiatry. 2007;78:93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jaeschke H, Schaarschmidt J, Gunther R, Mueller S. The hinge region of the TSH receptor stabilizes ligand binding and determines different signaling profiles of human and bovine TSH. Endocrinology. 2011;152:3986–3996 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor β gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969 [DOI] [PubMed] [Google Scholar]
- 37. Ferrara AM, Onigata K, Ercan O, Woodhead H, Weiss RE, Refetoff S. Homozygous thyroid hormone receptor β-gene mutations in resistance to thyroid hormone: three new cases and review of the literature. J Clin Endocrinol Metab. 2012;97:1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tran HA. Difficulties in diagnosing and managing coexisting primary hypothyroidism and resistance to thyroid hormone. Endocr Pract. 2006;12:288–293 [DOI] [PubMed] [Google Scholar]
- 39. Agrawal NK, Goyal R, Rastogi A, Naik D, Singh SK. Thyroid hormone resistance. Postgrad Med J. 2008;84:473–477 [DOI] [PubMed] [Google Scholar]
- 40. Beck-Peccoz P, Piscitelli G, Cattaneo MG, Faglia G. Successful treatment of hyperthyroidism due to nonneoplastic pituitary TSH hypersecretion with 3,5,3′-triiodothyroacetic acid (TRIAC). J Endocrinol Invest. 1983;6:217–223 [DOI] [PubMed] [Google Scholar]
- 41. Radetti G, Persani L, Molinaro G, et al. Clinical and hormonal outcome after two years of triiodothyroacetic acid treatment in a child with thyroid hormone resistance. Thyroid. 1997;7:775–778 [DOI] [PubMed] [Google Scholar]