

Abstract
Encephalocraniocutaneous lipomatosis (ECCL) is a rare sporadic neurocutaneous syndrome characterized by presence of central nervous system, ocular and cutaneous anomalies. The exact pathogenesis is still not known. We present the third case from the Indian subcontinent, who is a five year old girl with history of right sided seizures. Dermatological examination showed alopecia on right side of the scalp and ipsilateral limbal dermoid and nodular skin tags over the upper eyelid. The computerized tomography scan of the brain revealed porencephalic cyst, cerebral calcifications and atrophy of right brain. The histopathology of the skin lesions showed lipomatous hamartoma and features of non scarring alopecia. The constellation of these findings and in adherence to the diagnostic criteria of ECCL proposed in 2009, we consider this report as a definite case of ECCL.
Keywords: Brain anomalies, cutaneous lesions, Encephalocraniocutaneous lipomatosis
Introduction
What was known?
ECCL is a rare neurocutaneous syndrome of unknown etiopathogenesis that has an overlap with other neurocutaneous syndromes such as proteus and oculocerebrocutaneous syndromes.
Encephalocraniocutaneous lipomatosis (ECCL) or Haberland syndrome was first described by Haberland and Perou in 1970.[1] It is also referred as Fishman's syndrome.[2] ECCL is a rare syndrome that is largely characterized by unilateral cutaneous lesions like lipomas, connective tissue nevi and alopecia; in association with ipsilateral ophthalmologic and neurological malformations.[1,2] Since its first description in 1970, not more than 54 cases have been reported, and to the best of our knowledge this is the third case from the Indian subcontinent with complete evaluation.[3–6]
Case Report
A five year old girl born at term to non consanguineous parents after an uncomplicated delivery was brought by her parents with a complaint of right sided seizures since four years. On examination the child had frontal and parietal bony prominences on the right side along with a 13 × 6 cm smooth, soft, flat area of alopecia on the scalp. Soft nodular skin lesions were seen on right upper lid and periorbital area. Reddish nodular lesion at limbus encroaching onto cornea suggestive of limbal Dermoid, reddish lesion on bulbar conjunctiva suggestive of epibulbar Dermoid and coloboma of right upper lid were also noted [Figure 1]. Fundus examination showed myopic fundus on right side. Cardiovascular and respiratory system examinations were normal. Higher intellectual functions were normal on Central nervous system examination.
Figure 1.

Clinical photograph showing right frontal bony prominence, ipsilateral band of alopecia on the scalp and nodular skin lesions on upper eyelid
A plain non contrast computerized tomography (CT) scan and MRI of the brain showed dilatation of right ventricle, cortical dysplasia over right frontal lobe, a porencephalic cyst, cortical gyriform calcification of the right parietal lobe and occipital lobes [Figure 2a and b]. The histopathology examination of the alopecic area of the scalp showed absence of hair follicles, focal dermal fibrosis, and increased amounts of subcutaneous fat extending into the upper reticular dermis [Figure 3]. The nodular lesion on the eyelid submitted for histopathological examination showed a hamartoma made of disorganized elements of fibrous and adipose tissue [Figure 4].
Figure 2.
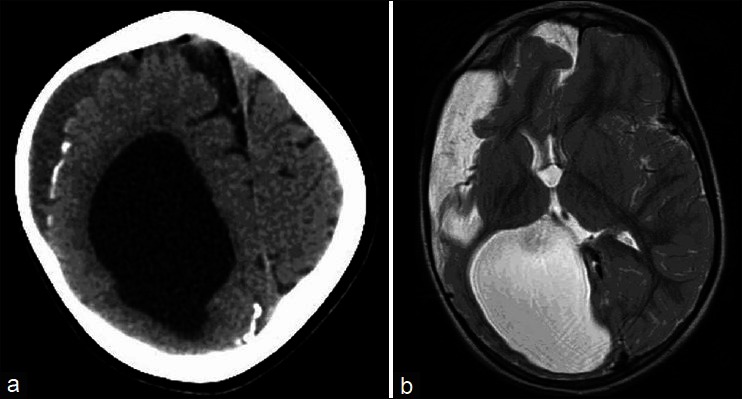
Non contrast computerized tomography Photograph of the brain showing (a) gyriform cortical calcifications over right parietal and frontal lobes along with dilatation of right lateral ventricle, (b) MRI of brain – T2 Weighed axial images at the level of ventricles showing dilatation of right lateral ventricle, arachanoid cyst and a small porencephalic cyst
Figure 3.
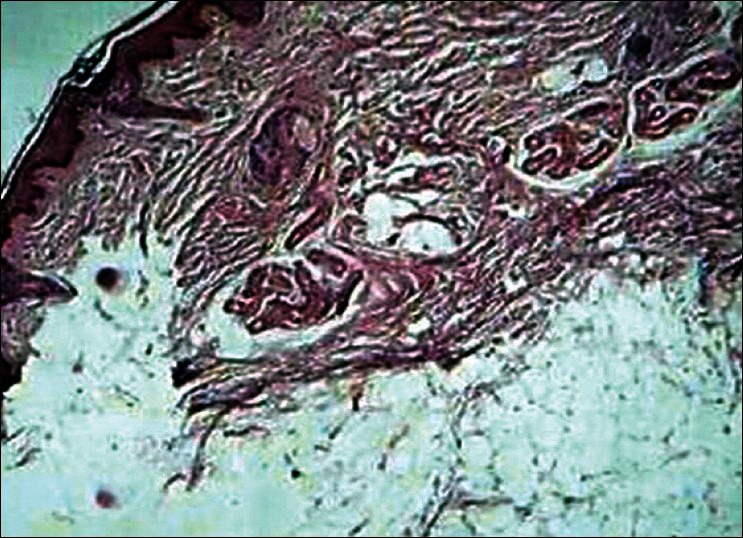
Photomicrograph from the scalp lesion showing increased amount of subcutaneous fat extending into upper reticular dermis suggestive of nevus psiloliparus (H and E, ×100)
Figure 4.
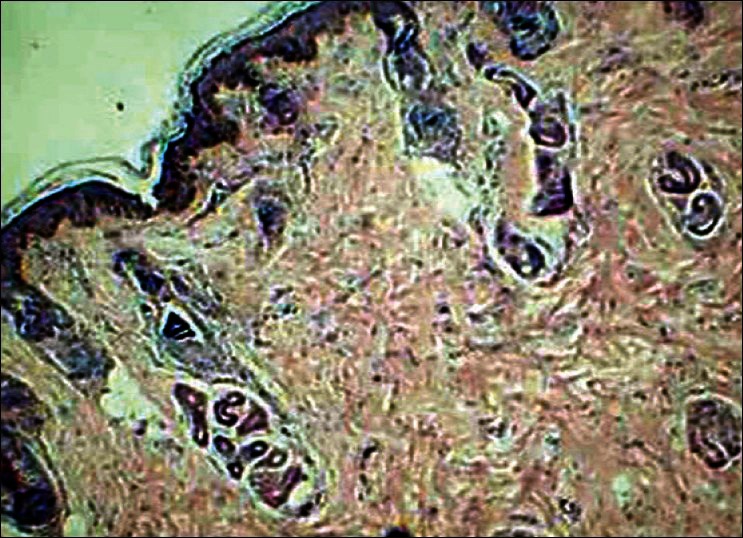
Photomicrograph from the eylelid lesion showing features of a hamartoma made of disorganized elements of fibrous and adipose tissue (H and E, ×100)
Discussion
ECCL is a rare clinical syndrome of unknown pathogenesis and the exact embryological defect responsible is yet to be determined. However certain mechanisms were hypothesized, such as ectodermal dysgenesis and mosaicism for a mutated autosomal gene involved in mesenchymal tumors and vasculogenesis with or without a second hit event.[1,4] The tissues affected in ECCL are neural crest derivatives, dermis and hypodermis of face and neck, head mesenchyme, dermal bones of the skull, truncoconal septum, odontoblasts and melanocytes. The eye anomalies (mainly epibulbar or limbar dermoids) and skin lesions (non scarring alopecia, nevus psiloliparis, subcutaneous fatty masses and skin tags) may be unilateral or bilateral and occur in a consistent pattern. About two-thirds of patients have a normal development or mild mental retardation and half have seizures.[4] Our patient had cutaneous lesions restricted to right side of the body and had normal development with right sided seizures.
Porencephalic cyst was the most frequently reported anomaly on radiological examination of the central nervous system.[6] The other abnormalities reported were enlargement of the lateral ventricle, arachnoid cyst, widening of the subarachnoid spaces, a lack of normal insular opercularization, a dysplastic cortex, and corticopial calcifications, thinning of the corpus callosum, intracranial lipoma and leptomeningeal angiomatosis.[7] Majority of which was seen in our case. In a comprehensive review of 52 patients with ECCL, 33 (63.4%) patients had intracranial lipoma.[8] Our case did not show either intracranial or intraspinal lipoma.
Histopathological examinations of the skin colored papules/nodular tags represent lipomas, fibromas, fibrolipomas, and connective tissue nevi or hamartomatous tissue consisting of cartilage, fat and connective tissue. The alopecic area of the scalp usually show focal dermal fibrosis associated with increased amounts of subcutaneous fat extending into the reticular dermis.[3,4] Similar changes were appreciated on the histopathological examination of skin lesions in our case. The histological examination of the brain in the case reported by Haberland and Perou showed a defective lamination of the cerebrum, polymicrogyria and calcification in the outer cerebral cortex overlying the porencephalic cyst.[1]
Clinically the main differential diagnosis includes sebaceous nevus syndrome, proteus syndrome and oculocerebrocutaneous (OCC) syndrome. Patients with sebaceous nevus syndrome are also associated with seizures and mental retardation, but the cutaneous lesions are mainly on the midline of the face. The facial midline was preserved in our case. The Proteus syndrome is progressive, bilateral, and asymmetric and involves the head, trunk and limbs. The brain alterations are also rare. ECCL is non progressive, unilateral and limited to the head. However some of the authors consider sebaceous nevus syndrome as a continuum of phenotypic expression of ECCL, and ECCL as a localized form of Proteus syndrome.[9] Absence of facial lipomas and scalp alopecia in OCC syndrome helps in differentiating it from ECCL.
The neuroradiological features of ECCL also overlap with certain conditions like Sturge-Weber syndrome (SWD) and unilateral macrencephaly. In SWD the mesodermal dysgenesis is limited to blood vessels, whereas in ECCL the fat tissue is primarily involved. In SWD the cortical calcifications are mainly in the occipito parietal areas, whereas in ECCL they are localized throughout the cortex. The affected hemisphere is clearly atrophic and only the lateral ventricle appears enlarged in ECCL, whereas in unilateral macrencephaly there is an overgrowth of one hemisphere. Neither intracranial lipomas nor arachnoids cysts occur in SWD or in unilateral macrencephaly.[7] In 2009, Moog proposed revised diagnostic criteria for ECCL. [4] In accord with that criteria and constellation of clinical, radiological, histopathological examination findings we diagnosed our case as ECCL. There is no specific treatment for ECCL. The management is usually symptomatic and surgical correction of cutaneous lesions is for cosmetic improvement.[6] Antenatal diagnosis is not usually made because the intracranial malformations noted on an antenatal sonogram are not specific for ECCL.[10]
Conclusion
ECCL is a very rare neurocutaneous syndrome. The clinical and radiological features of ECCL overlap with a number of other mosaic neurocutaneous clinical syndromes and can make a diagnosis of ECCL challenging. A multidisciplinary evaluation helps in diagnosing these rare cases of ECCL.
What is new?
The present case is the third case from Indian Subcontinent. A good account of clinical, radiological and histopathological features of ECCL was made. A thorough discussion of ECCL in comparison with other overlap neurocutaneous syndromes was made to have a better understanding of the disorder in adherence to the revised diagnostic criteria of ECCL.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Haberland C, Perou M. Encephalocraniocutaneous lipomatosis: A new example of ectomesodermal dysgenesis. Arch Neurol. 1970;22:144–55. doi: 10.1001/archneur.1970.00480200050005. [DOI] [PubMed] [Google Scholar]
- 2.Fishman MA, Chang CS, Miller JE. Encephalocraniocutaneous lipomatosis. Pediatrics. 1978;61:580–2. [PubMed] [Google Scholar]
- 3.Koishi GN, Yoshida M, Alonso N, Matushita H, Goldenberg D. Encephalocraniocutaneous lipomatosis (Haberland's syndrome): A case report of a neurocutaneous syndrome and a review of the literature. Clinics. 2008;63:406–8. doi: 10.1590/S1807-59322008000300020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet. 2009;46:721–9. doi: 10.1136/jmg.2009.066068. [DOI] [PubMed] [Google Scholar]
- 5.Rathoriya R, Shrivastava J. Encephalocraniocutaneous lipomatosis. Indian Pediatr. 2006;43:262–3. [PubMed] [Google Scholar]
- 6.Gokhale NR, Mahajan PM, Belgaumkar VA, Pradhan SN, Uttarwar NS. Encephalocraniocutaneous lipomatosis: A rare neurocutaneous syndrome. Indian J Dermatol Venereol Leprol. 2007;73:40–2. doi: 10.4103/0378-6323.30651. [DOI] [PubMed] [Google Scholar]
- 7.Parazzini C, Triulzi F, Russo G, Mastrangelo M, Scotti G. Encephalocraniocutaneous lipomatosis: Complete neuroradiologic evaluation and follow-up of two cases. Am J Neuroradiol. 1999;20:173–6. [PubMed] [Google Scholar]
- 8.Moog U, Jones MC, Viskochil DH, Verloes A, Van Allen MI, Dobyns WB. Brain anomalies in Encephalocraniocutaneous lipomatosis. Am J Med Genet Part A. 2007;143:2963–72. doi: 10.1002/ajmg.a.32074. [DOI] [PubMed] [Google Scholar]
- 9.Kokitsu-Nakata NM, Vendramini S, Versiani BR, Ramos ES, Guion-Almeida ML. Encephalocraniocutaneous Lipomatosis: Report on a Brazilian girl. Arq Cienc Saude. 2004;11:192–4. [Google Scholar]
- 10.Nowaczyk MJ, Mernagh JR, Bourgeois JM, Thompson PJ, Jurriaans E. Antenatal and postnatal findings in Encephalocraniocutaneous lipomatosis. Am J Med Genet. 2000;91:261–6. [PubMed] [Google Scholar]