

Summary
Accumulating preclinical and clinical evidence suggests the possibility of neurotoxicity from neonatal exposure to general anaesthetics. Here, we review the weight of the evidence from both human and animal studies and discuss the putative mechanisms of injury and options for protective strategies. Our review identified 55 rodent studies, seven primate studies, and nine clinical studies of interest. While the preclinical data consistently demonstrate robust apoptosis in the nervous system after anaesthetic exposure, only a few studies have performed cognitive follow-up. Nonetheless, the emerging evidence that the primate brain is vulnerable to anaesthetic-induced apoptosis is of concern. The impact of surgery on anaesthetic-induced brain injury has not been adequately addressed yet. The clinical data, comprising largely retrospective cohort database analyses, are inconclusive, in part due to confounding variables inherent in these observational epidemiological approaches. This places even greater emphasis on prospective approaches to this problem, such as the ongoing GAS trial and PANDA study.
Keywords: brain, anaesthesia, molecular effects; nerve, damage (postoperative); nerve, neurotransmitters; nerve, regeneration
Editor's key points.
Robust evidence from neonatal non-primate animal models indicates increased programmed brain cell death after exposure to general anaesthesia.
Emerging evidence from primate models confirms developmentally defined anaesthetic-induced neurotoxicity with associated neurocognitive deficits.
Currently available retrospective clinical data are inconclusive, and the results of prospective studies are pending, making drastic changes to current paediatric anaesthesia practice premature.
In this systematic review, we will update previous reviews of this topic published in the BJA1–3 by describing the current weight of preclinical evidence for anaesthetic-induced neurodegeneration in the neonatal brain, along with a discussion of putative mechanisms of injury and potential adverse effects of surgery on neurodevelopment. Then, we review the clinical literature and ongoing prospective investigations that should shed further light on the potential harm of anaesthesia, surgery, or both in the neonatal period.
Preclinical studies in rodents and primates have shown that anaesthesia is neurotoxic to the developing brain after exposure in the neonatal period.1,4–13 This neurotoxicity manifests as a pathological increase in apoptosis (programmed cell death), although other effects such as impaired neurogenesis and neuroinflammation likely also contribute. These studies randomized healthy neonatal animals to anaesthetic exposure or not; hence, a causal relationship has been established between the anaesthetic and neonatal brain injury. Furthermore, such neurotoxicity has been observed with exposure to drugs with similar mechanisms of action such as anti-epileptic medications14 and ethanol.15 Significant concern has arisen that present medical therapies could expose neonates to neurotoxicity.
Furthermore, in some animal studies, the anaesthetic injury is associated with impaired cognition that persists into adulthood. In primates, 24 h of ketamine exposure leads to apoptosis8 and cognitive impairment up to 3.5 yr after the insult.10 Several rodent studies also support this assertion of prolonged cognitive compromise associated with apoptosis after anaesthetic exposure.1,4,7,11,16 Thus, the brain injury adversely affects neurodevelopment and leads to long-term impairment in cognition, at least in animals.
The difficulties of replicating these studies in humans include the ethical obstacle of being unable to randomize neonates to having an anaesthetic or not; it is unacceptable to perform surgery without any anaesthetic and it is unacceptable to give a neonate anaesthesia for no reason.1,11 Both these options would likely provoke greater harm than current practice.1,11 Therefore, current clinical studies have been limited to an observational cohort design, with the incumbent issues of confounding factors. These accumulating clinical data cannot exclude a clinically important effect of neonatal and paediatric anaesthesia and surgery on cognition in later life.17–26 While these studies have used heterogeneous retrospective approaches (with varying degrees of confounding effects), they provide sufficient preliminary evidence to cause concern in clinicians and parents alike.
Search strategy methods
To provide an update on the weight of preclinical and clinical evidence of harm of anaesthetics and surgery on neurodevelopment, we performed literature searches for preclinical and clinical data. J.H. performed the search for preclinical data using the search terms: an(a)esth*, neuroapoptosis, neuronal cell death, develop* brain, and neurodevelop*. The results included in vivo studies identified in Tables 1 (rodents) and 2 (primates). Two piglet studies were identified but were not tabulated. This search was designed to highlight the scope of the preclinical literature pertaining to anaesthetic-induced neurodegeneration in the developing brain. This search revealed an initial 4379 titles that were reviewed for relevance. Fifty-five rodent, two piglet, and seven primate studies were identified. The search for clinical data was conducted by R.D.S. using the search terms: cognitive or behavioral disorder and anaesth* with the following limits: published in the last 10 yr, humans, English, infant: birth—23 months. To be included in the systematic review, some attempt to compare an anaesthetic exposed group with a non-exposed control group was required. However, we did include studies that used population controls, even if exposure status was not defined in this population. We only included studies of anaesthesia, not intensive care sedation, and only included studies from non-cardiac non-neurological surgery in term births. Our search revealed 3727 hits and the titles were then searched for relevant studies. The reference list of relevant studies and recent reviews were also hand searched for additional studies. Our formal search results are displayed in Table 3.
Table 1.
Rodent studies into the neurotoxic effects of anaesthetic agents. PND, post-natal day. Brain area codes: Am, amygdala; Th, thalamus (unspecified); ATh, thalamus anterior; VTh, thalamus ventral; ADTh, thalamus anterodorsal; LDTh, thalamus laterodorsal; AVTh, thalamus anteroventral; AMTh, thalamus anteromedial; Caud, caudate; Put, putamen; GP, globus pallidus; SN, substantia nigra; BF, basal forebrain; Cor, cerebral cortex (unspecified); RSC, retrosplenial cortex; CingC, cingulate cortex; PFC, prefrontal cortex; FroC, frontal cortex; ParC, parietal cortex; TemC, temporal cortex; OccC, occipital cortex; SomC, somasosensory cortex; PirC, piriform cortex; Hc, hippocampus; Sub, subiculum; Hyp, hypothalamus; StriaT, stria terminalis; Olf, olfactory bulb; SC, spinal cord. Studies identifying neuroprotective agents are identified in italics
| Reference | Species | Age | Anaesthetic | Duration (h) | Histology outcome | Histology results | Brain area | Neurodevelopmental outcome | Neurodevelopmental result | Other outcomes/comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Saito and colleagues, Anesthesiology 1993; 79: 1338–47 | Rats | PND1 | N2O 25–57% OR Halothane 0.5–1% in  25% 25% |
6 h then killed PND 2–5 | N/A | N/A | Whole | N/A | N/A | Whole brain homogenized, neuronal tip growth cone particles extracted by fractionation, polyacramide gel electrophoresis for 46 and 80 kDa proteins and protein kinase C activity assay—decreased protein-kinase C-mediated phosphorylation of 46 and 80 kDa proteins with increasing anaesthetic dose |
| Ikonomidou and colleagues, Science, 1999; 283: 70–4 | Rats | PND 0, 3, 7, 14, 21 | Dizocilpine 0.05–1 mg kg−1, single injection at 0, 8, or 16 h | Killed after 4, 8, 12, 16, 24, or 48 h |
|
Dose-dependent increased apoptosis throughout maximal in LDTh, FroC, ParC, CingC. More apoptosis seen after longer time elapsed post-dose. Peak vulnerability on PND 7. Regional variation in degree of apoptosis at different ages | Hc, Sub, LDTh, MDTh, VTh, FroC, ParC, RSC, CingC | N/A | N/A | Dizocilpine (MK801)=NMDA-R antagonist. Maximally affected on PND 7=(ascending order) FroC, Cau, Sub, RSC, ParC, CingC, LDTh |
| Thompson and colleagues, Brain Res Dev 2001; 24: 167–71 | Rats Sprague–Dawley | PND 6, 14, 21, 28, 56 | Urethane 1.25 g kg−1 single dose | Killed after 24, 24 or 96 h | Irreversible neuronal injury assessed by fluorescent LM after H&E staining | Age-dependent neuronal injury, max after urethane on PND 18–21 | PirC | N/A | N/A | |
| Jevtovic-Todorovic and colleagues, J Neurosci 2003; 23: 876–82 | Rats Sprague–Dawley | PND 7 | Midazolam 3–9 mg kg−1, N2O 50–150%, isoflurane 0.75–1.5%, alone, or in combination | 6 h |
|
Dose-dependent apoptotic degeneration with triple cocktail, less marked with isoflurane alone. No significant increase in apoptosis with midazolam or N2O alone. Most vulnerable=thalamus | All areas—see paper for results |
|
Triple anaesthetic induced long-term impairment in spatial/learning memory | Anaesthesia did not significantly affect arterial blood gases |
| Dziekto and colleagues, Neurobiol Dis 2004; 15: 177–87† | Rats Wistar | PND 7 | NMDA receptor antagonist MK801 0.5 μg kg−1 at 0, 8, and 16 h±rEPO 5–30 000 IU kg−1 at 0 h. Subgroup pretreated—16 h | Killed at 24 h from first dose MK801 |
|
Increased apoptosis with MK801. Apoptosis reduced with rEPO 5000 IU kg−1 co-treatment. No significant increase in protection with pre-treatment or increasing doses | Cor, Th, Sub | N/A | N/A | †Neuroprotection study. rEPO conferred 50% neuroprotection and partially restored MK801-enhanced neurotrophin-associated signalling pathways. RT-PCR for BDNF–MK801 reduced BDNF, partially restored by rEPO. Western blot for ERK1/2 and Akt–MK801 reduced phosphorlyated EKR and Akt, reversed by rEPO |
| Scallet and colleagues, J Chem Neuroanat 2004; 29: 71–80 | Rats Sprague–Dawley | PND 7 | Ketamine 10–20 mg kg−1 every 90 min, total 7 doses | Killed after seventh dose |
|
Increased apoptosis and necrosis with high dose (20 mg kg−1)* | DLTh, Am | N/A | N/A | *20 mg kg−1 produced blood levels 7× greater than anaesthetic blood level in humans. No increased cell death seen at therapeutic levels |
| Young and colleagues, Br J Pharmacol 2005; 146: 189–97 | Mice C57BL6 | PND 7 | Ketamine 10–40 mg kg−1 or midazolam 9 mg kg−1, single dose, alone or combination | Killed after 5 h |
|
Significant dose-dependent increase in apoptosis with ketamine or midazolam alone. Combined administration triggered significantly greater increase in neuroapoptosis | Cor, Cau, Put | N/A | N/A | Anaesthesia did not significantly affect arterial blood gases |
| Yon and colleagues, Neuroscience 2005; 135: 815–27 | Rats Sprague–Dawley | PND 1, 3, 7, 10, 14 | Midazolam 9 mg kg−1, N2O 75%, isoflurane 0.75% alone or combination | 2, 4, or 6 h |
|
Age-dependent anaesthesia-induced apoptosis (max PND 7) with triple anaesthesia. Intrinsic pathway activated at 2 h, extrinsic pathway activated at 6 h, but both remain inactive at PND 14. Thalamus vulnerable earlier, cortex later | OccC, CingC, ParC, RSC, Sub, ADTh, LDTh, AVTh, AMTh, Am | N/A | N/A | Peak vulnerability=PND 7=peak synaptogeneis. Western blot—Cytochrome c (intrinsic apoptotic pathway), Bcl-XL (intrinsic), Fas (extrinsic)—all increased with triple anaesthesia. Cerebral perfusion—laser-Doppler—no decrease in CBF with anaesthesia |
| Yonand colleagues, Neurobiol Dis 2006; 21: 522–39† | Rats | PND 7 | Combination: midazolam 9 mg kg−1, N2O 75%, isoflurane 0.75%± melatonin 1–20 mg kg−1 pre-/3 h into anaesthesia | 6 h |
|
Increased apoptosis in the cortex and thalamus with anaesthesia, counteracted by melatonin in a dose-dependent manner (35–40% reduction at 1 mg kg−1, 75–90% at 20 mg kg−1) | OccC, CingC, ParC, RSC, ADTh, LDTh, AVTh, AMTh | N/A | N/A | †Neuroprotection study. Melatonin causes dose-dependent decrease in severity of anaesthesia-induced apoptosis/neurodegeneration and induces up-regulation of Bcl-XL. Western blot—Cytochrome c, Bcl-XL (intrinsic apoptotic pathway)—results as for histology and mel upregegulated Bcl-XL |
| Lu and colleagues, Apoptosis, 2006; 11: 1603–15† | Rats Sprague–Dawley | PND 7 | Combination: midazolam 9 mg kg−1, N2O 75%, isoflurane 0.75%±β-oestradiol 300 μg kg−1 pre-/8 h post-anaesthesia | 2, 4, or 6 h | Apoptosis: caspase-3, -9, activated ceramide, Trk-B activation | Increased apoptosis via Trk-dependent and independent pathways in the cerebral cortex | Th, CingC, OccC, ParC, RS, Sub | N/A | N/A | †Neuroprotection study. β-oestradiol (known to up-regulate Akt) protects against anaesthesia-induced neuroapoptosis. Western blot—BDNF, p75NTR, Akt protein—results as for histology |
| Fredriksson and colleagues, Anesthesiology 2007; 107: 427–36 | Mice NMRI | PND 10 |
|
Killed 24 h post dose or tested PND 55–70 | Apoptosis: Fluoro-Jade B | Increased apotosis in all groups. Significant in ketamine+propofol/thiopental and high-dose propofol groups | Olf, StriaT | At PND 55–70
|
|
‘Both GABA agonist (thiopental/propofol) and NMDA antagonist (ketamine) potentiated neonatal brain cell death and resulted in functional deficits in adulthood’ |
| Ma and colleagues, Anesthesiology, 2007; 106: 746–53† | Rats Sprague–Dawley | PND 7 | 8 gas mixtures:
|
6 h | Apoptosis: caspase-3 | Isoflurane enhanced neuroapoptosis. Combination with N2O significantly increased apoptosis. Combination with xenon reduced apoptosis to that of controls (statistically greater reduction with 60% than 30% xenon). Neither N2O or xenon alone increased apoptosis | Hc, Cor | †Neuroprotection study. Xenon reduced isoflurane-induced apoptosis to that of controls. Unoblotting for caspase-3, -8, -9, and cytochrome c—no change in caspase-8 suggesting extrinsic apoptotic pathway not activated after 6 h. Capsase-3, -9, and cytochrome c (intrinsic apoptotic pathway) all increased with isoflurane±N2O, ameliorated by xenon | ||
| Nikizad and colleagues, Ann N Y Acad Sci 2007; 1122: 69–82 | Rats Sprague–Dawley | PND 7 | Combination: midazolam 9 mg kg−1, N2O 75%, isoflurane 0.75% | 6 h then killed after 0, 18, 42 h, 3 or 23 days | Neuronal density: Nissl | Decreased cell counts in the thalamus and subiculum by day 3 (PND 10) and in the cortex by day 23 (PND 30) post-anaesthesia | CingC, OccC, ParC, RSC, ADTh, LDTh, AVTh, AMTh, Subi | N/A | N/A | Western blot—synatophysin, synaptobrevin, amphiphysin, SNAP-25 (all presynaptic), CaM kinase II (postsynaptic)—decreased presynaptic proteins within 18 h, decreased SNAP and CaM at 42 h, recovered by day 23 |
| Rizzi and colleagues, Brain Pathol 2007; 18: 198–210 | Guinea-pigs, Hartley, pregnant | Gest age (days) 20–25, 35–40, >50 | Midazolam 1 mg kg−1, N2O 75%, isoflurane 0.55% alone or combination. Fentanyl 15 μg kg−1 h−1, rocuronium | 4 h |
|
|
Am, ATh, Subi, RSC, ParC, OccC, CingC, PirC | N/A | N/A | |
| Johnson and colleagues, J Neurosurg Anaesthesiol 2008; 20: 21–28 | Mice, infantile C57BL6 | PND 5–7 | Isoflurane 0.75%, 1.5%, 2% | 4, 2, 1 h | Apoptosis: caspase-3 | Increased apoptosis at all concentrations, min for 2%×1 h, max for 0.75%×4 h | Caud, Put | N/A | N/A | MAC for isoflurane in infant mice reported to be 2.26% (Loepke and colleagues 2006) |
| Sanders and colleagues, Anesth Analg 2008; 106: 1708–11 | RatsSprague–Dawley | PND 7 | N2O 75%+isoflurane 0.75% in  25% 25% |
6 h | Apoptosis: caspase-3 | Increased apoptosis in the spinal cord | SC |
|
No difference | |
| Cattano and colleagues, AnesthAnalg 2008; 106: 1712–14 | Mice infantile C57BL6 | PND 5–7 | Propofol 25–300 mg kg−1 single dose | Killed after 6 h | Apoptosis: caspase-3 | Increased apoptosis—linear dose–response curve | Cau, Put, Cor | N/A | N/A | |
| Cattano and colleagues, Can J Anaesth2008; 55: 403–7† | Mice infantile C57BL6 | PND 5–7 | Isoflurane 0.75%±xenon 70% | 4 h then killed after 2 h | Apoptosis: caspase-3 | Increased apoptosis with all: isoflurane> Xe+iso>xenon | Cau, Put, Cor | N/A | N/A | †Neuroprotection study. Xenon is apoptogenic used alone, but suppresses apoptogenic activity of isoflurane in combination |
| Xou and colleagues, Neuroscience 2008; 19: 1053–65† | Rats Sprague–Dawley | PND 7 | N2O 75%, isoflurane 0.55%, alone or combination, in  25% ± l-carnitine 300–500 mg kg−1 25% ± l-carnitine 300–500 mg kg−1
|
2, 4, 6, or 8 h then killed after 6 h |
|
No increase in apoptosis with single agent. Significantly increased apoptosis after 6 h combination anaesthesia. l-carnitine blocked apoptosis | FroC, Th, Am, Hc | N/A | N/A | †Neuroprotection study. l-carnitine known to attenuate neurological injury associated with mitochondira-related degenerative disorders—neuroprotective in this study. Western blot for Bax, Bcl-Xl, β-actin—down-regulated with combination anaesthsia, protected by l-carnitine |
| Zhang and colleagues, Neurosci Lett 2008; 12: 109–14 | Mice C57BL/6 | PND 7 | Sevoflurane 1.7% in  30% 30% |
2 h then killed after 0–12 h | Apoptosis: caspase-3, DAPI, electron microscopy | Significantly increased apoptosis at a subclinical dose of sevoflurane | Th, SomC, CingC | N/A | N/A | Sevoflurane 1.7%=MAC 0.75 in infants (subclinical dose). Blood glucose in the anaesthetized group was not different from the control group |
| Satomoto and colleagues, Anesthesiology 2009; 110: 528–37 | Mice C57BL/6 | PND 6 | Sevoflurane 3% in air | 6 h |
|
Increased apoptosis immediately after anaesthesia, most severe in RSC and Th | Hc, Sub, Cau, Put, Th, FroC, TemC, RSC, Am | Assessed at age 14–17 weeks:
|
General behaviour was normal in sevoflorane-exposed mice.
|
Exposure to sevoflurane did not cause significant changes in cerebral blood flow or arterial blood gas. Exposure of neonatal mice to inhaled sevoflurane could cause not only learning deficits but also abnormal social behaviours resembling autism spectrum disorder. Western blot for PARP (apoptotic biomarker)—increased with sevoflurane |
| Sanders and colleagues, Acta Anaesthesiol Scand 2009, 54: 710–16† | Rats Sprague–Dawley | PND 7 | Isoflurane 0.75% in  25%±
dexmedetomidine 25, 50, or 65 μg kg−1
at 0, 2, and 4 h 25%±
dexmedetomidine 25, 50, or 65 μg kg−1
at 0, 2, and 4 h
|
6 h | Apoptosis: caspase-3 | Isoflurane induced apoptosis, partially attenuated by dexmedetomidine 25 μg kg−1. Dose escalation of dex did not further increase neuroprotection | Hc, Thal, Cor | N/A | N/A | †Neuroprotection study. Dexmedetomidine protected against neuroapotosis in vitro. Western blot for Bcl-2 and p-ERK1/2—expression decreased by isofluorane, effect reversed by dex |
| Sanders RD and colleagues, Anesthesiology 2009; 110: 1077–85† | Rat Sprague–Dawley | PND 7 | Isoflurane 0.75% in  25% ±
dexmedetomidine 1, 10 or 25 μg kg−1
at 0, 2, and 4 h 25% ±
dexmedetomidine 1, 10 or 25 μg kg−1
at 0, 2, and 4 h
|
6 h | Apoptosis: caspase-3 | Dexmedetomidine provided dose-dependent protection against anaesthesia-induced neuroapoptosis. In Hc and Th but not Cor, 25 μg kg−1 reduced injury to baseline | Hc, Thal, Cor | Long-term memory: context fear conditioning test at P40 | Dexmedetomidine ameliorated neurocognitive impairment induced by anaesthesia | †Neuroprotection study. Dexmedetomidine protected against neuroapotosis and long-term memory impairment in this study |
| Straiko and colleagues, Anesthesiology 2009; 110: 862–8† | Mice C57/B16 | PND 5 | Ketamine 40 mg kg−1 or propofol 50–100 mg kg−1 single dose±lithium 3–6 mEq kg−1 | Killed after 2–5 h | Apoptosis: caspase-3 | Increased apoptosis with ketamine or propofol, significantly reduced by 3 and 6 mEq kg−1 lithium | Cau, Put | N/A | N/A | †Neuroprotection study. Lithium reduces neuroapoptosis-induced by ketamine or propofol. Western blot for phosphorylated ERK 1/2 and phosphorylated Akt—phosphorylation suppressed by ketamine or propofol; suppressant effect reversed by lithium for ERK1/2 only |
| Pesic and colleagues, Int J Dev Neurosci 2009; 27: 279–87 | Rats Wistar | PND 7 | Propofol 25 mg kg−1 single dose | Killed after 0, 1, 2, 4, 8, 16, or 24 h | Neuronal necrosis: Fluoro-Jade B staining | Increased apoptosis | Cor, Th | N/A | N/A | Western blot for TNFα, NGF, p-Akt1/2/3, cleaved caspase-3, β-actin; RT–PCR for NGFβ, caspase-8—results show extrinsic apoptotic pathway induced |
| Head and colleagues, Anesthesiology 2009; 110: 813–25 | Mice | PND 1–21 mice and PND 1neurones | Isoflurane 1.4% in air | 4 h |
|
Isoflurane increased apoptosis/reduced synapse number. Apoptosis mitigated in vitro by tPA, plasmin, or p75NTR inhibitor (Fc-p75NTR or TAT-Pep5) and in vivo by TAT-Pep5 | Hc | N/A | N/A | Supports hypothesis that isoflurane neurotoxicity in the developing rodent brain is mediated by reduced synaptic tPA release and enhanced proBDNF/p75NTR-mediated apoptosis |
| Bercker and colleagues, Neurotox Res 2009; 16: 140–7 | Rats Wistar | PND 6 | Sevoflurane 3–5% for 6 h or propofol 30 mg kg−1 every 90 min for 3 doses | 6 h then killed 24 h after start | Neurodegen: DeOlmos silver | Significant difference in neurodegeneration between control and propofol, but not sevoflurane | FroC, ParC, CingC, RSC, Cau, Thal Hc, Sub, Hyp | Spatial memory learning: Morris water maze | Persistent learning deficits in the propofol group but not in the sevoflurane group | |
| De Roo and colleagues, PLoS One 2009; 16: e7043 | Mice H-line | PND 15–30 | Midazolam 25 mg kg−1 or propofol 50 mg kg−1 or ketamine 40 mg kg−1 | 5 h then killed | Spine dynamics assessed on hippocampal slice cultures | All agents increased spine protrusion density on tufted apical dendrites, more marked on PND 15 than 30 | Cor, Hc | N/A | N/A | Exposure to general anaesthetic agents rapidly induces dendritic spine growth (up to double) and the formation of functional synapses within a few hours |
| Wang and colleagues, Pediatr Res 2009; 66: 435–40 | Rats Sprague–Dawley pregnant | Gest age (days) 21 | Isoflurane 1.3–3% plus  100% Controls in 100% Controls in  100% 100% |
1 h then delivery after 6 h | Apoptosis: caspase-3 | Increased apoptosis significant with 3% isoflurane, not with 1.3% | Hc, RSC | N/A | N/A | Plasma S100β levels significantly increased with 3% isoflurane, not with 1.3%. Blood S100β may be a useful predictor of fetal brain damage during pregnancy |
| Zhao and colleagues, J Pharmacol Exp Ther 2010; 333: 14–22 | Rats | PND 7 plus in vitro study | Isoflurane 1–1.5%±xestospongin C 30 min prior | 6 h | Apoptosis: caspase-3 | Increased apoptosis with isoflurane, abolished by xestospongin C | Hc | Memory and learning ability evaluated through Morris water maze at P42–46 | No significant learning impairments noted in either group | Xestospongin C=InsP3R inhibitor. Inhibition of InsP3R significantly inhibited isoflurane-induced neurodegeneration in this study. Western blot for ClCsp3, BACE, BaxBcl-XL, and Bax—isoflurane increased BACE, ameliorated by xestospongin. Isoflurane did not change expression of BaxBcl-XL or Bax |
| Shu and colleagues., Anesthesiology 2010; 113: 360–8† | Rats Sprague–Dawley | PND 7 | 70% N2O+0.75% isoflurane. Preconditioning: 70% xenon, 70% N2O or
 8% for 2 h
8% for 2 h
|
6 h, given 24 h after precondition | Apoptosis: caspase-3 | Anaesthesia caused increased apoptosis. Pretreatment with hypoxia exacerbated anaesthesia-induced apoptosis. Pretreatment with N2O had no effect. Pretreatment with xenon prevented apoptosis | Hc, Cor | Trace fear conditioning at P40 | Impaired cognitive function with anaesthesia±hypoxic preconditioning. With xenon preconditioning, no significant difference in cognitive function cf control | †Neuroprotection study. Xenon protected against anaesthesia-induced apoptosis and learning deficits in this study. Western blot for Bcl-2. ClCsp3, cytochrome c, p53, mitogen-activated protein kinases—results as for histology. Xenon pretreatment increased Bcl-2 expression and decreased cytochrome c release and p53 expression |
| Milanovic and colleagues, Dev Neurosci 2010; 32: 288–301 | Rats Wistar | PND 7 | Propofol 20 mg kg−1 hourly | 2, 4, or 6 h then killed after 0, 4, 16, or 24 h |
|
Increased apoptosis and necrosis 24 h after 6 h long exposure but not after 2 or 4 h long exposure | RSC, ParC, CingC, OccC, PirC, ATh | N/A | N/A | Western blot for proteolytic cleavage products of α-II-spectrin, protein kinase C and poly(ADP-ribose) polymerase 1—showed increased caspase-3 and calpain activity, in a time-dependent and brain structure-specific manner |
| Cattano and colleagues, Minerva Anestesiol 2010; 76: 420–2 | Rats Sprague–Dawley | PND 7 | N2O 75%+ 25% 25% |
2 h | N/A | N/A | N/A | N/A | Total RNA isolated from each brain. Real-Time PCR showed up-regulation of nNOS (but not iNOS) and p53, providing evidenve of pro-apoptotic action of N2O | |
| Briner and colleagues, Anesthesiology 2010; 112: 546–56 | Rats Wistar | PND 16 | Isoflurane 1.5% or sevoflurane 2.5% or desflurane 7% | 30, 60, or 120 min |
|
|
Cor | N/A | N/A | These data strongly suggest that exposure to volatile anaesthesia does not induce cell death in the developing the cerebral cortex at PND 16 |
| Liang and colleagues, Anesthesiology 2010; 112: 1325–34 | Mice C57BL/6 | PND 7 | Isoflurane 0.75% or sevoflurane 1.1% (to achieve MAC 0.5)+  30% 30% |
6 h | Apoptosis: caspase-3 | Increased apoptosis isoflurane>sevoflurane | Hc, Cor |
|
No statistically significant difference in learning or memory with either agent | Plasma S100β significantly increased with isoflurane but not sevoflurane. Western blot for ClCsp3 and PARP (apoptosis biomarkers) and CDK4 and cyclin D1 (cell cycle regulatory proteins)—showed increased apoptosis results as for histology, but no change in cell cycle biomarkers |
| Yeude and colleagues, PLoS One 2010; 5: e11374 | Mice C57BL/6 | PND 2 or 7 | Phencyclidine 35 mg kg−1 on P2, or 50 mg kg−1 on P7, or both—single injections | Killed 10 h after last drug dose | Apoptosis: caspase-3 | Increased apoptosis with phencyclidine P2+7>P7>P2. Dense staining of Th, Cau, and Put in all groups. More apoptosis in Hc and Amy in the P2 group, more apoptosis in the neocortex in the P7 group | CingC, RSC, Cau, Put, LDTh, Hc, Amy |
|
The P2 group showed mild retention impairments in water maze test. The P7 group showed impaired acoustic startle response. P2+7 mice showed significant impairment in all tests, deficits persistent at P170 | Phencyclidine=NMDA antagonist. Phencyclidine induced different patterns of neuroapoptosis depending on developmental age at the time of exposure. Two separate exposures caused more severe neuropathological and neurobehavioural consequences |
| Lunardi and colleagues, Neurotox Res 2010; 17: 179–88 | Rats Sprague–Dawley | PND 7 | Combination: midazolam 9 mg kg−1, N2O 75%, isoflurane 0.75% | 6 h then killed on PND 21 | Quantitative and morphometric analysis of synapses by EM | Decreased neuropil and synapse density and morphology indicative of mitochondrial degeneration in the anaesthesia group | Sub | N/A | N/A | |
| Liu and colleagues, Curr Neuropharmacol 2011; 9: 256–61 | Rats Sprague–Dawley | PND 7 | Ketamine 5–20 mg kg−1 in one, three, or six injections at 2 h intervals | Max 12 h then killed at 5 min or 1, 2, 4, 6, or 18 h |
|
|
FroC | N/A | N/A | Quantitative PCR for NR1, NR2A, and NR2C subunits of NMDA receptor—significantly increased in ketamine-treated rats. Microarray analysis showed altered expression of apoptotic-relevant genes and increased NMDA-R gene expression with ketamine. Suggests ketamine causes compensatory up-regulation of NMDA-R, subsequently triggering apoptosis |
| Istaphanous and colleagues, Anesthesiology 2011; 114: 578–87 | Mice C57BL6/J | PND 7–8 | Desflurane 12.2% or isoflurane 2.7% or sevoflurane 5.4% to achieve MAC 0.6 | 6 h | Apoptosis: caspase-3, NeuN | Significantly increased neuronal cell death and apoptotic activity irrespective of anaesthetic agent | Cor | N/A | N/A | Colorimetric assay for activated caspase-3—increased after anaesthetic exposure. Increased neuroapotosis with all three agents, no significant difference between agents |
| Kodama and colleagues, Anesthesiology, 2011; 115: 579–91 | Mice C57BL/6 | PND 6 | Desflurane 4% or desflurane 8% or sevoflurane 3% or isoflurane 2% | 3 or 6 h | Apoptosis: caspase-3, TUNEL | Significanty increased apoptosis after 6 h, desflurane >isoflurane> sevoflurane. No significant difference cf. controls after 3 h | SomC, RSC, Hc | At 6 weeks age:
|
|
Western blot for PARP (biomarker for apoptosis)—increased after 6 h desflurane >isoflurane >sevoflurane. Results suggest that neurotoxicity of desflurane >isoflurane >sevoflurane |
| Zhou and colleagues, Neuroscience 2011; 174: 64–70 | Rats Sprague–Dawley | PND 7 | N2O 70% + Isoflurane 0.75% in  25% 25% |
6 h |
|
Increased apoptosis in the cingulate and hippocampus (GABA/glut) and substantia nigra (dop) but not basal forebrain (chol) | BF, SN, Hc, CingC | N/A | N/A | Suggests anaesthetic exposure significantly increases neuroapoptosis of glutamatergic, GABAergic and dopaminergic but not cholinergic neurones in developing brain |
| Cui and colleagues, Indian J Pharmacol 2011; 43: 648–51 | Mice Kunming | PND 5–7 | Propofol 50–150 mg kg−1, 4 injections at 90 min intervals | 6 h | Apoptosis: caspase-3, c-Fos | Increased apoptosis at 100–150 mg kg−1 propofol | Hc | N/A | N/A | Western blot for ClCsp3 and c-Fos—results as for histology |
| Tu and colleagues, Brain Res Bull 2011; 86: 29–35‡ | Rats Sprague–Dawley | PND 7 |
|
7 days |
|
Significant apoptosis in propofol+air/hypoxia groups. No significant apoptosis in propofol+oxygen and all vehicle groups | Hc | N/A | N/A |
‡Plus hypoxia. Propofol-induced attenuation of respiration may produce lower blood oxygen concentrations in air, resulting in neuronal degeneration.  50% rescued propofol-induced hypoxia. Western blot for ClCsp3—increased apoptosis results as for histology 50% rescued propofol-induced hypoxia. Western blot for ClCsp3—increased apoptosis results as for histology |
| Briner and colleagues, Anesthesiology 2011; 115: 282–93 | Rats Wistar | PND 5–90, male only for >PND 20 | Propofol 40 mg kg−1 then 4× 20 mg kg−1, injections at hourly intervals (total five injections) | 6 h |
|
Propofol decreased dendritic spine density when administered at PND 5–10, but increased spine density and synapse number when administered at PND 15–30 | PFC | N/A | N/A | Propofol-induced modifications in dendritic spine density persisted up to PND 90 |
| Tsuchimoto and colleagues, Paediatr Anaesth2011; 21: 1209–13† | Mice ddN strain | PND 7 | Isoflurane 1%±rEPO 50 000 IU kg−1 | 6 h | Neuronal density: Nissl | EPO attenuated isoflurane-induced neurodegeneration | Hc | Spatial learning/memory assessed by radial arm maze on PND 56–59 | EPO improved spatial learning/memory of mice exposed to isoflurane | †Neuroprotection study. EPO attenuated isoflurane-induced neurodegeneration |
| Sanchez and colleagues, Anesthesiology 2011; 115: 992–1002 | Rats Sprague–Dawley | PND 7 | Combination: midazolam 9 mg kg−1, N2O 75%, isoflurane 0.75% in  24% 24% |
6 h then killed at PND 21–28 | Regional distribution and ultrastructural properties of mitochindria: electron microscopy | Anaesthesia caused protracted injury to mitochondria including disturbance of regional distribution and ultrastructural appearance | Sub | N/A | N/A | Spectrophotometric analyses of mitochondrial electron transport chain activity—anaesthesia increased complex IV activity (known to increase free radical production) |
| Shu and colleagues, Neurobiol Dis 2012; 45: 743–50‡ | Rats Sprague–Dawley | PND 7 | 6 groups:
|
Killed after 0 or 6 h or DVT assessment at 40 days |
|
6 h anaesthesia caused widespread CNS injury; both nociceptive stimuli (formalin, surgery) increased this injury | Cor, Th, Hc, SC | Trace fear conditioning 40 days post-insult | 6 h anaesthesia caused long-term detriment in cognitive function. Both nociceptive stimuli exacerbated this | ‡Plus noxious stimuli. Nociceptive stimulation and prolonged anaesthesia produced significantly more apoptosis than anaesthesia alone. Western blot for TNFα and IL1β—anaesthesia plus nociceptive stimuli increased IL1β but not TNFα |
| Boscolo and colleagues, Neurobiol Dis 2012; 45: 1031–41 | Rats Sprague–Dawley | PND 7 | 6 groups:
|
6 h |
|
GA caused:
|
Subi |
|
|
*EUK-134=synthetic ROS scavenger, PPX=pramipexole, a synthetic aminobezothiazol derivative=mitochondrial protector. ELISA for pg 8-isoprostane, the most abundant prostaglandin-like compound formed in vivo from free radical-catalysed peroxidation of essential fatty acids—increased with GA, ameliorated by EUK |
| Huang and colleagues, Brain Res 2012; 2: 164–71 | Rats Sprague–Dawley | PND 7 | Ketamine 25–75 mg kg−1 daily for 3 days | 3 days (3 doses) | Apoptosis: TUNEL | Ketamine 75 mg kg−1 (but not 25–50 mg kg−1) significantly increased TUNEL+ cells | Hc | Morris water maze test, assessed on PND 60 | Ketamine 75 mg kg−1 caused learning and memory impairment in adulthood | Western blot for t-PKCγ, p-PKCγ, t-ERK1/2, p-ERK1/2, Bcl-2—ketamine decreased p-PKCγ, p-ERK1/2, Bcl-2 (pro-apoptotic effect). No change in t- PKCγ, t-EK1/2 |
| Fang and colleagues, Neurosci Bull 2012; 28: 499–508 | Rats Sprague–Dawley male | PND 7 | Sevoflurane 2% with O2 2 litre min−1 | 4 h |
|
Increased cell death and decreased progenitor proliferation until at least 4 days post-sevoflurane | Hc | Morris water maze test assessed at 2 and 6 weeks postsevoflurane | Spatial reference memory was not affected at 2 weeks but was affected at 6 weeks postsevoflurane | Western blot for MKI67 (mRNA Ki-67 protein, present during all active phases of cell cycle, absent from resting cells)—decreased post-sevoflurane |
| Liu and colleagues, Anesthesiology 2012; 117: 64–71‡ | Rats Sprague–Dawley male | PND 7 | Ketamine 2 mg ml−1, 5 injections at 90 min intervals±0.01 ml CFA (peripheral noxious stimulation) | 6 h |
|
Increased apoptosis with ketamine, attenuated by concomitant noxious stimulation with CFA | CingC, SomC | N/A | N/A | ‡Plus noxious stimulus. Concurrent noxious stimulation with ketamine attenuated neurapoptosis. Western blot for Cl-Csp3, t-AKT, p-AKT, GSK-3β, cyclin D1 and β-actin—ketamine minimally decreased p-AKT and GSK-3β. Ketamine increased cyclin D1, response suppressed with concomitant noxious stimulus |
| Zhou and colleagues, Mol Med Rep 2012; 6: 543–6 | Rats Sprague–Dawley | PND 7 | Sevoflurane 2.3% | 6 h, then killed 6 h post-sevoflurane |
|
Sevofluorane-exposed rats showed disorganized neuronal architecture, increased apoptosis and decreased nNOS expression | Hc | N/A | N/A | Previous studies have shown that sustained nNOS inhibition triggers neuroapoptosis |
| Popic and colleagues, PLoS One 2012; 7: e34396 | Rasts Wistar male | PND 14 | Propofol 25 mg kg−1 single injection | Killed after 0, 1, 2, 4, 8, 16, or 24 h | Neuronal necrosis: Fluoro-Jade B staining | No increase in neuronal necrosis seen at any time point | Cor, Thal | N/A | N/A | Western blot for BDNF, NGF, TrkA (=NGF-R), TrkB (=BDNF-R), Akt, ERK, Cl-Csp3—showed mostly down-regulation of neurotrophins, their receptors, and downstream effector kinases |
| Ponten and colleagues, Acta Anaesthesiol Scand 2012; 56: 1058–65† | NMRI | PND 10 | Ketamine 50 mg kg−1± clonidine 10 or 40 mg kg−1 pretreatment 30 min prior | Killed 24 h-post dose or tested PND 55 | Apoptosis: Fluoro-Jade B | Increased apoptosis with ketamine. Pre-treatment with 40 mg kg−1 but not 10 mg kg−1 clonidine reversed ketamine-induced apoptosis | FroC, ParC, Hc | Spontaneous activity in a novel environment at PND 55 | Reduced spontaneous activity after ketamine. Pre-treatment with 40 mg kg−1 but not 10 mg kg−1 clonidine reversed this | †Neuroprotection study. Clonidine pre-treatment reversed the adverse affects of ketamine in this mouse model |
| Wang, Neurol Sci 2012; 33: 535–44 | Rats Sprague–Dawley pregnant | Adult pre-gest or preg | Sevofluorane 2.4% MAC in oxygen. Exposed pre-gest or at gest age 6, 10, 14, and 18 days | 6 h then killed on PND 0, 7, 14, 28 |
|
Pre-pregnancy anaesthesia did not cause increased apoptosis. Sevofluorane in pregnancy resulted in disorganized architecture, increased apoptosis, and decreased nNOS expression at PND 0, 7, 14 but not PND 28 | Hc | N/A | N/A | RT–PCR for NMDA-R 1/2 expression—pre-pregnancy anaesthesia had no effect. Sevofluorane in pregancny resulted in increased expression on PND 0, 7, 14 but no significant difference at PND 28. Western blot for PKCα, p-JNK, p-ERK, and FOS showed no effect with pre-pregnancy anaesthesia. Sevoflurane in pregnancy resulted in up-regulation of PKCα and p-JNK and down-regulation of p-ERK and FOS on PND 0, 7, 14 but no significant difference on PND 28 |
Table 2.
Primate studies of anaesthetic-induced neuroapoptosis. PND, post-natal day; GA, gestational age; MAC, mean alveolar concentration
| Reference | Species | Age | Anaesthetic | Duration (h) | Histology outcome | Neurodevelopmental outcome |
|---|---|---|---|---|---|---|
| Slikker and colleagues, Toxicol Sci 2007; 98: 145–58 | Rhesus macaques (Macaca mulatta) | GA 122 days or PND 5 or PND 35 | Ketamine 20 mg kg−1 i.m. followed by infusion of 20–50 mg kg−1 h−1 i.v. to maintain light surgical plane of anaesthesia | 3 or 24 h | Increased apoptosis, necrosis, and NMDA-receptor NR1 subunit expression in neonatal brains after 24 h ketamine at GA 122 or PND 5 but not PND 35 or with shorter duration ketamine | N/A |
| Zou and colleagues, Int J Dev Neurosci 2009; 27: 727–31 | Rhesus macaques (Macaca mulatta) | PND 5–6 | Ketamine 20 mg kg−1 i.m. followed by infusion 20–50 mg kg−1 h−1 i.v. to maintain light surgical plane of anaesthesia | 3, 9, or 24 h | Time-dependent increased apoptosis and necrosis seen after ketamine 9 or 24 h, not seen at 3 h. Apoptosis prominent in neocortical areas, especially frontal cortex | N/A |
| Brambink and colleagues, Anesthesiology 2010; 112: 834–41 | Rhesus macaques | PND 5–6 | Isoflurane, end-tidal 0.7–1.5 vol% to maintain light surgical plane of anaesthesia | 5 h | Increased apoptosis. Temporal and somatosensory cortices most severely affected | N/A |
| Zou and colleagues, Neurotox Teratol 2011; 33: 592–7 | Rhesus macaques | PND 5–6 | N2O 70%, isoflurane 1% alone or combination | 8 h | Increased apoptosis with combination anaesthesia but not with N2O or isoflurane alone. Frontal and temporal cortices and hippocampus most severely affected | N/A |
| Paule and colleagues, Neurotoxicol Teratol 2011; 33: 220–30 | Rhesus macaques | PND 5–6 | Ketamine 20 mg kg−1 i.m. followed by infusion 20–50 mg kg−1 h−1 i.v. to maintain light surgical plane of anaesthesia | 24 h | N/A | At 7 months and persistent at 3.5 yr, the ketamine group had poorer performance in learning, colour, and position discrimination tasks; slower response speed; lower motivation |
| Brambink and colleagues, Anesthesiology 2012; 116: 372–84 | Rhesus macaques | GA 120 days or PND 6 | Ketamine i.v. 10–20 mg kg−1 bolus then 10–85 mg−1 kg−1 h−1 infusion to maintain intermediate surgical plane of anaesthesia | 5 h | Widespread apoptosis in all groups. Loss of neurones 2.2× greater in fetuses than in neonates. Damage throughout fetal brain; damage maximal in basal ganglia in neonatal brain | N/A |
| Brambink and colleagues, Ann Neurol 2012; 72: 525–35 | Rhesus macaques | PND 6 | Isoflurane, end-tidal 0.7–1.5 vol% to maintain light surgical plane of anaesthesia | 5 h | Significant apoptosis seen throughout CNS in both white and grey matter. Fifty-two per cent dying cells were glia, 48% were neurones. Oligodendrocytes engaged in myelinogenesis were selectively vulnerable | N/A |
Table 3.
Clinical observational studies of neurocognitive impairment after infant anaesthesia and surgery (IAS). NOS, Newcastle–Ottawa Score
| Reference | Design | Study period | Follow-up | Sample size | Operations | Anaesthetic | Confounder adjustment | Outcomes with exposure to anaesthesia and surgery | Grade of evidence | NOS (max=9*) |
|---|---|---|---|---|---|---|---|---|---|---|
| DiMaggio and colleagues19 | Birth case–control | 1991–2001 | 1–2 yr | 5050 control, 383 exposed | Inguinal hernia repair | N/A | Yes | Behavioural/developmental delay after IAS | B | 8* |
| Hansen and colleagues17 | Birth cohort | 1986–1990 | 25–16 yr | 14 575 control, 2689 exposed | Inguinal hernia repair | N/A | Yes | Academic performance: No difference but increased non-attainment of test scores with IAS | B | 8* |
| Ing and colleagues21 | Birth cohort | 1989–1992 | 10 yr | 2287 control, 321 exposed | Varied | N/A | Yes | Neuropsychological testing: increased expressive and receptive language deficits after IAS | B | 8* |
| Wilder and colleagues24 | Birth cohort | 1976–1982 | 19 yr | 4764 control, 593 exposed | Varied | Known | Yes | Increased learning difficulties with increased exposure to IAS | B | 8* |
| Sprung and colleagues23 | Birth cohort | 1976–1982 | 19 yr | 5357 control, 531 exposed | Varied | Known | Yes | Increased ADHD with increased exposure to IAS | B | 8* |
| Flick and colleagues22 | Case–control | 1976–1982 | 19 yr | 5007 controls, 350 exposed | Varied | Known | Yes | Increased learning difficulties after IAS | B | 8* |
| DiMaggio and colleagues20 | Sibling cohort | 1999–2005 | 10146 control, 304 exposed | Varied | N/A | No | Increased developmental and behavioral disorders after IAS. Effect not found in more tightly controlled analysis | B | 8* | |
| Block and colleagues25 | Treatment cohort | 1990–2008 | 7–17.9 yr | 287 exposed | Inguinal hernia, orchidopexy, pyloromyotomy | Known | No | Academic achievement tests: mean scores lower than population norm after IAS | B | 5* |
| Bartels and colleagues26 | MCMA twin cohort | 1986–1995 | 12 yr | 1890 control, 1248 exposed | Varied | N/A | Yes | Educational achievement: scores significantly lower than population norm for males, non-significantly lower for females after IAS. However, no effect observed between monozygotic twins | B | 8* |
Preclinical evidence for anaesthetic-induced neurodegeneration: rodent studies
After the seminal work of Jevtovic-Todorovic and colleagues4 and Ikonomidou and colleagues,13 numerous studies have now demonstrated that exposure of neonatal rodents to anaesthetics induces apoptosis in the developing brain (Table 1). This injury is most readily observed in the first 2 weeks of life in rodents when synaptogenesis peaks. The exact clinical correlate for this injury is unknown. Many factors influence the translation of these findings to humans, one being that synaptogenesis extends for many years post-partum in humans, suggesting that the window of vulnerability in humans may be longer.27 Most rodent studies have also used relatively long exposures to anaesthetics, although isoflurane has been shown to induce apoptosis after exposure for just 1 h at concentrations of <1 MAC.28 While ketamine and other N-methyl-d-aspartate (NMDA) antagonist drugs were initially implicated,13 different combinations of agents have also been shown to induce apoptosis such as midazolam–nitrous oxide–isoflurane,4 nitrous oxide–isoflurane,29 or combinations of ketamine and thiopental or propofol.30 Similarly, benzodiazepine drugs, such as midazolam,31 have been shown to be toxic.
Perhaps of most significance to modern neonatal clinical practice is that all volatile anaesthetics, and propofol,32 have been shown to induce the injury.7,33–35 The apoptosis is widespread among central nervous system loci, with cortical, thalamic, basal ganglia, and hippocampal injury prominent; the injury is also present in the spinal cord.4,6,7,36 Significant effects on neurogenesis have also been noted, with suppressed proliferation of neural progenitors observed for 5 days after isoflurane exposure to 7-day-old rat pups.37 This is paralleled by effects of isoflurane38 (but not propofol)39 on neuronal growth in in vitro culture, suggesting direct effects on these progenitor cells. Neurogenesis is important for both cognitive function and particularly brain repair; therefore, the inhibitory effects of isoflurane on neurogenesis likely play an important part in the development of impaired cognition in rodents.
When long-term follow-up of neonatal anaesthetic exposure have been performed, the majority of studies have identified some cognitive impairment persisiting into adulthood. This relatively consistent finding is of particular interest, given the wide range of different tests used. Rodent studies suggest that general anaesthetics do not induce long-term motor impairment or altered nociceptive processing6 but do impair memory function, likely through injury in both the hippocampus and frontal cortex.4,6,7 Another study noted that anaesthetic exposure leads to behaviours that may be akin to autism;34 this is particularly interesting as many clinical studies have focused on behavioural abnormalities rather than overt cognition. Of course, sophisticated cognitive function is difficult to test in rodents, limiting our knowledge about the exact effect on cognitive and other neurobehavioural domains. Rodent studies are also difficult to interpret due to the limited ability to control and monitor the physiology of the animal during anaesthesia. Indeed, in some of the studies, some of the newborn rodents die during anaesthesia implying that physiological control is not comparable with that in humans.
Spinal anaesthesia also exerts some neurotoxic effects (recently reviewed by Walker and Yaksh)40 with ketamine again implicated. Intrathecal ketamine induced-apoptosis in the spinal cord was associated with abnormal gait and altered nociceptive processing.41 Further information is needed on the effects of local anaesthetics, although recent data do not indicate harm.36 Similarly, morphine and clonidine do not induce apoptosis when administered intrathecally to rats.42
In utero exposure to isoflurane also leads to apoptosis in the rat fetal hippocampus and retrosplenial cortex.43 In a separate study, exposure to 1.4% isoflurane for 4 h during 1 day of pregnancy led to abnormal spatial memory acquisition and reduced anxiety in rats in adulthood.44 Hence, there is evidence that the rodent fetus, like the neonates, is vulnerable to developmental neurotoxicity.
Preclinical evidence for anaesthetic-induced neurodegeneration: piglets and primates
Non-rodent mammals, including piglets and non-human primates, are also vulnerable to injury. Piglets are of particular interest as their brain development closely parallels humans, with synaptogenesis occurring pre- and post-natally. Isoflurane–nitrous oxide induced apoptosis in a single 5-day-old piglet.12 In a second piglet study, isoflurane–nitrous oxide was shown to cause more damage in intrauterine growth restricted piglets than in normal birth weight piglets (though there were no non-anaesthetized controls).45 Further useful information could be gleaned by studying piglets as their brain growth spurt, myelination, and development of electrical activity are thought to be similar to humans.46
Importantly, anaesthetic injury is also observed in the primate brain (Table 2). The initial studies involved ketamine exposure for 3, 5, 9, and 24 h at a ‘surgical plane’ of anaesthesia. Unlike many of the rodent studies, in primates, there was good control of physiological variables.8,10,47,48 The doses used were much higher than would be used in humans. However, it is well known that animals require higher doses (on a mg kg−1 basis) of some anaesthetics such as ketamine to produce anaesthesia, and these doses were necessary to simulate ‘clinical’ conditions. Exposure to 5, 9, or 24 h of ketamine produced robust apoptosis in the frontal cortex in 5-day-old monkeys; however, a 3 h exposure was not toxic.8,47 Twenty-four hours of ketamine produced no apoptosis in 35-day-old monkeys. Although the lack of effect seen with 3 h of exposure is reassuring, the limited number of animals studied and focus on only one brain injury somewhat limits conclusions about the relative safety of just a 3 h exposure. Ketamine administration for 24 h has also been associated with long-term cognitive impairment up to 3.5 yr later10 using the Operant Test Battery, a test that correlates with intelligence in children. The cognitive impairments induced in rhesus monkeys are relatively ‘high level’ involving both learning and concept formation, consistent with significant apoptosis in the frontal cortex. Ketamine injury is also apparent in primate fetus with exposure during pregnancy. In fact, the rhesus fetus is more vulnerable than the neonate.47 The fetal stage examined in the rhesus macaque appears to correlate with the last trimester of human pregnancy or premature birth.27
Isoflurane has also been shown to induce apoptosis in the neonatal rhesus monkey brain after a 5 h exposure at an anaesthetic dose very similar to that required in humans (0.7–1.5%).48 The areas particularly affected include layers II, V, and VI in the frontal, somatosensory (primary visual areas), and temporal cortices. This indicates that diverse neuronal types, in widely distributed regions throughout the cerebral cortex, are vulnerable. The authors argue that, in terms of brain development, these monkeys are comparable with 6-month-old infants, and suggesting that humans up to that age might be vulnerable.48 A similar study has shown that in addition to neurones, isoflurane also produces degeneration of oligodendrocytes, cells that play a key role in myelinating axons.49 A single exposure to 5 h of isoflurane led to loss of around 6.3% of the total population of myelinating oligodendrocytes; the biological consequences of reducing myelinating oligodendrocytes from the developing brain may include disruption of myelinogenesis and pathological changes in axonal structure or function. The oligodendrocyte degeneration induced by isoflurane in neonates differs from that in the preterm brain where it is loss of the premyelinating oligodendrocytes that occurs due to their increased vulnerability at this developmental stage of brain development.
Data are accumulating regarding the diverse range of injuries induced by anaesthetics; however, significant questions remain in how to translate these findings to humans. Critical issues include the different doses required, how to compare periods of development across species, how to interpret neurobehavioural outcomes in animals and humans, and the degree of physiological control during anaesthesia. The relatively long exposure time used in animal models to induce injury also entails some uncertainty. The rodent brain develops over a much shorter period than humans, so the relative period of anaesthesia is considerably greater in rodents. Lastly, the influence of surgery itself is unknown. An understanding of the putative mechanisms of injury is essential to knowing how to translate these findings to humans.
Putative mechanism of the anaesthetic injury
One hypothesis for the mechanism underlying the neurotoxic effect of anaesthesia is that suppression of neurotrophic synaptic signalling leads to apoptosis of the postsynaptic neurone,50 via both the intrinsic and extrinsic apoptotic pathways16,29 (Fig. 1). This hypothesis directly links the putative mechanisms of anaesthesia51,52 to the mechanisms of neurotoxicity in the newborn. In terms of neurodevelopment, the anaesthetic suppression of synaptic activity might make the dying neurone redundant and lead to programmed cell death, known as apoptosis. This is consistent with the known effects of synaptic signalling in neurodevelopment;50 the abundance of rapid-eye-movement sleep in the neonatal period (providing an active brain in a restful baby and presumably avoiding neurone-suppressing effects of non-rapid-eye-movement sleep);53 and the known effects of anaesthesia on synaptic activity.52,54 One possibility is that the anaesthetic-induced suppression of synaptic activity leads to altered processing of the neurotrophin brain-derived neurotrophic factor (BDNF), leading to the cell death promoting form of the neurotrophin.55 Manipulating this pathway might be one way to prevent post-synaptic neuronal apoptosis.55
Fig 1.
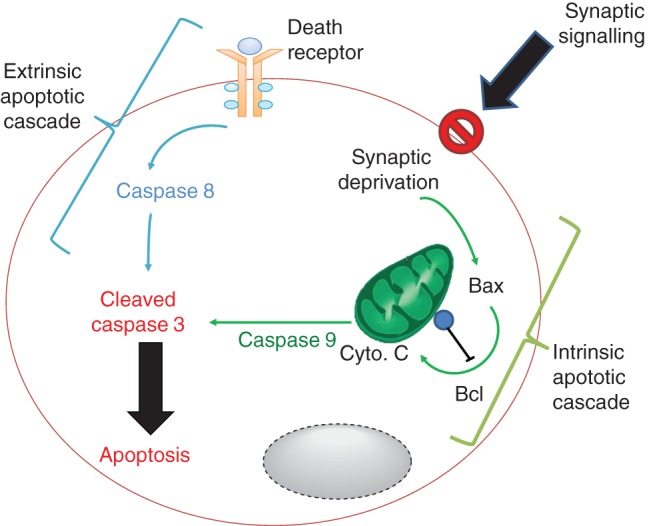
Proposed mechanisms of anaesthetic-induced neurodegeneration in the newborn involving the intrinsic and extrinsic apoptotic cell death pathways. Suppression of synaptic signalling is proposed to attenuate neurotrophic survival signals leading to activation of the intrinsic apoptotic cascade. This involves mobilization of the pro-apopotic protein Bax, which in turn induces cytochrome c (Cyto. C) release from mitochondria, activation of caspase-9, and subsequent induction of caspase 3 cleavage to provoke apoptosis. Anti-apoptotic effectors such as Bcl oppose this pathway. The extrinsic pathway is stimulated by ligands binding to death receptors located on the cell membrane that cause caspase 8 activation, which then cleaves and activates caspase 3. These ligands include cytokines such as TNF-α.
Neuronal apoptosis typically occurs through two distinct signalling pathways, the intrinsic and extrinsic apoptotic pathways16,29 (Fig. 1). The intrinsic pathway involves internal signals activating a mitochondrial-dependent pathway with the release of cytochrome c, activation of the pro-apoptotic factor BAX, and ultimately DNA cleavage and cell death. The loss of synaptic activity can lead to apoptosis through direct activation of the intrinsic apoptotic cascade.56 The exact molecular mechanism through which anaesthetics suppress synaptic signalling is unclear, although effects on glutamate (e.g. NMDA receptors) and γ-aminobutyric acid (GABA) synaptic transmission seem likely.1 Interestingly, the GABAA antagonist gabazine does not reverse the apoptotic effects of isoflurane in vitro, suggesting that chloride permeable GABAA receptors might not be solely responsible.7 However, given that several GABAergic drugs (such as midazolam, propofol, thiopental, and the volatile anaesthetics) induce apoptosis, it is premature to dismiss this mechanism. A role for chloride gradients has been implicated in the toxicity, since the Na+–K+–2Cl− cotransporter 1 inhibitor bumetanide reverses sevoflurane-induced apoptosis.57 Beyond any putative role of chloride gradients, there are several synaptic and intracellular targets that still require evaluation before the mechanism is more clearly understood. For example, suppression of pro-survival extracellular-related kinase (ERK) signalling has been associated with apoptosis from ketamine, propofol, and/or isoflurane; rescue of this pathway with lithium58 or dexmedetomidine35 protects against neuroapoptosis.
The extrinsic pathway is typically triggered by external stimuli such as the cytokine tumour necrosis factor (TNF)-α. While anaesthetic and sedative agents typically exert anti-inflammatory effects in the adult,59 in the young, the effects seem largely pro-inflammatory.60 This pro-inflammatory effect might not only stimulate the extrinisic apoptotic cascade but could also synergize with other inflammatory stimuli such as pain and surgery. This hypothesis requires further testing. The reason for this pro-inflammatory effect is unclear, although it might relate to altered GABAA signalling with neurodevelopment. At birth, GABAA receptor activation can be excitatory, and this switches to the adult inhibitory effect with ageing. This might mean that the immune effects of GABAA signalling switch from a pro- to anti-inflammatory effect with development.59,61
Interestingly, not all neurones are vulnerable to injury. Glutamatergic, GABAergic, and dopaminergic neurones die after anaesthetic exposure to neonatal rats; however, cholinergic neurones are resistant.62 Hence, it can be hypothesized that neurones innervated by cholinergic neurones are vulnerable to anaesthetic injury as neurotrophic cholinergic signalling is suppressed. The distributed nature of these neurones implies that this does not account for all cell death. Nonetheless, as anaesthesia is known to suppress cholinergic signalling,63 it is possible that cholinergic manipulation is another way of altering apoptosis, particularly in the cortex where cholinergic innervation is extensive. Given the distinct type of anaesthesia induced by ketamine, it may not be prudent to assume that similar pathways are involved in its neuroapoptotic mechanism compared with volatile agents.
Another hypothesis that has emerged is that anaesthetics induce seizure activity to cause neurotoxicity based on work with sevoflurane in neonatal rats.57 Studies are required to understand if a similar activity occurs in the human neonatal brain under anaesthesia. Despite suggestions that suppression of neural activity or the opposite—seizure activity—underpins the anaesthetic brain injury, there are limited data correlating EEG or neuroimaging activity with injury in vivo.57 Further mechanistic studies investigating non-invasive translational markers of anaesthetic injury, such as EEG, are required.
The effect of anaesthetic exposure on hypoxic–ischaemic brain injury in the neonate is unclear. One study demonstrated that a 2 h hypoxic exposure the day before anaesthetic injury increased the subsequent apoptosis.64 This suggests that neonates with prior, even mild, brain injury may be more susceptible to anaesthetic injury and thus may represent a vulnerable cohort of patients. Further preclinical study is required.
Agents that reduce the injury
Insights might also be afforded by examining the accumulating data for agents that can protect against anaesthetic neurotoxicity, which comes entirely from rodent studies (relevant studies are highlighted in italic in Table 1). One of the more promising class of agents that might provide neuroprotection against anaesthetic-induced apoptosis is the α2-adrenergic agonists, which can exert cognitive protection.7,35 α2-Adrenoceptors are known to play a key role in neurodevelopment underlying the rationale for targeting this receptor. Dexmedetomidine and clonidine are the only drugs that have been shown to reduce anaesthetic-induced apoptosis and also prevent subsequent decline in cognition (although neither are completely attenuated).7,35,65 The protective effect of dexmedetomidine was shown against isoflurane injury, and was attenuated in the hippocampus by an α2-adrenoceptor antagonist indicating involvement of α2-adrenoceptors.7 Dexmedetomidine also reversed the cognitive impairment induced by isoflurane. Furthermore, clonidine protects against ketamine neuroapoptosis and behavioural abnormalities.65 Importantly, α2-agonists do not themselves provoke apoptosis or produce neurological abnormalities by either systemic or spinal injection.7,42,65
Xenon is another candidate neuroprotective drug; xenon itself shows minimal toxicity but also can attenuate isoflurane-induced apoptosis when given with isoflurane29 or before isoflurane.64 This effect of xenon is particularly intriguing as it is an NMDA antagonist, similar to ketamine, but is protective rather than toxic. The expense and difficulties in administering xenon may mitigate against its development for neonatal anaesthesia however.
Melatonin5,66 and β-estradiol14,67 provide protection against anaesthetic and anti-epileptic drug-induced apoptosis in rats. Both are known to act as free radical scavengers, anti-inflammatory, and anti-apoptotic agents. They target the intrinsic apoptotic pathway,5,14,66,67 which is up-regulated in anaesthetic injury,29 and hence they are plausible candidates of neuroprotective drugs. Other agents such as lithium,58,68 hypothermia,69 l-carnitine,70 xestospongin-C,71 bumetanide,57 and erythropoeitin72 can also provide protection, but further studies are required to confirm these observations and to allow clinical translation. The possibility of modulating the BDNF pathway to promote cell survival, rather than cell death, is also of particular interest,55 especially if combined with anaesthetic drugs that up-regulate BDNF expression.5,29,67 Most of the signalling pathways that have been manipulated so far to reduce anaesthetic-induced apoptosis involve the intrinsic apoptotic cascade, particularly modulating the Bcl-2 or Bcl-XL to Bax ratio (Fig. 1). The potential to modulate other contributing factors, including the extrinsic pathway, needs further attention. Furthermore, all of these proposed therapies need to be trialled in animal models other than rodents, given the limited clinical translation from rodent models to humans.
Environmental enrichment can improve cognitive function after neonatal exposure to sevoflurane in rats, suggesting that it might be possible to compensate for the injury.73 However, anaesthetic-exposed rodents might not learn as fast as non-exposed rats,73 similar to primates.10 Hence, it is unclear whether this really constitutes a therapeutic non-pharmacological protective strategy.
Potential impact of surgery on neurodevelopment
Neonatal pain, induced by surgery, is known to induce long-term behavioural effects in animals and humans,1,74 emphasizing the need for adequate analgesia in the perioperative period. In neonatal rodents, brief inflammatory pain leads to increased neuronal activation and cell death, particularly in immature animals.75 The combination of glial cell activation, pro-inflammatory cytokine release, and prolonged pain is likely to produce prolonged neuronal hyperexcitability in cortical and subcortical areas. Cell death might result from this prolonged excitation.75
Anaesthesia is given in the context of surgery to prevent harmful effects of painful stimulation; the impact of surgery on anaesthetic injury is thus of great clinical significance. There are only limited, and conflicting, data on the influence of surgical stimulus on the anaesthetic injury. Nociceptive stimuli (surgical incision or formalin) provided during 6 h of nitrous oxide–isoflurane anaesthesia exacerbated neuroapoptosis and long-term cognitive impairment in rat pups.76 This might be driven by exacerbation of extrinsic apoptotic signalling through inflammation (Fig. 1). However, limitations of this animal model include the lack of physiological control and realistic surgical stimulation. Others have shown that tissue injury, induced by tail clamping, has no influence on sevoflurane-induced apoptosis,73 while another group found that ketamine-induced neuroapoptosis was antagonized by noxious stimulation.77
The apparent inconsistencies in these studies might be explained by the differing molecular targets affected by ketamine, sevoflurane, and isoflurane–nitrous oxide. For example, ketamine is known to possess anti-inflammatory properties and does not target GABAA receptors, so the difference between the effect of noxious stimulation on the two anaesthetic regimens could relate to the different anaesthetic drugs studied. Another difference is in the degree of tissue injury and surgical stimulus. The relative toxic or protective effects of the anaesthetic might depend on the size of the injury relative to the duration and dose of anaesthetic. Hence, studies are required to address how surgical stimulation affects anaesthetic-induced injury in a large animal model where physiological variables can be better controlled.
The impact of surgery, in addition to anaesthesia, on perinatal brain injury also requires further investigation as inflammatory stimulation could exacerbate the previously incurred hypoxic–ischaemic injury.78 Surgery induces significant pain that might exacerbate the excitotoxic injury of hypoxia–ischaemia by augmenting neuronal activity. Furthermore, surgical trauma is characterized by pro-inflammatory cytokine release, microcirculatory disturbance, and cell-mediated immune dysfunction, followed by a compensatory anti-inflammatory response. Also directly altering neuronal function,79 these inflammatory changes might provoke further degeneration,80 including activation of the extrinsic apoptotic cascade leading to further neuronal death.
Epidemiology evidence for anaesthetic and surgical effects on neurodevelopment
There are numerous cohort and cross-sectional studies that have investigated the association between surgery, anaesthesia, and cognitive or neurobehavioural outcome, with conflicting results. A recent systematic review and Bayesian meta-analysis identified 12 retrospective epidemiology studies relevant to anaesthetic neurotoxicity in children. Seven of these were included in the analysis, involving 40 685 children.18 Bayesian meta-analysis differs from the standard meta-analytic approach as it seeks to identify the odds ratio of the mean effect size and calculate the likelihood that any future study would identify the same mean effect size. A measure of uncertainty in the finding is also produced: the credible interval. The model takes into account the prior beliefs of the investigators (in this case, that anaesthesia exerts no toxic effect). These data can be ‘synthesized’ to produce a summary of the data, but also can be used to form a ‘predictive’ model for a future study and hence has value in the planning of research. Importantly, there was significant heterogeneity between the studies for both the unadjusted and adjusted outcome of adverse neurodevelopmental and behavioural outcome of the analysis for ‘exposed to any anaesthesia’. The unadjusted odds ratio was 1.9 (95% credible interval 1.2–3.0) for the synthesized data, although for the predictive model, the odds ratio was 2.2 (95% credible interval 0.6–6.1). The adjusted odds ratio was 1.4 (95% credible interval 0.9–2.2) for the data synthesis, and for the predictive model, the odds ratio was 1.5 (95% credible interval 0.5–4.0). Using this technique, the authors concluded that there is a ‘modestly elevated risk of adverse behavioral or developmental outcomes in children who were exposed to anesthesia/surgery during early childhood. The evidence, however, is considerably uncertain’.
Systematic review of epidemiology studies
We have conducted our own systematic review of the literature and provide a descriptive table to the reader where we have evaluated the methodology and grade of evidence for each study (Table 3). This information is provided, so the reader can appreciate the accumulating clinical evidence that anaesthetics and surgery could induce harm. We identified nine studies in total,17,19–25,81 and we systematically graded the evidence82 from each study, and also detailing their strengths and weaknesses.
Excluded studies
To be included in the systematic review, some attempt to compare an exposed group with a non-exposed control group was required; however, we did include studies using population controls.17,25 One excluded study (due to a lack of a non-exposed control group) found that the point estimate for neurobehavioural impairment was non-significantly higher for operations at a younger age (under 24 months of age); however, the confidence intervals were wide, overlapping with one.83
One study, which was incorporated in the Bayesian meta-analysis,18 was excluded as it pertained to intensive care sedation rather than anaesthesia and surgery.84 This study did not find an association between the duration of sedation for mechanical ventilation in premature neonates and adverse cognitive outcome.84 In contrast, another small study found that anaesthesia and surgery in premature neonates was associated with impaired mental development index and reduced brain volumes and increased white matter injury on magnetic resonance imaging at 2 yr of age.85 This study was also not included in our Table 3 as it focused on preterm brain injury. It is important to note that morphine use in the neonatal intensive care unit has been associated with impaired cognitive function at 5 yr of age as assessed by the visual analysis IQ subtest.86 We did not include studies that found that neuraxial labour analgesia87 or anaesthesia for Caesarean section81 were not associated with adverse cognitive outcomes.
Included studies
The majority of studies included in our review had robust methodology (indicated by a Newcastle–Ottawa score of >8*; Table 3). Nonetheless, there is heterogeneity in the approaches, degree of confounding, and endpoints analysed. Despite these limitations, the cumulative data suggest a small increase in the risk of adverse neurodevelopmental outcome with exposure to anaesthesia and surgery. All nine studies provided some evidence that anaesthesia and surgery might be associated with impairments in cognition or behaviour (Table 3). Furthermore, there is some evidence that the risk increases with increased cumulative exposure to anaesthesia and surgery.22–24 Of course, given the observational nature of these studies, it is difficult to dissect the effect of anaesthesia from the effect of surgery, or the underlying pathology driving the need for surgery, or patient comorbidities. Another limitation of these studies is the limited information about perioperative care, including anaesthetic drugs, haemodynamics, postoperative care, complications, and comorbidities. Furthermore, the relevance of the anaesthetic techniques and perioperative care used in these retrospective studies to modern practice is unclear (as many of the studies date back decades to allow sufficient patient follow-up). Therefore, there is significant reason to be uncertain about the clinical data, as suggested by the Bayesian meta-analysis.
One study is worth particular attention as it compared twins where one twin was exposed to anaesthesia and surgery and the other was not.26 This study found that educational achievement scores were significantly lower than the population norm for males exposed to anaesthesia but non-significantly lower for females. However, no difference was detected between monozygotic twins where one twin was exposed and another was not. The authors use these data to argue that there is not a causal relationship between anaesthesia and surgery and adverse cognitive outcomes.26 However, the limited sample size precludes any definitive conclusions. Another limitation of this study and other similar studies is the relatively coarse outcome measure. Finding no effect in a summary score such as composite school score does not exclude effects in neurobehavioural subdomains. A similar criticism can be made of another study that reported no effect of anaesthesia and surgery on academic achievement;17 more subtle tests might have identified a difference. Against this, the same study suggested that anaesthesia and surgery increases the risk of not obtaining any test score, consistent with a profound deficit in cognition. Therefore, even within studies, interpretation of the data can be difficult. The other seven studies largely provide point estimates that suggest there might be harm from anaesthesia and surgery, but with confidence intervals that are too wide to allow confidence in their findings.
Given the limitations of these observational studies, it is difficult to draw any firm conclusions. At best, they suggest that an association between anaesthesia exposure and adverse outcome is possible, but this cannot be definitely confirmed or ruled out. This emphasizes the need for further high-quality prospective approaches to this problem.
Prospective studies of the effect of anaesthetics and surgery on neurodevelopment
The significant limitations of the retrospective observational epidemiology studies mandate prospective approaches to studying the effects of anaesthesia and surgery on neurodevelopment.11,88 Two important studies underway include the GAS trial and PANDA cohort study. The GAS trial is a randomized controlled trial of general anaesthesia compared with spinal anaesthesia for hernia surgery in more than 700 neonates.89 Randomization is stratified by age and by site to ensure balanced groups. The aim is to discern whether general anaesthesia and awake regional anaesthesia have similar effects on cognitive function at 2 and 5 yr of age. The GAS trial is likely to complete recruitment in February 2013. PANDA (Pediatric Anesthesia and NeuroDevelopment Assessment) is a cohort study that will enrol 500 sibling matched pairs (1000 children) who underwent hernia surgery under general anaesthesia (ASA I–II) before 36 months of age.2 The children will undergo a series of neuropsychological tests between 8 and 15 yr of age. The results of a recent pilot study for PANDA90 suggest that it will have adequate power to detect statistically significant differences in total IQ (mean difference of four points, standard deviation of 17 points; α = 0.05; β = 0.8).
Future investigations into the neurodevelopmental effects of perioperative care
There are numerous outstanding questions, not least the clinical relevance of the large amount of preclinical evidence for harm of anaesthetic exposure. It remains to be seen whether neuroprotective agents, effective in rodents, can provide clinical protection. Here, an important step will be translation via large animal models such as the piglet or primate that share greater similarities to human neurodevelopment.91 We are a long way from clinical trials of these putative neuroprotective agents. Rather we emphasize the need for further prospective studies and translational research approaches to the problem. The prospect of ‘in human’ neuroimaging to detect injury must be explored further,85 and also non-invasive biomarkers of injury such as the EEG. In the short term, we are not able to issue clinical guidelines.11 We would not recommend reducing or increasing the anaesthetic dose and there is a lack of evidence to support neuroprotective interventions unless they are indicated for other purposes (e.g. premedication or analgesia).
Conclusions
The increasing public exposure of research in this area is making parents increasingly aware that anaesthetic exposure could affect neurodevelopment in children. We suggest that is currently only weak evidence that anaesthesia is harmful but that withholding or significantly altering established anaesthesia techniques is likely more harmful.1,11 In some circumstances, delaying surgery might be appropriate; however, surgery in children is rarely ever elective and delay can also cause harm. We recommend that any discussion over delaying an operation should involve the surgeon.
Declaration of interest
R.D.S. has acted as a consultant for Air Liquide, Paris, France, concerning the development of medical gases and has received speaker fees from Orion (Turku, Finland) and Hospira (Chicago, USA).
Funding
R.D.S. is supported by the Wellcome Trust, Action Medical Research, Association of Anaesthetists of Great Britain and Ireland, and the National Institute of Health (USA). J.H. is supported by the National Institute of Health Research. A.J.D. is supported by the Australian National Health and Medical Research Council and the Victorian Government Operational Infrastructure Support Program. N.J.R. is supported by the Wellbeing of Women, Medical Research Council and Action Medical Research. D.M. is supported by the Medical Research Council, Action Medical Research, Alzheimer's Society and SPARKS. This work was undertaken at University College Hospital/University College London, which received a proportion of funding from the UK Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme.
References
- 1.Sanders RD, Ma D, Brooks P, Maze M. Balancing paediatric anaesthesia: preclinical insights into analgesia, hypnosis, neuroprotection, and neurotoxicity. Br J Anaesth. 2008;101:597–609. doi: 10.1093/bja/aen263. doi:10.1093/bja/aen263. [DOI] [PubMed] [Google Scholar]
- 2.Sun L. Early childhood general anaesthesia exposure and neurocognitive development. Br J Anaesth. 2010;105(Suppl. 1)):i61–8. doi: 10.1093/bja/aeq302. doi:10.1093/bja/aeq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hudson AE, Hemmings HC., Jr Are anaesthetics toxic to the brain? Br J Anaesth. 2011;107:30–7. doi: 10.1093/bja/aer122. doi:10.1093/bja/aer122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yon JH, Carter LB, Reiter RJ, Jevtovic-Todorovic V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis. 2006;21:522–30. doi: 10.1016/j.nbd.2005.08.011. doi:10.1016/j.nbd.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Sanders RD, Xu J, Shu Y, Fidalgo A, Ma D, Maze M. General anesthetics induce apoptotic neurodegeneration in the neonatal rat spinal cord. Anesth Analg. 2008;106:1708–11. doi: 10.1213/ane.0b013e3181733fdb. doi:10.1213/ane.0b013e3181733fdb. [DOI] [PubMed] [Google Scholar]
- 7.Sanders RD, Xu J, Shu Y, et al. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110:1077–85. doi: 10.1097/ALN.0b013e31819daedd. doi:10.1097/ALN.0b013e31819daedd. [DOI] [PubMed] [Google Scholar]
- 8.Slikker W, Jr, Zou X, Hotchkiss CE, et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–58. doi: 10.1093/toxsci/kfm084. doi:10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- 9.Wang C, Sadovova N, Hotchkiss C, et al. Blockade of N-methyl-d-aspartate receptors by ketamine produces loss of postnatal day 3 monkey frontal cortical neurons in culture. Toxicol Sci. 2006;91:192–201. doi: 10.1093/toxsci/kfj144. doi:10.1093/toxsci/kfj144. [DOI] [PubMed] [Google Scholar]
- 10.Paule MG, Li M, Allen RR, et al. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol. 2011;33:220–30. doi: 10.1016/j.ntt.2011.01.001. doi:10.1016/j.ntt.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanders RD, Davidson A. Anesthetic-induced neurotoxicity of the neonate: time for clinical guidelines? Paediatr Anaesth. 2009;19:1141–6. doi: 10.1111/j.1460-9592.2009.03141.x. doi:10.1111/j.1460-9592.2009.03141.x. [DOI] [PubMed] [Google Scholar]
- 12.Rizzi S, Ori C, Jevtovic-Todorovic V. Timing versus duration: determinants of anesthesia-induced developmental apoptosis in the young mammalian brain. Ann N Y Acad Sci. 2010;1199:43–51. doi: 10.1111/j.1749-6632.2009.05173.x. doi:10.1111/j.1749-6632.2009.05173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. doi:10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 14.Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA. 2002;99:15089–94. doi: 10.1073/pnas.222550499. doi:10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikonomidou C, Bittigau P, Ishimaru MJ, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. doi:10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 16.Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–27. doi: 10.1016/j.neuroscience.2005.03.064. doi:10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 17.Hansen TG, Pedersen JK, Henneberg SW, et al. Academic performance in adolescence after inguinal hernia repair in infancy: a nationwide cohort study. Anesthesiology. 2011;114:1076–85. doi: 10.1097/ALN.0b013e31820e77a0. [DOI] [PubMed] [Google Scholar]
- 18.Dimaggio C, Sun LS, Ing C, Li G. Pediatric anesthesia and neurodevelopmental impairments: a Bayesian meta-analysis. J Neurosurg Anesthesiol. 2012;24:376–81. doi: 10.1097/ANA.0b013e31826a038d. doi:10.1097/ANA.0b013e31826a038d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiMaggio C, Sun LS, Kakavouli A, Byrne MW, Li G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J Neurosurg Anesthesiol. 2009;21:286–91. doi: 10.1097/ANA.0b013e3181a71f11. doi:10.1097/ANA.0b013e3181a71f11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiMaggio C, Sun LS, Li G. Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesth Analg. 2011;113:1143–51. doi: 10.1213/ANE.0b013e3182147f42. doi:10.1213/ANE.0b013e3182147f42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ing C, Dimaggio C, Whitehouse A, et al. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics. 2012;130:e476–85. doi: 10.1542/peds.2011-3822. doi:10.1542/peds.2011-3822. [DOI] [PubMed] [Google Scholar]
- 22.Flick RP, Katusic SK, Colligan RC, et al. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011;128:e1053–61. doi: 10.1542/peds.2011-0351. doi:10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sprung J, Flick RP, Katusic SK, et al. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clinic Proc. 2012;87:120–9. doi: 10.1016/j.mayocp.2011.11.008. doi:10.1016/j.mayocp.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilder RT, Flick RP, Sprung J, et al. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. doi:10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Block RI, Thomas JJ, Bayman EO, Choi JY, Kimble KK, Todd MM. Are anesthesia and surgery during infancy associated with altered academic performance during childhood? Anesthesiology. 2012;117:494–503. doi: 10.1097/ALN.0b013e3182644684. doi:10.1097/ALN.0b013e3182644684. [DOI] [PubMed] [Google Scholar]
- 26.Bartels M, Althoff RR, Boomsma DI. Anesthesia and cognitive performance in children: no evidence for a causal relationship. Twin Res Hum Genet. 2009;12:246–53. doi: 10.1375/twin.12.3.246. doi:10.1375/twin.12.3.246. [DOI] [PubMed] [Google Scholar]
- 27.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. doi:10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 28.Johnson SA, Young C, Olney JW. Isoflurane-induced neuroapoptosis in the developing brain of nonhypoglycemic mice. J Neurosurg Anesthesiol. 2008;20:21–8. doi: 10.1097/ANA.0b013e3181271850. doi:10.1097/ANA.0b013e3181271850. [DOI] [PubMed] [Google Scholar]
- 29.Ma D, Williamson P, Januszewski A, et al. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology. 2007;106:746–53. doi: 10.1097/01.anes.0000264762.48920.80. doi:10.1097/01.anes.0000264762.48920.80. [DOI] [PubMed] [Google Scholar]
- 30.Fredriksson A, Ponten E, Gordh T, Eriksson P. Neonatal exposure to a combination of N-methyl-d-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology. 2007;107:427–36. doi: 10.1097/01.anes.0000278892.62305.9c. doi:10.1097/01.anes.0000278892.62305.9c. [DOI] [PubMed] [Google Scholar]
- 31.Young C, Jevtovic-Todorovic V, Qin YQ, et al. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146:189–97. doi: 10.1038/sj.bjp.0706301. doi:10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cattano D, Young C, Straiko MM, Olney JW. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth Analg. 2008;106:1712–4. doi: 10.1213/ane.0b013e318172ba0a. doi:10.1213/ane.0b013e318172ba0a. [DOI] [PubMed] [Google Scholar]
- 33.Liang G, Ward C, Peng J, Zhao Y, Huang B, Wei H. Isoflurane causes greater neurodegeneration than an equivalent exposure of sevoflurane in the developing brain of neonatal mice. Anesthesiology. 2010;112:1325–34. doi: 10.1097/ALN.0b013e3181d94da5. doi:10.1097/ALN.0b013e3181d94da5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satomoto M, Satoh Y, Terui K, et al. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–37. doi: 10.1097/ALN.0b013e3181974fa2. doi:10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- 35.Sanders RD, Sun P, Patel S, Li M, Maze M, Ma D. Dexmedetomidine provides cortical neuroprotection: impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta Anaesthesiol Scand. 2010;54:710–6. doi: 10.1111/j.1399-6576.2009.02177.x. [DOI] [PubMed] [Google Scholar]
- 36.Yahalom B, Athiraman U, Soriano SG, et al. Spinal anesthesia in infant rats: development of a model and assessment of neurologic outcomes. Anesthesiology. 2011;114:1325–35. doi: 10.1097/ALN.0b013e31821b5729. doi:10.1097/ALN.0b013e31821b5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stratmann G, Sall JW, May LD, et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 2009;110:834–48. doi: 10.1097/ALN.0b013e31819c463d. doi:10.1097/ALN.0b013e31819c463d. [DOI] [PubMed] [Google Scholar]
- 38.Sall JW, Stratmann G, Leong J, et al. Isoflurane inhibits growth but does not cause cell death in hippocampal neural precursor cells grown in culture. Anesthesiology. 2009;110:826–33. doi: 10.1097/ALN.0b013e31819b62e2. doi:10.1097/ALN.0b013e31819b62e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sall JW, Stratmann G, Leong J, Woodward E, Bickler PE. Propofol at clinically relevant concentrations increases neuronal differentiation but is not toxic to hippocampal neural precursor cells in vitro. Anesthesiology. 2012;117:1080–90. doi: 10.1097/ALN.0b013e31826f8d86. doi:10.1097/ALN.0b013e31826f8d86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker SM, Yaksh TL. Neuraxial analgesia in neonates and infants: a review of clinical and preclinical strategies for the development of safety and efficacy data. Anesth Analg. 2012;115:638–62. doi: 10.1213/ANE.0b013e31826253f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker SM, Westin BD, Deumens R, Grafe M, Yaksh TL. Effects of intrathecal ketamine in the neonatal rat: evaluation of apoptosis and long-term functional outcome. Anesthesiology. 2010;113:147–59. doi: 10.1097/ALN.0b013e3181dcd71c. doi:10.1097/ALN.0b013e3181dcd71c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker SM, Grafe M, Yaksh TL. Intrathecal clonidine in the neonatal rat: dose-dependent analgesia and evaluation of spinal apoptosis and toxicity. Anesth Analg. 2012;115:450–60. doi: 10.1213/ANE.0b013e3182501a09. doi:10.1213/ANE.0b013e3182501a09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Peretich K, Zhao Y, Liang G, Meng Q, Wei H. Anesthesia-induced neurodegeneration in fetal rat brains. Pediatr Res. 2009;66:435–40. doi: 10.1203/PDR.0b013e3181b3381b. doi:10.1203/PDR.0b013e3181b3381b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palanisamy A, Baxter MG, Keel PK, Xie Z, Crosby G, Culley DJ. Rats exposed to isoflurane in utero during early gestation are behaviorally abnormal as adults. Anesthesiology. 2011;114:521–8. doi: 10.1097/ALN.0b013e318209aa71. doi:10.1097/ALN.0b013e318209aa71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schubert H, Eiselt M, Walter B, Fritz H, Brodhun M, Bauer R. Isoflurane/nitrous oxide anesthesia and stress-induced procedures enhance neuroapoptosis in intrauterine growth-restricted piglets. Intensive Care Med. 2012;38:1205–14. doi: 10.1007/s00134-012-2576-2. doi:10.1007/s00134-012-2576-2. [DOI] [PubMed] [Google Scholar]
- 46.Jelsing J, Nielsen R, Olsen AK, Grand N, Hemmingsen R, Pakkenberg B. The postnatal development of neocortical neurons and glial cells in the Gottingen minipig and the domestic pig brain. J Exp Biol. 2006;209(Pt 8):1454–62. doi: 10.1242/jeb.02141. doi:10.1242/jeb.02141. [DOI] [PubMed] [Google Scholar]
- 47.Brambrink A, Back S, Avidan M, Creeley C, Olney J. Ketamine and isoflurane anesthesia triggers neuronal and glial apoptosis in the neonatal macaque. Anesthesiology. 2010;A375 [Google Scholar]
- 48.Brambrink AM, Evers AS, Avidan MS, et al. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–41. doi: 10.1097/ALN.0b013e3181d049cd. doi:10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brambrink AM, Back SA, Riddle A, et al. Isoflurane-induced apoptosis of oligodendrocytes in the neonatal primate brain. Ann Neurol. 2012;72:525–35. doi: 10.1002/ana.23652. doi:10.1002/ana.23652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olney JW, Young C, Wozniak DF, Ikonomidou C, Jevtovic-Todorovic V. Anesthesia-induced developmental neuroapoptosis. Does it happen in humans? Anesthesiology. 2004;101:273–5. doi: 10.1097/00000542-200408000-00004. doi:10.1097/00000542-200408000-00004. [DOI] [PubMed] [Google Scholar]
- 51.Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–86. doi: 10.1038/nrn2372. doi:10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- 52.Alkire MT, Hudetz AG, Tononi G. Consciousness and anesthesia. Science. 2008;322:876–80. doi: 10.1126/science.1149213. doi:10.1126/science.1149213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siegel JM. Clues to the functions of mammalian sleep. Nature. 2005;437:1264–71. doi: 10.1038/nature04285. doi:10.1038/nature04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanders RD, Tononi G, Laureys S, Sleigh JW. Unresponsiveness≠unconsciousness. Anesthesiology. 2012;116:946–59. doi: 10.1097/ALN.0b013e318249d0a7. doi:10.1097/ALN.0b013e318249d0a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110:813–25. doi: 10.1097/ALN.0b013e31819b602b. doi:10.1097/ALN.0b013e31819b602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leveille F, Papadia S, Fricker M, et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci. 2010;30:2623–35. doi: 10.1523/JNEUROSCI.5115-09.2010. doi:10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edwards DA, Shah HP, Cao W, Gravenstein N, Seubert CN, Martynyuk AE. Bumetanide alleviates epileptogenic and neurotoxic effects of sevoflurane in neonatal rat brain. Anesthesiology. 2010;112:567–75. doi: 10.1097/ALN.0b013e3181cf9138. doi:10.1097/ALN.0b013e3181cf9138. [DOI] [PubMed] [Google Scholar]
- 58.Straiko MM, Young C, Cattano D, et al. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology. 2009;110:862–8. doi: 10.1097/ALN.0b013e31819b5eab. doi:10.1097/ALN.0b013e31819b5eab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanders RD, Hussell T, Maze M. Sedation & immunomodulation. Crit Care Clin. 2009;25:551–70. doi: 10.1016/j.ccc.2009.05.001. , ix. [DOI] [PubMed] [Google Scholar]
- 60.Pesic V, Milanovic D, Tanic N, et al. Potential mechanism of cell death in the developing rat brain induced by propofol anesthesia. Int J Dev Neurosci. 2009;27:279–87. doi: 10.1016/j.ijdevneu.2008.12.005. doi:10.1016/j.ijdevneu.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanders RD, Godlee A, Fujimori T, et al. Benzodiazepine augmented γ-amino-butyric acid signaling increases mortality from pneumonia in mice. Crit Care Med. 2013 doi: 10.1097/CCM.0b013e31827c0c8d. doi:10.1097/CCM.0b013e31827c0c8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou ZW, Shu Y, Li M, et al. The glutaminergic, GABAergic, dopaminergic but not cholinergic neurons are susceptible to anaesthesia-induced cell death in the rat developing brain. Neuroscience. 2011;174:64–70. doi: 10.1016/j.neuroscience.2010.10.009. doi:10.1016/j.neuroscience.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 63.Lydic R, Baghdoyan HA. Sleep, anesthesiology, and the neurobiology of arousal state control. Anesthesiology. 2005;103:1268–95. doi: 10.1097/00000542-200512000-00024. doi:10.1097/00000542-200512000-00024. [DOI] [PubMed] [Google Scholar]
- 64.Shu Y, Patel SM, Pac-Soo C, et al. Xenon pretreatment attenuates anesthetic-induced apoptosis in the developing brain in comparison with nitrous oxide and hypoxia. Anesthesiology. 2010;113:360–8. doi: 10.1097/ALN.0b013e3181d960d7. doi:10.1097/ALN.0b013e3181d960d7. [DOI] [PubMed] [Google Scholar]
- 65.Ponten E, Viberg H, Gordh T, Eriksson P, Fredriksson A. Clonidine abolishes the adverse effects on apoptosis and behaviour after neonatal ketamine exposure in mice. Acta Anaesthesiol Scand. 2012;56:1058–65. doi: 10.1111/j.1399-6576.2012.02722.x. doi:10.1111/j.1399-6576.2012.02722.x. [DOI] [PubMed] [Google Scholar]
- 66.Forcelli PA, Janssen MJ, Vicini S, Gale K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann Neurol. 2012;72:363–72. doi: 10.1002/ana.23600. doi:10.1002/ana.23600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu LX, Yon JH, Carter LB, Jevtovic-Todorovic V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis. 2006;11:1603–15. doi: 10.1007/s10495-006-8762-3. doi:10.1007/s10495-006-8762-3. [DOI] [PubMed] [Google Scholar]
- 68.Young C, Straiko MM, Johnson SA, Creeley C, Olney JW. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol Dis. 2008;31:355–60. doi: 10.1016/j.nbd.2008.05.009. doi:10.1016/j.nbd.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Creeley CE, Olney JW. The young: neuroapoptosis induced by anesthetics and what to do about it. Anesth Analg. 2010;110:442–8. doi: 10.1213/ANE.0b013e3181c6b9ca. doi:10.1213/ANE.0b013e3181c6b9ca. [DOI] [PubMed] [Google Scholar]
- 70.Zou X, Sadovova N, Patterson TA, et al. The effects of l-carnitine on the combination of, inhalation anesthetic-induced developmental, neuronal apoptosis in the rat frontal cortex. Neuroscience. 2008;151:1053–65. doi: 10.1016/j.neuroscience.2007.12.013. doi:10.1016/j.neuroscience.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 71.Zhao Y, Liang G, Chen Q, et al. Anesthetic-induced neurodegeneration mediated via inositol 1,4,5-trisphosphate receptors. J Pharmacol Exp Ther. 2010;333:14–22. doi: 10.1124/jpet.109.161562. doi:10.1124/jpet.109.161562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dzietko M, Felderhoff-Mueser U, Sifringer M, et al. Erythropoietin protects the developing brain against N-methyl-d-aspartate receptor antagonist neurotoxicity. Neurobiol Dis. 2004;15:177–87. doi: 10.1016/j.nbd.2003.10.006. doi:10.1016/j.nbd.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 73.Shih J, May LD, Gonzalez HE, et al. Delayed environmental enrichment reverses sevoflurane-induced memory impairment in rats. Anesthesiology. 2012;116:586–602. doi: 10.1097/ALN.0b013e318247564d. doi:10.1097/ALN.0b013e318247564d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taddio A, Katz J, Ilersich AL, Koren G. Effect of neonatal circumcision on pain response during subsequent routine vaccination. Lancet. 1997;349:599–603. doi: 10.1016/S0140-6736(96)10316-0. doi:10.1016/S0140-6736(96)10316-0. [DOI] [PubMed] [Google Scholar]
- 75.Anand KJ, Garg S, Rovnaghi CR, Narsinghani U, Bhutta AT, Hall RW. Ketamine reduces the cell death following inflammatory pain in newborn rat brain. Pediatr Res. 2007;62:283–90. doi: 10.1203/PDR.0b013e3180986d2f. doi:10.1203/PDR.0b013e3180986d2f. [DOI] [PubMed] [Google Scholar]
- 76.Shu Y, Zhou Z, Wan Y, et al. Nociceptive stimuli enhance anesthetic-induced neuroapoptosis in the rat developing brain. Neurobiol Dis. 2012;45:743–50. doi: 10.1016/j.nbd.2011.10.021. doi:10.1016/j.nbd.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 77.Liu JR, Liu Q, Li J, et al. Noxious stimulation attenuates ketamine-induced neuroapoptosis in the developing rat brain. Anesthesiology. 2012;117:64–71. doi: 10.1097/ALN.0b013e31825ae693. doi:10.1097/ALN.0b013e31825ae693. [DOI] [PubMed] [Google Scholar]
- 78.Sanders RD, Manning HJ, Robertson NJ, et al. Preconditioning and postinsult therapies for perinatal hypoxic–ischemic injury at term. Anesthesiology. 2010;113:233–49. doi: 10.1097/ALN.0b013e3181dc1b84. doi:10.1097/ALN.0b013e3181dc1b84. [DOI] [PubMed] [Google Scholar]
- 79.Krueger JM, Clinton JM, Winters BD, et al. Involvement of cytokines in slow wave sleep. Prog Brain Res. 2011;193:39–47. doi: 10.1016/B978-0-444-53839-0.00003-X. doi:10.1016/B978-0-444-53839-0.00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–44. doi: 10.1038/35094583. doi:10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 81.Sprung J, Flick RP, Wilder RT, et al. Anesthesia for cesarean delivery and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;111:302–10. doi: 10.1097/ALN.0b013e3181adf481. doi:10.1097/ALN.0b013e3181adf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stang A. Critical evaluation of the Newcastle–Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25:603–5. doi: 10.1007/s10654-010-9491-z. doi:10.1007/s10654-010-9491-z. [DOI] [PubMed] [Google Scholar]
- 83.Kalkman CJ, Peelen L, Moons KG, et al. Behavior and development in children and age at the time of first anesthetic exposure. Anesthesiology. 2009;110:805–12. doi: 10.1097/ALN.0b013e31819c7124. doi:10.1097/ALN.0b013e31819c7124. [DOI] [PubMed] [Google Scholar]
- 84.Roze JC, Denizot S, Carbajal R, et al. Prolonged sedation and/or analgesia and 5-year neurodevelopment outcome in very preterm infants: results from the EPIPAGE cohort. Arch Pediatr Adolesc Med. 2008;162:728–33. doi: 10.1001/archpedi.162.8.728. doi:10.1001/archpedi.162.8.728. [DOI] [PubMed] [Google Scholar]
- 85.Filan PM, Hunt RW, Anderson PJ, Doyle LW, Inder TE. Neurologic outcomes in very preterm infants undergoing surgery. J Pediatr. 2012;160:409–14. doi: 10.1016/j.jpeds.2011.09.009. doi:10.1016/j.jpeds.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 86.de Graaf J, van Lingen RA, Simons SH, et al. Long-term effects of routine morphine infusion in mechanically ventilated neonates on children's functioning: five-year follow-up of a randomized controlled trial. Pain. 2011;152:1391–7. doi: 10.1016/j.pain.2011.02.017. doi:10.1016/j.pain.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 87.Flick RP, Lee K, Hofer RE, et al. Neuraxial labor analgesia for vaginal delivery and its effects on childhood learning disabilities. Anesth Analg. 2011;112:1424–31. doi: 10.1213/ANE.0b013e3181f2ecdd. doi:10.1213/ANE.0b013e3181f2ecdd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McCann ME, Bellinger DC, Davidson AJ, Soriano SG. Clinical research approaches to studying pediatric anesthetic neurotoxicity. Neurotoxicology. 2009;30:766–71. doi: 10.1016/j.neuro.2009.02.013. doi:10.1016/j.neuro.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 89.Davidson AJ, McCann ME, Morton NS, Myles PS. Anesthesia and outcome after neonatal surgery: the role for randomized trials. Anesthesiology. 2008;109:941–4. doi: 10.1097/ALN.0b013e31818e3f79. doi:10.1097/ALN.0b013e31818e3f79. [DOI] [PubMed] [Google Scholar]
- 90.Sun LS, Li G, DiMaggio CJ, et al. Feasibility and pilot study of the Pediatric Anesthesia NeuroDevelopment Assessment (PANDA) project. J Neurosurg Anesthesiol. 2012;24:382–8. doi: 10.1097/ANA.0b013e31826a0371. doi:10.1097/ANA.0b013e31826a0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Faulkner S, Bainbridge A, Kato T, et al. Xenon augmented hypothermia reduces early lactate/N-acetylaspartate and cell death in perinatal asphyxia. Ann Neurol. 2011;70:133–50. doi: 10.1002/ana.22387. doi:10.1002/ana.22387. [DOI] [PubMed] [Google Scholar]