

Abstract
BACKGROUND
The response to treatment for asthma is characterized by wide interindividual variability, with a significant number of patients who have no response. We hypothesized that a genomewide association study would reveal novel pharmacogenetic determinants of the response to inhaled glucocorticoids.
METHODS
We analyzed a small number of statistically powerful variants selected on the basis of a family-based screening algorithm from among 534,290 single-nucleotide polymorphisms (SNPs) to determine changes in lung function in response to inhaled glucocorticoids. A significant, replicated association was found, and we characterized its functional effects.
RESULTS
We identified a significant pharmacogenetic association at SNP rs37972, replicated in four independent populations totaling 935 persons (P = 0.0007), which maps to the glucocorticoid-induced transcript 1 gene (GLCCI1) and is in complete linkage disequilibrium (i.e., perfectly correlated) with rs37973. Both rs37972 and rs37973 are associated with decrements in GLCCI1 expression. In isolated cell systems, the rs37973 variant is associated with significantly decreased luciferase reporter activity. Pooled data from treatment trials indicate reduced lung function in response to inhaled glucocorticoids in subjects with the variant allele (P = 0.0007 for pooled data). Overall, the mean (± SE) increase in forced expiratory volume in 1 second in the treated subjects who were homozygous for the mutant rs37973 allele was only about one third of that seen in similarly treated subjects who were homozygous for the wild-type allele (3.2 ± 1.6% vs. 9.4 ± 1.1%), and their risk of a poor response was significantly higher (odds ratio, 2.36; 95% confidence interval, 1.27 to 4.41), with genotype accounting for about 6.6% of overall inhaled glucocorticoid response variability.
CONCLUSIONS
A functional GLCCI1 variant is associated with substantial decrements in the response to inhaled glucocorticoids in patients with asthma. (Funded by the National Institutes of Health and others; ClinicalTrials.gov number, NCT00000575.)
Asthma is a complex genetic syndrome that affects 300 million persons worldwide.1 The response to treatment is also genetically complex and is characterized by high intraindividual repeatability2 and high interindividual variability,3 with up to 40% of patients with asthma having no response to therapy. Inhaled glucocorticoids are the most widely prescribed medications for controlling asthma. Levels of endogenous glucocorticoids are heritable and vary, both at baseline and in response to environmental perturbation.4–6 Moreover, studies in families with conditions other than asthma have shown both familial segregation and heritability in responses to glucocorticoid medications.7,8 Given the heritability within the therapeutic class of glucocorticoids, as well as the high degrees of between-patient variability and within-patient repeatability in the response to inhaled glucocorticoids for the treatment of asthma, it is likely that this response has a genetic basis.
To date, pharmacogenetic investigations in asthma have focused on candidate genes.9–13 A powerful family-based screening algorithm14 for genomewide association studies has identified novel genetic loci that contribute to obesity15,16 and Alzheimer’s disease.17 The algorithm uses parental genotype information to rank single-nucleotide polymorphisms (SNPs) that have the greatest potential power for association. A small subset of statistically powerful SNPs can then be tested in the probands with the use of the family-based association test (FBAT).18 Identifying markers in this fashion limits the potential for false-positive associations and increases the likelihood of replication in subsequent studies.
We hypothesized that a genomewide association study would identify novel variants associated with the response to inhaled glucocorticoids for asthma. We tested this hypothesis with the use of the family-based screening algorithm in subjects randomly assigned to inhaled glucocorticoids in the Childhood Asthma Management Program (CAMP).19,20 Through screening, we identified SNPs that offered the greatest power for a replicable association with the longitudinal response to inhaled glucocorticoids, measured as a change in forced expiratory volume in 1 second (FEV1). After screening, we tested the association of the highest-powered SNPs in four additional, independent populations drawn from clinical trials involving subjects with asthma.
METHODS
STUDY DESIGN AND SCREENING AND REPLICATION COHORTS
Figure 1 provides an overview of the study design. Detailed descriptions of the methods (including the population descriptors, functional characterization, and statistical approaches used) can be found in the Supplementary Appendix, available with the full text of this article at NEJM.org.
Figure 1. Study Design.
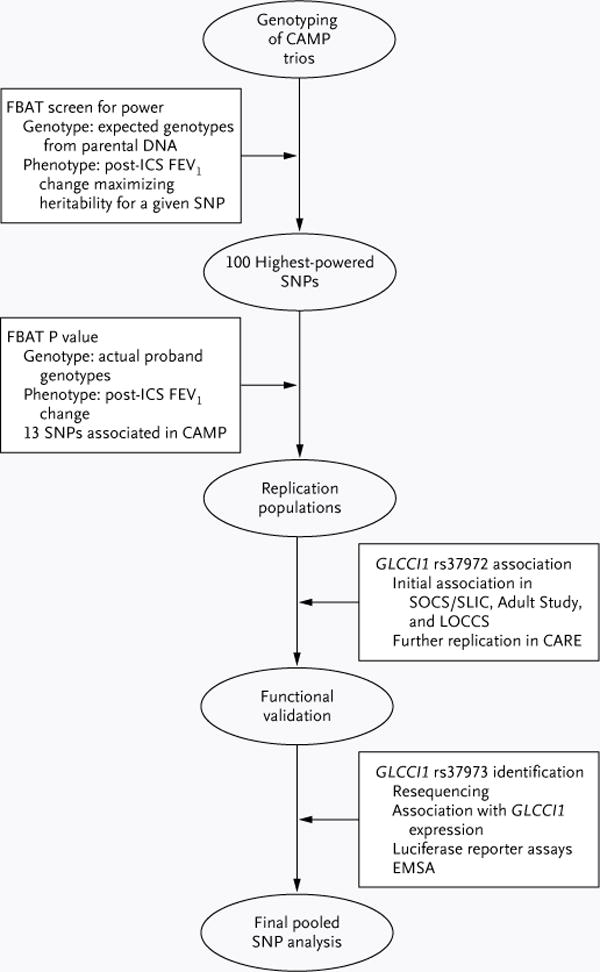
The family-based association test (FBAT) screening algorithm was applied to genomewide genotyping data in parent–child trios corresponding to probands randomly assigned to treatment with inhaled glucocorticoids (ICS) in the Childhood Asthma Management Program (CAMP), in order to identify the top 100 single-nucleotide polymorphisms (SNPs) in terms of power for replication (heritability) in association with changes in forced expiratory volume in 1 second (FEV1). These SNPs were tested for association in the CAMP population, and 13 variants were genotyped in three independent replication populations (in the Salmeterol or Corticosteroids [SOCS] and Salmeterol ± Inhaled Corticosteroids [SLIC] trials, the Adult Study, and the Leukotriene Modifier or Corticosteroid or Corticosteroid–Salmeterol [LOCCS] trial). One variant, rs37972, was associated in each population. The potential for function of this variant and a closely correlated variant, rs37973, was assessed, and rs37973 was confirmed to alter expression of the glucocorticoid-induced transcript 1 gene (GLCCI1); rs37973 was then tested for overall association with the response to ICS therapy in four independent clinical-trial populations. CARE denotes Childhood Asthma Research and Education, and EMSA electrophoretic mobility-shift assay.
In the CAMP,19,20 a randomized, controlled trial, we followed 1041 children with asthma who were 5 to 12 years of age at the onset of the study and who received treatment for a mean period of 4.6 years. The children were randomly assigned to treatment with inhaled budesonide, nedocromil sodium, or placebo. From this study, we selected 422 white, non-Hispanic participants and their parents for genotyping on the HumanHap550 v3 BeadChip (Illumina); of this group, 118 trios (consisting of a child and his or her two parents) were randomly assigned to budesonide. These trios constituted the family-based screening cohort in which we assessed the longitudinal response to glucocorticoid therapy. All research involving data collected from the CAMP Genetics Ancillary Study was conducted at the Channing Laboratory of the Brigham and Women’s Hospital according to the appropriate CAMP policies and regulations for human-subjects protection.
To replicate our initial findings, we genotyped DNA obtained from subjects with asthma who were enrolled in three clinical trials: the 6-week common run-in period during which the subjects were using inhaled glucocorticoids in the Salmeterol or Corticosteroids (SOCS) trial21 and the Salmeterol ± Inhaled Corticosteroids (SLIC) trial,22 the Adult Study,23 and the Leukotriene Modifier or Corticosteroid or Corticosteroid–Salmeterol (LOCCS) trial (ClinicalTrials.gov number, NCT00156819).24
After the initial replication phase, an additional replication was performed that was limited to the variant associated in each of the populations with the use of data from the Childhood Asthma Research and Education (CARE) Network trials,25,26 archived on the database of Genotypes and Phenotypes (dbGaP) (www.ncbi.nlm.nih.gov/gap) within the SNP Health Association Resource (SHARe) Asthma Resource Project (SHARP).
Table 1 summarizes these populations, most of which have been described in detail in reports on previous pharmacogenetic studies.27,28 All subjects or their legal guardians provided written informed consent to participate in the study protocols and ancillary genetic testing.
Table 1.
Characteristics of Screening and Replication Cohorts.*
| Characteristic | CAMP, Screening† | SOCS–SLIC Trials, First Replication | Adult Study, Second Replication | LOCCS Trial, Third Replication | CARE Network Trials, Fourth Replication‡ |
|---|---|---|---|---|---|
| No. of subjects | 118 | 264 | 385 | 185 | 101 |
| Inhaled glucocorticoid | Budesonide | Triamcinolone | Flunisolide | Fluticasone | Fluticasone |
| Age — yr | 9.0 ± 2.1 | 34.0 ± 11.6 | 39.2 ± 13.5 | 34.2 ± 15.5 | 10.9 ± 3.3 |
| Sex — no. of subjects (%) | |||||
| Male | 72 (61.0) | 109 (41.3) | 165 (42.9) | 67 (36.2) | 62 (61.4) |
| Female | 46 (39.0) | 155 (58.7) | 220 (57.1) | 118 (63.8) | 39 (38.6) |
| Baseline FEV1 — % of predicted | 93.5 ± 14.6 | 78.0 ± 15.9 | 71.7 ± 12.8 | 90.6 ± 9.6 | 99.6 ± 12.9 |
| Change in FEV1 —%§ | 7.4 ± 15.4 | 7.5 ± 20.1 | 7.2 ± 20.0 | 9.4 ± 12.5 | 7.7 ± 9.9 |
Plus–minus values are mean ± SD. Analyses included only white subjects to limit the possibility of associations due to population stratification. There were no significant differences between groups. CAMP denotes Childhood Asthma Management Program, CARE Childhood Asthma Research and Education, LOCCS Leukotriene Modifier or Cortico steroid or Corticosteroid–Salmeterol, SLIC Salmeterol ± Inhaled Corticosteroids, and SOCS Salmeterol or Cortico steroids.
This group of subjects represents a composite phenotype.
The CARE analysis included data on the response to inhaled glucocorticoids from the Characterizing the Response to a Leukotriene Receptor Antagonist and an Inhaled Corticosteroid (CLIC) trial and the Pediatric Asthma Controller Trial (PACT), archived on the dbGaP Web site (www.ncbi.nlm.nih.gov/gap) as part of the SNP Health Association Resource Asthma Resource Project. CARE data were evaluated for the sole purpose of determining associations with the glucocorticoid-induced transcript 1 gene (GLCCI1) single-nucleotide polymorphisms.
For comparability, values for the change in forced expiratory volume in 1 second (FEV1) are shown as the percent of the predicted value after 4 to 8 weeks of inhaled glucocorticoid therapy.
DRUG RESPONSES AND OUTCOMES
In the CAMP, the greatest differences in FEV1 that were attributable to budesonide were seen during the first 16 months of treatment.20 We calculated the change in FEV1 as the difference between FEV1 at baseline and during the five follow-up visits that took place within the 16 months after randomization (FEV1treatment–FEV1baseline). Residuals of each of these differences (adjusted for age, sex, and height) were used in the genomewide association study. For each replication cohort, a single measure of the change in FEV1 after 4 to 8 weeks of therapy with inhaled glucocorticoids was used as part of the clinical trial (i.e., the Adult Study and the CARE Network trials) or during a closely monitored runin period (i.e., the SOCS and SLIC trials and the LOCCS trial).
GENOTYPING AND QUALITY CONTROL
Genomewide HumanHap550v3 BeadChip SNP genotyping was performed by Illumina. Data were cleaned as previously described29 (Table 1 in the Supplementary Appendix), and 547,645 markers (97.54%) passed quality-control metrics. Genotyping was successful in 1169 CAMP participants, including 403 probands and their parents; for 11 probands, only 1 parent was available. The average rate of genotyping completion per study participant was 99.75%. Thirteen SNPs that met the screening criteria were genotyped with the use of Sequenom in the replication cohorts; the functional rs37973 SNP was genotyped with the use of TaqMan (Applied Biosystems). Average completion rates were higher than 95%. Genomewide Affymetrix SNP 6.0 Array genotype data were available for the SHARP and CARE Network samples. Because rs37972 and rs37973 were not genotyped on that platform, corresponding genotypes were inferred by means of imputation with the Markov Chain Haplotyping software30 on the basis of HapMap Phase 2 Release 22 data.31 The ratio of empirically observed to expected (binomial) dosage variance for these imputed SNPs was higher than 0.9, indicating good-quality imputation.
FUNCTIONAL VALIDATION
The methods described below were used for data acquisition (Fig. 3 in the Supplementary Appendix). Detailed descriptions of the methods used for functional analyses can be found in the Supplementary Appendix.
GLCCI1 Expression Profiles in Lymphoblastoid Cells
We correlated rs37972 and rs37973 with dexamethasone-mediated changes in glucocorticoid-induced transcript 1 gene (GLCCI1) expression in lymphoblastoid cell lines derived from 147 white probands in the CAMP population. Expression profiles were measured after stimulation for 6 hours with 10−6 M dexamethasone or a sham treatment with the use of the HumanRef-8v2 BeadChip and were adjusted for background, were log-transformed, and underwent variance stabilization and normalization. Associations of GLCCI1 expression with genotype were determined by means of linear regression analysis.
Luciferase Assay
We constructed luciferase reporter plasmids by cloning human DNA fragments amplified by means of polymerase chain reaction (PCR). The pGL4.23 vector contains a minimal promoter that facilitates the investigation of response-element activity. Products for the enhancer assay containing the wild-type sequence, the rs37972 or rs37973 variant, or the rs37972–rs37973 haplotype were subcloned into the upstream region of the pGL4.23–luciferase vector (Promega). We confirmed allelic differences in promoter activity in the human B-cell line Raji with the use of the pGL3-basic vector. Cells were transfected with these reporter constructs and with pRL-TK renilla luciferase vector as a normalization control with the use of the FuGENE 6 transfection reagent (Roche Diagnostics). Relative luciferase activity of mock and GLCCI1 reporter constructs was calculated as the ratio of firefly luciferase activity to renilla luciferase. Reporter assays were analyzed with the use of Student’s t-test.
Electrophoretic Mobility-Shift Assay
An electrophoretic mobility-shift assay was performed with the use of Gel Shift Assay Systems (Promega) according to the manufacturer’s instructions. Double-stranded rs37973 oligonucleotides end-labeled with [γ-32P] ATP with the use of T4 polynucleotide kinase were mixed with nuclear extracts from Jurkat, Raji, and human acute monocytic leukemia (THP-1) cells.
STATISTICAL ANALYSIS
FBAT is robust with respect to population stratification. The FBAT–Principal Components (FBAT-PC) method uses the five repeated measures of change in FEV1 in response to inhaled glucocorticoids for the CAMP population in a linear combination to generate an overall composite phenotype that maximizes the potential genetic contribution for each SNP locus, thereby yielding the highest power for a subsequent association analysis.32 This composite phenotype was used in the genomewide association study. We analyzed the data from that study with the use of the family-based screening algorithm, which ranks SNPs according to the power to detect an association without biasing the subsequent FBAT.14,18 We selected SNPs that met two criteria: they were among the 100 highest-powered SNPs and had a P value of 0.05 or less on FBAT. Thirteen SNPs met both criteria.
For the replication populations, we used generalized linear models to evaluate the association between SNPs and the change in FEV1. The analysis was adjusted for age, sex, and height under the same genetic model identified during screening, with the use of SAS software, version 9.1 (SAS Institute). A replication was defined as a result with a nominal P value of less than 0.05. A P value that combined evidence across all replication populations was calculated with the use of Liptak’s approach33,34 and was adjusted by means of the Bonferroni correction for multiple testing of the 12 genotyped SNPs in the replication populations. After genotyping the functional variant, we performed a final pooled analysis of the overall change in FEV1 and for association with the lowest versus highest quartile of the change in FEV1 by means of linear and logistic-regression analyses, adjusted for age, sex, height, and study.
RESULTS
INITIAL GENOMEWIDE ASSOCIATION STUDY AND REPLICATION ANALYSES
Table 1 summarizes the populations in the five clinical trials. As compared with the other three replication populations, the subjects in the CAMP and CARE Network trials were younger and predominantly male and had better lung function at baseline. In each trial, FEV1 significantly increased after the administration of inhaled glucocorticoids for 4 to 8 weeks but showed wide interindividual variability, as evidenced by the large standard deviations in the population response.
We performed the genomewide association screening in the CAMP trial, given its family-based design.14 Among 403 parent–child trios, 534,290 autosomal SNPs were available for analysis (Table 1 in the Supplementary Appendix). Trios containing 118 probands randomly assigned to treatment with budesonide (an inhaled glucocorticoid) underwent the family-based screening. Quantile–quantile plots (not shown) comparing the P values for FBAT allelic associations with those expected for a null distribution revealed a lambda value of 1.02 and were conservative in nature. After screening, the 100 highest-powered SNPs were evaluated for association. Table 2 shows the 13 SNPs selected for replication; 12 were successfully genotyped. Each SNP was in Hardy–Weinberg equilibrium.
Table 2.
Thirteen of the 100 Highest-Powered Single-Nucleotide Polymorphisms (SNPs), According to a Genomewide Screening Analysis with the Family-Based Association Test (FBAT).*
| SNP | Power Rank† | Model‡ | No. of Informative Families | P Value on FBAT | Chromosomal Position | Gene |
|---|---|---|---|---|---|---|
| rs6993479 | 5 | Dominant | 54 | 0.004 | chr8:71108153 | |
| rs1320125 | 26 | Additive | 84 | 0.006 | chr2:240936900 | |
| rs956133 | 32 | Additive | 84 | 0.043 | chr2:215531591 | ABCA12 |
| rs37972 | 38 | Additive | 88 | 0.010 | chr7:7974034 | GLCCI1 |
| rs10933595 | 43 | Additive | 80 | 0.021 | chr2:240940495 | |
| rs4282162 | 49 | Recessive | 55 | 0.028 | chr4:21426819 | KCNIP4 |
| rs2804311 | 61 | Recessive | 47 | 0.011 | chr9:545631 | ANKRD15 |
| rs2644645 | 83 | Dominant | 57 | 0.021 | chr5:174828792 | SFXN1 |
| rs10496195 | 92 | Recessive | 43 | 0.042 | chr2:74928440 | HK2 |
| rs7498886 | 93 | Recessive | 53 | 0.049 | chr16:60636703 | CDH8 |
| rs12446238 | 94 | Recessive | 53 | 0.049 | chr16:60632639 | CDH8 |
| rs2172706 | 95 | Additive | 72 | 0.003 | chr1:152935918 | |
| rs624964 | 100 | Dominant | 69 | 0.018 | chr10:11271837 | CUGBP2 |
Replication results for the 12 SNPs successfully genotyped in the initial three replication cohorts can be found in Table 2 in the Supplementary Appendix; data from the fourth replication cohort, participants in the Childhood Asthma Research and Education Network trials, were made available only after the initial replication analysis and were limited to the rs37972 variant from this table.
The data from the genomewide association study were analyzed with the use of the family-based screening algorithm, which ranks SNPs according to power to detect an association without biasing the subsequent FBAT results. After the ranking by power, the traditional FBAT was used to generate the reported P values.
Model refers to the type of genetic model identified as top-powered for the SNP power rank. Since the screening step did not limit the number of statistical comparisons, all three genetic models were ranked according to statistical power for subsequent association analyses.
One SNP, rs37972, was associated with the change in FEV1 in three of the four replication populations at a canonical P value of 0.05 or less under the same genetic model (additive) as in the CAMP population (P = 0.03 in the SOCS and SLIC trials, P = 0.03 in the LOCCS trial, P = 0.08 in the Adult Study, and P = 0.04 in the CARE Network trials), yielding a Liptak combined P value of 0.0007. Allowing for testing of 11 other genotyped SNPs, we found that the adjusted P value remained significant (P = 0.0085). In each population, the minor (T) rs37972 allele frequency was about 0.40. In a comparison of subjects who were homozygous for the wild-type allele (CC) with those who were homozygous for the mutant allele (TT), the mean (± SE) change in FEV1 after 4 to 8 weeks of treatment with inhaled glucocorticoids was 11.7 ± 2.6% versus 2.7 ± 3.8%, 9.5 ± 1.7% versus 3.1 ± 2.7%, 11.8 ± 1.8% versus 3.1 ± 2.7%, and 9.8 ± 1.6% versus 4.5 ± 2.8% for the populations in the SOCS and SLIC trials, the Adult Study, the LOCCS trial, and the CARE Network trials, respectively (Fig. 2).
Figure 2. Changes in Lung Function with Therapy According to rs37972 Genotype.
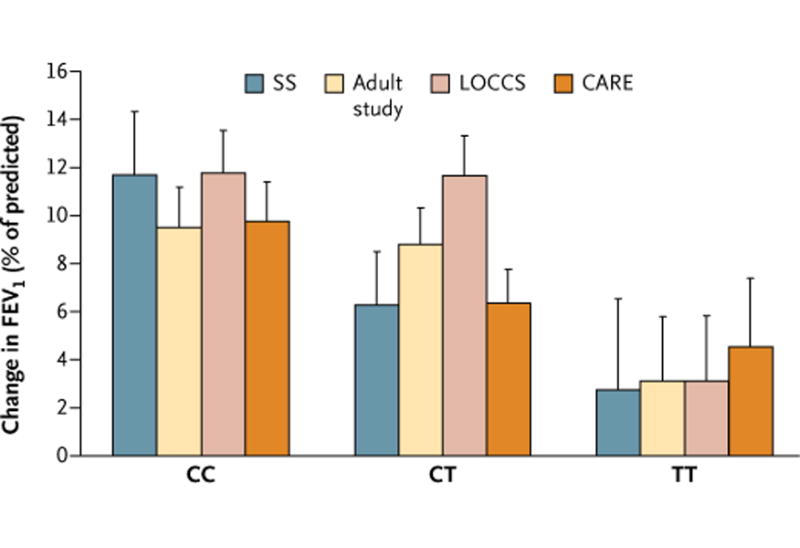
The association of genotypes from GLCCI1 rs37972 with changes in lung function is shown as the mean (± SE) change in forced expiratory volume in 1 second (FEV1), expressed as the percent of the predicted value, after 4 to 8 weeks of therapy with inhaled glucocorticoids in four replication populations: participants in the run-in periods of the SOCS and SLIC trials (SS) (264 participants), the Adult Study (385 participants), the LOCCS trial (185 participants), and the CARE Network trials (101 participants). For each population, the additive model showed a significant association, with poorer responses being associated with the mutant (T) allele (Liptak combined P = 0.0007). The rs37972 minor-allele frequencies for the SS, Adult Study, LOCCS, and CARE populations were 0.40, 0.38, 0.38, and 0.37, respectively. Since rs37972 correlates perfectly (is in complete linkage disequilibrium) with rs37973, the within-trial clinical response was identical for the latter variant (data not shown). CC denotes homozygous wild-type allele, CT heterozygous wild-type and mutant alleles, and TT homozygous mutant allele.
FUNCTIONAL CHARACTERIZATION
The rs37972 SNP is located on GLCCI1, 1473 bp in the 5′ direction from the ATG start site. To characterize the function of this SNP, we resequenced GLCCI1 and identified an allelic basis for transcriptional regulation. Resequencing identified five novel variants (Table 3 in the Supplementary Appendix); none were in significant linkage disequilibrium (i.e., highly correlated) with rs37972. We did, however, confirm that another GLCCI1 promoter variant, rs37973, was in complete linkage disequilibrium with rs37972 (r2 = 0.99) (Fig. 1 in the Supplementary Appendix).
GLCCI1 expression was measured in a panel of human tissues with the use of a reverse-transcriptase PCR assay, with glyceraldehyde-3-phosphate dehydrogenase serving as a nonvariant control. GLCCI1 was highly expressed in lung and lymphoid tissues, including T cells, B cells, and natural killer cells. Moreover, GLCCI1 was specifically induced in primary B cells treated with glucocorticoids under asthmalike conditions (Fig. 2 in the Supplementary Appendix).
In lymphoblastoid B cells derived from probands in the CAMP study, GLCCI1 expression was significantly increased by dexamethasone (β = 0.17 ± 0.05; P < 0.0001 for log2GLCCI1 expression). Moreover, subjects who were homozygous for the mutant allele, as compared with those who were homozygous for the wild-type allele, had significantly lower expression in sham-treated cells (additive model β = −0.16 ± 0.05; P = 0.003 for log2GLCCI1 expression) and dexamethasone-treated cells (additive model β = −0.16 ± 0.05; P = 0.002 for log2GLCCI1 expression) for both rs37972 and rs37973 (Fig. 3 in the Supplementary Appendix). Genotypic associations with GLCCI1 expression were confirmed on the basis of data from HapMap subjects35,36 (Fig. 4 in the Supplementary Appendix). Dexamethasone-stimulated GLCCI1 expression in the CAMP B-cell lines was associated with a robust response to glucocorticoids (highest quartile; odds ratio, 3.22; 95% confidence interval [CI], 1.41 to 7.38), a finding that was consistent with altered expression as the functional basis for the observed genotypic associations.
To clarify which SNP directly alters GLCCI1 expression, we constructed clones of luciferase reporter plasmids containing DNA fragments of rs37972, rs37973, or both (Fig. 3A). In the enhancer assay, the rs37972T–rs37973G (minor) haplotype clone had significantly less transcriptional activity than did the rs37972C–rs37973A (major) haplotype clone in all three cell lines studied (Jurkat, Raji, and THP-1) (Fig. 3B). Luciferase reporter activities of the rs37973G clones were reduced, a finding that was consistent with the haplotypes and indicated that rs37973 can modulate transcriptional activity (Fig. 3B). We confirmed the allelic differences in rs37973 transcriptional activity with the use of an independent promoter assay (Fig. 5 in the Supplementary Appendix).
Figure 3. Functional Analysis of GLCCI1 Allelic Variation.
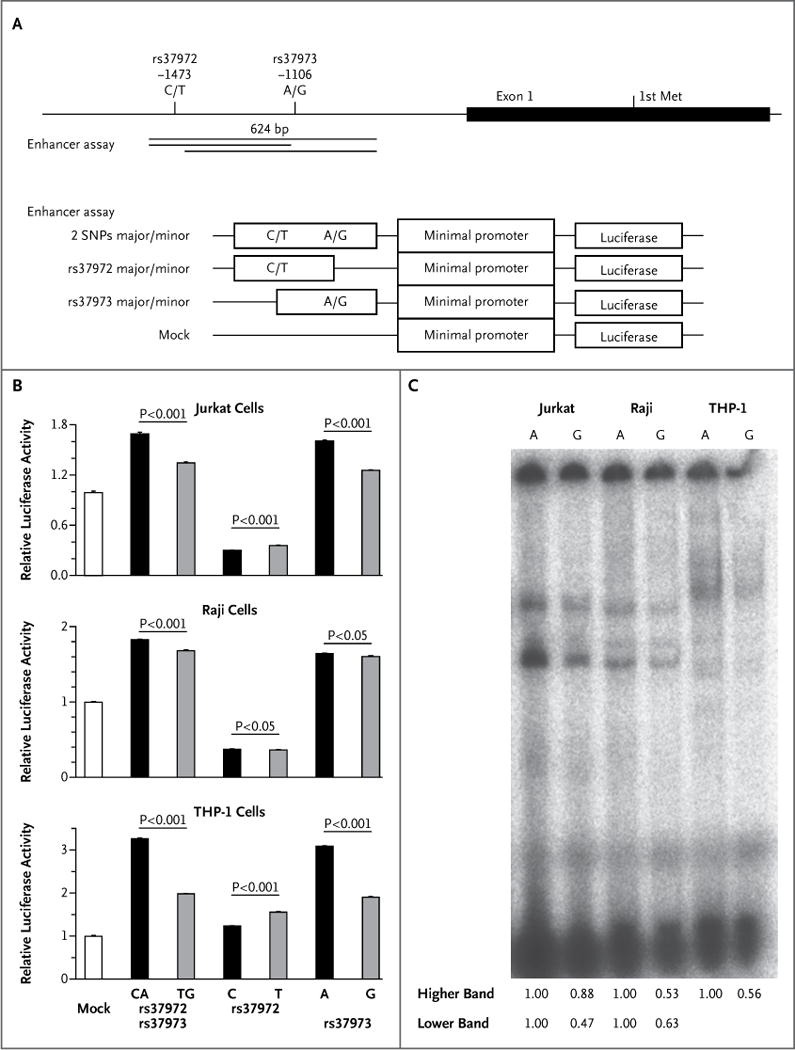
Panel A shows a graphical overview of the rs37972 and rs37973 single-nucleotide polymorphisms in relation to the first exon structure of GLCCI1. The position of the first methionine (Met), the start codon of GLCCI1, is shown. Indicated DNA fragments were cloned into luciferase vectors for the enhancer assay. Panel B shows the allelic differences in relative luciferase activity in Jurkat cells, Raji cells, and human acute monocytic leukemia (THP-1) cells, obtained with the use of a pGL4.23–luciferase vector. Data represent mean (± SD) values from one experiment performed in triplicate and are expressed relative to the luciferase activity of the mock transfectant, which was arbitrarily set at 1. P values were calculated with the use of Student’s t-test. Similar results were obtained in three independent experiments. Panel C shows results of an electrophoretic mobility-shift assay (EMSA) of rs37973 in Jurkat, Raji, and THP-1 cells. The intensity of each band on EMSA was analyzed by densitometry, and the intensity ratio of each G-allele band to each A-allele band is shown. Two independent experiments were performed with similar results. The genomic region containing the A allele of rs37973 was predicted to bind two transcription factors, CCAAT/enhancer binding protein (C/EBP) alpha and zinc finger E-box binding homeobox 1 (ZEB1). Although the signal intensity of the DNA–protein complex derived from the A allele was higher than that from the G allele in the presence of nuclear extract of each of the cell lines on EMSA, we could not confirm the binding of C/EBP alpha and ZEB1 to the sequence by supershift assay (data not shown). It is possible that other, unidentified transcription factors might bind to this sequence.
Electrophoretic mobility-shift assay of rs37973 also showed lower signal intensities of DNA–protein complexes for the G allele than for the A allele, in the presence of Jurkat, Raji, and THP-1 nuclear extracts (Fig. 3C). Although TRANSFAC (www.biobase-international.com/product/transcription-factor-binding-sites) predicted putative transcription-factor–binding sites at rs37973 CCAAT/enhancer binding protein (C/EBP) alpha (ACTTT-GTTCAATGC) and zinc finger E-box binding homeobox 1 (ZEB1) (ATGCAGGTTCCAG), we could not identify any specific binding of nuclear extracts to oligonucleotides containing rs37973.
FUNCTIONAL VARIANT ASSOCIATION
We genotyped the functional rs37973 in our replication populations and performed a pooled analysis of the change in FEV1 after therapy with inhaled glucocorticoids. The rs37973 SNP was associated with significant decrements in FEV1 (pooled P = 0.0007). Overall, FEV1 improved in response to inhaled glucocorticoids in subjects who were homozygous for the wild-type allele, as compared with those who were homozygous for the mutant allele (9.4 ± 1.1% vs. 3.2 ± 1.6%), consistent with the initial rs37972 association. We evaluated the association between rs37973 and response to inhaled glucocorticoids as defined by the highest and lowest quartiles of change in FEV1 (greater than 13.8% for the highest quartile and less than −1.7% for the lowest quartile) — that is, for the subjects in the lowest quartile of FEV1 response, improvement was less than 0%. After adjustment for age, sex, height, and study, the additive genetic model yielded an odds ratio for a poor response with rs37973 of 1.52 (95% CI, 1.13 to 2.03). Thus, subjects who were GG homozygotes were about two and a half times as likely to have a response in the lowest quartile (odds ratio, 2.36; 95% CI, 1.27 to 4.41) as were those who were AA homozygotes, with genotype accounting for an estimated 6.6% of overall response variability.
DISCUSSION
In our genomewide association study of more than 530,000 SNP markers with the use of a family-based screening algorithm, we identified 13 SNPs that were within the top 100 in statistical power for replication and associated with changes in FEV1 after the administration of inhaled glucocorticoids in the CAMP population. One GLCCI1 SNP, rs37972, was associated with the same phenotype with the use of the same genetic model in three of four independent populations of clinicaltrial populations with asthma, resulting in marked attenuation of the response to treatment with inhaled glucocorticoids (Fig. 2). This SNP is in complete linkage disequilibrium (i.e., perfectly correlated) with the functional rs37973, which down-regulates GLCCI1 expression — an effect augmented by exogenous glucocorticoids (Fig. 3, and Fig. 3 in the Supplementary Appendix). In turn, glucocorticoid-induced GLCCI1 expression in B-cell lines was significantly associated with the response to inhaled glucocorticoids in the subjects with asthma from whom the cells were obtained. Thus, it is fully plausible that rs37973 causes a decremental response to inhaled glucocorticoids in patients with asthma through changes in GLCCI1 expression.
In addition to the functional role of rs37973, we noted significant associations between this variant and clinical responsiveness to inhaled glucocorticoids. In subjects who were homozygous for the wild-type allele, FEV1 improved by a factor of three after therapy with inhaled glucocorticoids, as compared with those who were homozygous for the mutant allele (pooled P = 0.0007). Moreover, subjects who were homozygous for the mutant allele were about two and a half times as likely to have a poor response to inhaled glucocorticoids (lowest quartile) as were those who were homozygous for the wild-type allele. Overall, this variant accounted for 6.6% of the variability related to the lowest-quartile response, indicating that variation at this locus may account for a substantial proportion of patients who have a poor response to therapy with inhaled glucocorticoids.
In this study, we identified a consistent genetic association that was replicated in three of four independent clinical trials with a combined total of 935 subjects. The family-based screening method used to identify the associated variant is ideally suited to pharmacogenetic studies in which sample sizes are limited and P values for true associations are likely to be modest. In our study, the statistical power was further enhanced by the use of repeated measures of the outcome variable during FBAT screening of the CAMP population.32
GLCCI1 maps to chromosome 7p21.3 and contains eight exons. Its role in glucocorticoid signaling was first described by Chapman et al., who identified several differentially expressed sequenced tags in glucocorticoid-sensitive and glucocorticoid-resistant thymoma-derived cell lines after the administration of dexamethasone.37 One of these, GIG18 (glucocorticoid-induced gene 18), showed marked up-regulation. Now called GLCCI1, it is expressed in both lung cells and immune cells, and its expression is significantly enhanced by the presence of glucocorticoids in asthmalike conditions (Fig. 2 in the Supplementary Appendix). GLCCI1 may be an early marker of glucocorticoid-induced apoptosis.38 Apoptosis is a key mechanism through which glucocorticoids resolve lymphocytic and eosinophilic inflammation in asthma.39 Therefore, decreased GLCCI1 expression, as a result of rs37973, may reduce inflammatory-cell apoptosis, leading to a diminished clinical response to inhaled glucocorticoids.
Our study has several limitations. First, we specifically focused on 100 top-powered SNPs; the vast majority of SNPs in the genomewide association study were not tested. Clearly, a more comprehensive interrogation is warranted. Second, at enrollment, 85% of SOCS and SLIC participants reported recent use of inhaled glucocorticoids. Nonetheless, we noted a robust response to inhaled glucocorticoids during the common run-in period. Third, although our results would suggest that rs37973 has a functional role, other potential functional variants (e.g., alternative splice or intronic variants) may be present within GLCCI1. Although further mechanistic studies are needed to clarify the precise role of GLCCI1 in the response to asthma treatment, our elucidation of a functional variant to support the association finding far exceeds that of the typical genomewide study. Finally, owing to the small numbers of subjects in other racial groups, our analyses were restricted to whites, thereby limiting the generalizability of the results.
In conclusion, our genomewide association study of more than 530,000 SNPs revealed a novel functional SNP, rs37973, that decreases the expression of GLCCI1, a gene influencing the pharmacologic response to inhaled glucocorticoids in asthma.
Supplementary Material
Acknowledgments
Supported by grants from the National Institutes of Health (NIH U01 HL65899, P01 HL083069, R01 HG003646, R01 HL092197, and K23 HG3983), the NIH Pharmacogenomics Research Network, and the Ministry of Education, Culture, Sports, Science, and Technology in Japan. The Childhood Asthma Management Program (CAMP) Genetics Ancillary Study is supported by grants from the National Heart, Lung, and Blood Institute (NHLBI) (U01 HL075419, U01 HL65899, P01 HL083069, R01 HL086601, and T32 HL07427); the Single-Nucleotide Polymorphism Health Association Asthma Resource Project (SHARP) was funded by the NHLBI and was carried out by researchers from the Asthma Clinical Research Network, the CAMP, and the Childhood Asthma Research and Education (CARE) Network; the Asthma Clinical Research Network (ACRN) investigators and research teams were supported by grants from the NHLBI (U01 HL51510, U01 HL51834, U01 HL51831, U01 HL51845, U01 HL51843, M01 RR00079, and M01 RR03186); and the Leukotriene Modifier or Corticosteroid or Corticosteroid–Salmeterol (LOCCS) trial was supported by an unrestricted grant to the American Lung Association from GlaxoSmithKline. SHARP genotyping services were provided by Affymetrix under a contract (N02-HL-6-4278) from the NHLBI.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank all the CAMP subjects for their ongoing participation in this study; the CAMP investigators and research team for collection of CAMP Genetic Ancillary Study data; the American Lung Association Asthma Clinical Research Centers for the conduct of the LOCCS trial; and Drs. Kiego Nishida and Ji-Yang Wang of the Research Center for Allergy and Immunology for their technical advice and suggestions.
Footnotes
This article (10.1056/NEJMoa0911353) was published on September 26, 2011, at NEJM.org.
References
- 1.Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59:469–78. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 2.Drazen JM, Silverman EK, Lee TH. Heterogeneity of therapeutic responses in asthma. Br Med Bull. 2000;56:1054–70. doi: 10.1258/0007142001903535. [DOI] [PubMed] [Google Scholar]
- 3.Szefler SJ, Martin RJ, King TS, et al. Significant variability in response to inhaled corticosteroids for persistent asthma. J Allergy Clin Immunol. 2002;109:410–8. doi: 10.1067/mai.2002.122635. [DOI] [PubMed] [Google Scholar]
- 4.Inglis GC, Ingram MC, Holloway CD, et al. Familial pattern of corticosteroids and their metabolism in adult human subjects — the Scottish Adult Twin Study. J Clin Endocrinol Metab. 1999;84:4132–7. doi: 10.1210/jcem.84.11.6146. [DOI] [PubMed] [Google Scholar]
- 5.Ober C, Abney M, McPeek MS. The genetic dissection of complex traits in a founder population. Am J Hum Genet. 2001;69:1068–79. doi: 10.1086/324025. [Erratum, Am J Hum Genet 2002;70:284.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steptoe A, van Jaarsveld CH, Semmler C, Plomin R, Wardle J. Heritability of daytime cortisol levels and cortisol reactivity in children. Psychoneuroendocrinology. 2009;34:273–80. doi: 10.1016/j.psyneuen.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armaly MF. The heritable nature of dexamethasone-induced ocular hypertension. Arch Ophthalmol. 1966;75:32–5. doi: 10.1001/archopht.1966.00970050034007. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz JT, Reuling FH, Feinleib M, Garrison RJ, Collie DJ. Twin heritability study of the effect of corticosteroids on intraocular pressure. J Med Genet. 1972;9:137–43. doi: 10.1136/jmg.9.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drazen JM, Yandava CN, Dubé L, et al. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat Genet. 1999;22:168–70. doi: 10.1038/9680. [DOI] [PubMed] [Google Scholar]
- 10.Israel E, Chinchilli VM, Ford JG, et al. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet. 2004;364:1505–12. doi: 10.1016/S0140-6736(04)17273-5. [DOI] [PubMed] [Google Scholar]
- 11.Tantisira KG, Lake S, Silverman ES, et al. Corticosteroid pharmacogenetics: association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum Mol Genet. 2004;13:1353–9. doi: 10.1093/hmg/ddh149. [DOI] [PubMed] [Google Scholar]
- 12.Tantisira KG, Hwang ES, Raby BA, et al. TBX21: a functional variant predicts improvement in asthma with the use of inhaled corticosteroids. Proc Natl Acad Sci USA. 2004;101:18099–104. doi: 10.1073/pnas.0408532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tantisira KG, Silverman ES, Mariani TJ, et al. FCER2: a pharmacogenetic basis for severe exacerbations in children with asthma. J Allergy Clin Immunol. 2007;120:1285–91. doi: 10.1016/j.jaci.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Van Steen K, McQueen MB, Herbert A, et al. Genomic screening and replication using the same data set in family-based association testing. Nat Genet. 2005;37:683–91. doi: 10.1038/ng1582. [DOI] [PubMed] [Google Scholar]
- 15.Herbert A, Gerry NP, McQueen MB, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2006;312:279–83. doi: 10.1126/science.1124779. [DOI] [PubMed] [Google Scholar]
- 16.Lasky-Su J, Lyon HN, Emilsson V, et al. On the replication of genetic associations: timing can be everything! Am J Hum Genet. 2008;82:849–58. doi: 10.1016/j.ajhg.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertram L, Lange C, Mullin K, et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83:623–32. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM. Using the noninformative families in family-based association tests: a powerful new testing strategy. Am J Hum Genet. 2003;73:801–11. doi: 10.1086/378591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Childhood Asthma Management Program Research Group. The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Control Clin Trials. 1999;20:91–120. [PubMed] [Google Scholar]
- 20.Idem. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med. 2000;343:1054–63. doi: 10.1056/NEJM200010123431501. [DOI] [PubMed] [Google Scholar]
- 21.Lazarus SC, Boushey HA, Fahy JV, et al. Long-acting beta2-agonist monotherapy vs continued therapy with inhaled corticosteroids in patients with persistent asthma: a randomized controlled trial. JAMA. 2001;285:2583–93. doi: 10.1001/jama.285.20.2583. [DOI] [PubMed] [Google Scholar]
- 22.Lemanske RF, Jr, Sorkness CA, Mauger EA, et al. Inhaled corticosteroid reduction and elimination in patients with persistent asthma receiving salmeterol: a randomized controlled trial. JAMA. 2001;285:2594–603. doi: 10.1001/jama.285.20.2594. [DOI] [PubMed] [Google Scholar]
- 23.Bielory L, Picone F, Rabinowitz P, et al. Multicentre, randomised, parallel-group study of the efficacy and tolerability of flunisolide administered once daily via AeroChamber in the treatment of mild to moderate asthma. Clin Drug Investig. 2000;19:93–101. [Google Scholar]
- 24.Peters SP, Anthonisen N, Castro M, et al. Randomized comparison of strategies for reducing treatment in mild persistent asthma. N Engl J Med. 2007;356:2027, 39. doi: 10.1056/NEJMoa070013. [Erratum, N Engl J Med 2007;357:728.] [DOI] [PubMed] [Google Scholar]
- 25.Sorkness CA, Lemanske RF, Jr, Mauger DT, et al. Long-term comparison of 3 controller regimens for mild-moderate persistent childhood asthma: the Pediatric Asthma Controller Trial. J Allergy Clin Immunol. 2007;119:64–72. doi: 10.1016/j.jaci.2006.09.042. [Erratum, J Allergy Clin Immunol 2007;120:285.] [DOI] [PubMed] [Google Scholar]
- 26.Szefler SJ, Phillips BR, Martinez FD, et al. Characterization of within-subject responses to fluticasone and montelukast in childhood asthma. J Allergy Clin Immunol. 2005;115:233–42. doi: 10.1016/j.jaci.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Litonjua AA, Lasky-Su JA, Schneiter K, et al. ARG1 is a novel bronchodilator response gene: screening and replication in four asthma cohorts. Am J Respir Crit Care Med. 2008;178:688–94. doi: 10.1164/rccm.200709-1363OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tantisira KG, Lake S, Silverman ES, et al. Corticosteroid pharmacogenetics: association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum Mol Genet. 2004;13:1353–9. doi: 10.1093/hmg/ddh149. [DOI] [PubMed] [Google Scholar]
- 29.Himes BE, Hunninghake GM, Baurley JW, et al. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am J Hum Genet. 2009;84:581–93. doi: 10.1016/j.ajhg.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lange C, van Steen K, Andrew T, et al. A family-based association test for repeatedly measured quantitative traits adjusting for unknown environmental and/or polygenic effects. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1067. Article17. [DOI] [PubMed] [Google Scholar]
- 33.Liptak T. On the combination of independent tests. Magyar Tudomanyos Akademia Matematikai Kutato Intezetenek Kozlemenyei. 1958;3:171–97. [Google Scholar]
- 34.Won S, Morris N, Lu Q, Elston RC. Choosing an optimal method to combine P-values. Stat Med. 2009;28:1537–53. doi: 10.1002/sim.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stranger BE, Forrest MS, Clark AG, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1(6):e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stranger BE, Nica AC, Forrest MS, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–24. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chapman MS, Qu N, Pascoe S, et al. Isolation of differentially expressed sequence tags from steroid-responsive cells using mRNA differential display. Mol Cell Endocrinol. 1995;108:R1–R7. doi: 10.1016/0303-7207(95)03481-l. [DOI] [PubMed] [Google Scholar]
- 38.Chapman MS, Askew DJ, Kuscuoglu U, Miesfeld RL. Transcriptional control of steroid-regulated apoptosis in murine thymoma cells. Mol Endocrinol. 1996;10:967–78. doi: 10.1210/mend.10.8.8843413. [DOI] [PubMed] [Google Scholar]
- 39.Ho CY, Wong CK, Ko FW, et al. Apoptosis and B-cell lymphoma-2 of peripheral blood T lymphocytes and soluble fas in patients with allergic asthma. Chest. 2002;122:1751–8. doi: 10.1378/chest.122.5.1751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.