

Abstract
Background & Aims
Increased vascular density has been associated with progression of human inflammatory bowel diseases (IBDs) and animal models of colitis. Pathologic angiogenesis in chronically inflamed tissues is mediated by several factors that are regulated at specialized lipid rafts known as caveolae. Caveolin-1 (Cav-1), the major structural protein of caveolae in endothelial cells, is involved in the regulation of angiogenesis, so we investigated its role in experimental colitis.
Methods
Colitis was induced by administration of dextran sodium sulfate to wild-type and Cav-1−/− mice, as well as Cav-1−/− mice that overexpress Cav-1 only in the endothelium. Colon tissues were analyzed by histologic analyses. Leukocyte recruitment was analyzed by intravital microscopy; angiogenesis was evaluated by immunohistochemistry and in vivo disk assays.
Results
Cav-1 protein levels increased after the induction of colitis in wild-type mice. In Cav-1−/− mice or mice given a Cav-1 inhibitory peptide, the colitis histopathology scores, vascular densities, and levels of inflammatory infiltrates decreased significantly compared with controls. Lower levels of leukocyte and platelet rolling and adhesion colitis also were observed in Cav-1−/− mice and mice given a Cav-1 inhibitory peptide, compared with controls. Cav-1−/− mice that received transplants of wild-type bone marrow had a lower colitis score than wild-type mice. Data from mice that overexpress Cav-1 only in the endothelium indicated that endothelial Cav-1 is the critical regulator of colitis. Genetic deletion or pharmacologic inhibition of endothelial Cav-1 also significantly decreased vascular densities and angiogenesis scores, compared with controls.
Conclusions
Endothelial Cav-1 mediates angiogenesis in experimental colitis. Modulation of Cav-1 could provide a novel therapeutic target for IBD.
The inflammatory bowel diseases (IBDs) consisting of Crohn’s disease (CD) and ulcerative colitis (UC) are chronic inflammatory pathologies of the small and large intestine that affect millions of people worldwide. Study of IBD in clinical settings has led to observations of increased vascular density that have been verified and quantitated in human disease and experimental models of colitis.1,2 Recently, these increases in vascular density have been shown to correlate with disease activity, suggesting that pathologic angiogenesis influences the progression of IBD and experimental colitis.2 As such, pathologic angiogenesis refers to the growth of new vasculature in chronically inflamed tissue in a dysregulated manner that perpetuates the chronic nature of disease as we have described previously.2 There are multiple mediators of this angiogenic response, which include growth factors, cytokines and chemokines, targets of cell signaling, and others. Interactions between angiogenic mediators and differential regulation of them during different stages of disease are believed to regulate the pathologic angiogenic response. Specialized lipid rafts known as caveolae are the site of regulation for many known angiogenesis mediators including vascular endothelial growth factor, endothelial nitric oxide synthase (eNOS), and various inflammatory cytokines that elicit angiogenic responses. These cholesterol-rich domains formed by caveolin protein aggregates are well suited to support signal transduction; and, recently, interactions between caveolins, eNOS, and vascular endothelial growth factor receptors have been implicated as a critical part of the angiogenesis signaling cascade.3
Caveolins are structural proteins that aggregate to form flask-shaped caveolae on the plasma membrane of multiple cell types. These caveolae are involved in cell signaling and protein trafficking, and play a role in many diseases including cancer, atherosclerosis, diabetes, cardiovascular disease, fibrosis, and others.4 There are 3 caveolin proteins, caveolin-1 (Cav-1), caveolin-2 (Cav-2), and caveolin-3 (Cav-3), of these, Cav-1 is the major structural component of caveolae in endothelial cells. Cav-1, in addition to being a structural protein, has its own activity, which has led to some confusion. For example, Cav-1 is an inhibitor of eNOS and thus increased expression of Cav-1 decreases endothelial NO production; however, NO production by endothelial cells during angiogenesis depends on Cav-1’s structural function colocalizing eNOS within caveolae.3,5 This is because the structural signaling platform created by Cav-1 provides for proper signal integration to activate angiogenic responses. Thus, if Cav-1 is lost the signaling components are no longer organized correctly and endothelial cell signaling is perturbed.6,7 As such, Cav-1 is likely to be a necessary component of cytokine-induced angiogenesis. Recently, Cav-1 has been shown to participate in tumor necrosis factor receptor-1 signaling, which is involved in inflammation and angiogenesis during IBD, possibly through receptor cross-talk and in mediating tumor necrosis factor receptor-1 internalization.8 Thus, Cav-1 is clearly important to both angiogenic and inflammatory responses, but the role it plays in colitis is unknown.
Although there are no current models that reproduce human IBD exactly, the 3% dextran sodium sulfate (DSS) model of experimental colitis is used widely as an effective tool for studying crucial aspects of acute disease. Importantly, this model emulates several key pathophysiologic features of inflammation and angiogenesis known to be prominent in human IBD, and is useful for studying pathologic angiogenesis during colitis. We previously reported increased vascular density correlating with colitis disease severity, as is known to occur in human IBD, in this model.2 In addition, our previous data show that angiogenic stimulation during DSS colitis involves complex regulation of many pro-angiogenic and anti-angiogenic molecules.1 The relationship between Cav-1 and angiogenesis suggests that Cav-1 may mediate pathologic angiogenesis during DSS colitis. Here we report that inhibition of endothelial Cav-1 function results in attenuation of DSS-induced colitis either through genetic deletion or pharmacologic inhibition. These findings show an important role for Cav-1 in regulating colitis initiation and progression, and suggest that inhibition of Cav-1 function may be a useful option for the treatment of IBD.
Materials and Methods
Mice
Animals used in these studies were bred and housed at the Association for Assessment and Accreditation of Laboratory Animal Care, internationally accredited Louisiana State University Health Sciences Center–Shreveport animal resource facility, and maintained according to the National Research Council’s Guide for Care and Use of Laboratory Animals. Cav-1+/+ (Cav-1 wild type [WT]), Cav-1−/− (Cav-1 knockout), and Cav-1−/− mice that overexpress Cav-1 in the endothelium (Cav-1 RC) were bred in-house.9 The Cav-1+/+ and Cav-1−/− mice used herein were backcrossed 6 generations on the C57BL/6J mouse strain. All experiments were performed with approval of the Institutional Animal Care and Use Committee.
Three Percent DSS Model of Experimental Colitis
The 3% DSS colitis model was performed as we have reported previously.2 Briefly, 3% DSS was administered for 6 days in the drinking water of experimental animals. Animals were monitored for signs of disease including weight loss, stool consistency, occult blood, and gross rectal bleeding, which were factored together to establish a disease activity index as we have reported previously.2 Consumption of water was monitored and no significant differences in DSS water consumption was observed throughout the experiments between the experimental cohorts (supplementary Figure 1A; see supplementary material online at www.gastrojournal.org). Unpublished data examining the nature of DSS colitis and comparison of our data from all experimental groups used in these studies have shown that the disease activity index used to track progression of colitis is more informative than weight loss alone; however, weight loss data for all groups throughout the experimental period is reported in supplementary Figure 1B–D (see supplementary material online at www.gastrojournal.org). Measurements of mice colon length were taken immediately after death as an additional indicator of disease. Mice were given 3 mg/kg antennapedia peptide (AP) control or AP-Cav peptides via intraperitoneal injection once daily beginning on day 0 and continuing throughout the experiments.
Histopathologic Scoring of Three Percent DSS Colitis
Paraffin-embedded, H&E-stained, cross-sections of the distal colons were scored for pathology. Histopathologic scoring was performed in a double-blinded manner as we have reported previously; on a scale of 0–3 for no, slight, moderate, and severe inflammation, respectively; depth of injury on a scale of 0–3 for no, mucosal, mucosal and submucosal, and transmural injury, respectively; and crypt damage on a scale of 0–4 for no damage, basal one-third damaged, basal two-thirds damaged, only surface epithelium intact, and entire crypt epithelium lost, respectively.2 A score of 0 – 40 then was given to each sample based on multiplying the score of each parameter by the percentage of the tissue involved (1×, 0%–25%; 2×, 25%–50%; 3×, 50%–75%; and 4×, 75%–100%) and adding the totals together. The numbers of neutrophils and monocytes (via F4/80 immunohistochemical staining) were counted as the number of cells present per 40× field and averaged as we have reported previously.10
Tissue Angiogenic Index
Cross-sections of colon tissue from each animal were analyzed as we have reported previously.2 Slides were fixed and stained for PECAM-1 (CD31) and a Eclipse TE2000-S epiflourescent scope (Nikon, Melville, NY) was used to capture images as previously described.2 The total pixel area stained for PECAM-1 was divided by that stained for 4′,6-diamidino-2-phenylindole (DAPI) to calculate an angiogenic index.
Bone Marrow Chimeras
Bone marrow chimeras were made by irradiating recipient mice and replacing the bone marrow with that taken from donor mice. The mice were irradiated twice, 3 hours apart, at 500 rads in a gamma irradiator. In between irradiations the donor marrow was prepared. Briefly, femurs from donor mice were isolated and the bones were flushed with 10 mL of sterile Hank’s balanced salt solution into a 50-mL conical vial. This solution was allowed to settle, then filtered through a 100-μm cell strainer, spun down, and resuspended. The cells then were counted, spun down, and resuspended in an appropriate amount of sterile phosphate-buffered saline, allowing for 2 × 106 cells per tail-vein injection. After the second irradiation, donor marrow cells were injected into the recipient mice. Mice then were placed on antibiotic water for 2 weeks. The DSS models were started after 6 weeks.
Intravital Fluorescence Microscopy
Intravital microscopy was performed as we have described previously.11 Mice were anesthetized and given rhodamine-6G, and then carboxyflourescein diacetate succinimudyl ester-labeled platelets were injected. Interactions of leukocytes and platelets in colonic postcapillary venules (diameters, 20–40 μm) were recorded. Video analysis was performed and leukocytes were classified based on their interactions with the vascular wall by 3 criteria: free flowing, rolling (cells move more slowly than centerline blood flow), and adherent (cells remain stationary for >30 seconds). Rolling of platelets and leukocytes was expressed as the number of rolling cells per second per mm of blood vessel diameter and adherence was expressed as the number of cells per mm2 of venular surface area, as calculated assuming cylindric vessel shape.
In Vivo Angiogenesis Disk Assay
Angiogenesis disk assays were performed using Millipore 14-mm Plexiglas rings with a 0.59-mm hole and 13-mm, 0.45-μm filters (Millipore Inc, Billerica, MA) as we have described previously.12 Disks were assembled and colon lysates from either Cav-1+/+ control or DSS-treated mice were injected into the disk (100 μL of 16.75 μg/mL total protein cell lysate), sealed, and implanted subcutaneously into Cav-1−/− and Cav-1+/+ mice proximal to the left hind limb. Disks were left in place for 7 days, at which time the mice were killed and the skin surrounding the disks was removed. The skin then was imaged with a WPI minitron digital camera (MTV7266ND; WPI Inc, Sarasota, FL) and images were analyzed for the number of branch points emanating from major vessels. The skin also was scored for angiogenesis based on a score of 1–3 for normal skin with mild reactive serosa, mild to moderate reactive serosa with some increase in vascularity, and moderate to severe reactive serosa with increased vascularity, respectively.
Statistical Analyses
Statistical analyses were performed using the Student t test or analysis of variance with a Bonferroni posttest among all groups and are presented as the mean ± standard error of the mean (SEM). P values of less than .05, less than .01, or less than .0001 are significant as indicated in the figure legends. All experiments were replicated at least twice and experimental n values are reported in the figure legends.
Supplementary Data
Supplementary Figure 1 (see supplementary material online at www.gastrojournal.org) contains water consumption and weight loss data for all experimental groups. Supplementary Figure 2 (see supplementary material online at www.gastrojournal.org) contains sample images of normal colonic histology for Cav-1+/+, Cav-1−/−, and Cav-1 RC mice (supplementary Figure 2A–C, respectively). Supplementary Figure 2D (see supplementary material online at www.gastrojournal.org) shows a sample image of the histopathology of Cav-1 RC DSS-treated mice.
Results
Loss of Cav-1 Attenuates DSS Colitis
We examined Cav-1 expression by Western blot in Cav-1+/+ mice to determine if Cav-1 up-regulation occurs during DSS colitis (Figure 1A). Densitometric analysis of these data showed that Cav-1 expression significantly increased over time throughout the DSS model suggestive of Cav-1 involvement during colitis (Figure 1B). To determine what effect genetic deletion of Cav-1 has on experimental colitis, the DSS model was performed with Cav-1+/+ and Cav-1−/− mutant mice. The results of the disease activity index acquired during the model are shown in Figure 2A. Cav-1+/+ mice treated with DSS showed severe disease typical of this model with significant disease activity by day 3, with progressive severity throughout the model. Cav-1−/− mice showed significant protection against DSS colitis when compared with Cav-1+/+ mice, resulting in a much lower disease activity index. Moreover, characteristic colon shortening was significantly prevented in Cav-1−/− mice treated with DSS (Figure 2A, inset). Likewise, genetic deficiency of Cav-1 significantly prevented DSS-induced weight loss (supplementary Figure 1B). Enzyme-linked immunosorbent cytokine assays for expression of tumor necrosis factor-α, interferon-γ, interleukin (IL)-1β, IL-4, and IL-10 were performed between Cav-1+/+ and Cav-1−/− mice and are reported in Table 1. Genetic deficiency of Cav-1 did not significantly alter the expression of the typical inflammatory cytokines tumor necrosis factor-α, interferon-γ, or IL-1β. However, loss of Cav-1 expression did enhance IL-10 expression during DSS colitis. Figure 2B shows representative examples of DSS colon histopathology from Cav-1−/− and Cav-1+/+ DSS-treated mice. The top row of images show photomicrographs of least severe (left panel) and most severe, including frank ulceration (right panel), disease in Cav-1+/+ colons; whereas the bottom row shows the same for Cav-1−/− colons. Histopathologic scoring of these specimens revealed that the Cav-1−/− mice were significantly protected against inflammatory tissue damage vs Cav-1+/+ mice (Figure 2C). Histopathology scores for control mice were 0 and are not shown; images of the normal histology of Cav-1+/+ and Cav-1−/− colons are shown in supplementary Figure 2A and B, respectively. Figure 2D and E report the numbers of infiltrating neutrophils and monocytes, respectively, during DSS colitis. Genetic deficiency of Cav-1 significantly decreased the numbers infiltrating neutrophils and monocytes in response to DSS treatment. Together, these data show that Cav-1 expression plays a key role in modulating DSS-induced disease activity and tissue histopathology.
Figure 1.

Western blot of Cav-1 expression in Cav-1+/+ mice during colitis. (A) Western blot for Cav-1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control at days 0, 2, 4, and 6 of the DSS model. (B) Densitometry analysis of Cav-1 expression over time. n =4, **P >.01.
Figure 2.

Genetic deletion of Cav-1 protects against experimental colitis. (A) Disease activity index for Cav-1−/− and Cav-1+/+ DSS-treated mice. Top: colon lengths for Cav-1+/+ and Cav-1−/− experimental mice. (B) Sample images of H&E sections of more severe (left) and less severe (right) colitis for both groups. (C) Graph of the histology scores for both groups. (D and E) Number of neutrophils and monocytes counted per 40× field for Cav-1+/+ and Cav-1−/− mice. Statistics performed with the Student t test and comparisons for the disease activity index were made at each time point. n = 10, *P < .05.
Table 1.
Cytokine Profile for Cav-1+/+ and Cav-1−/− Experimental Groups
| Experimental cohorts | pg TNF-α/μg protein | pg IFN-γ/μg protein | pg IL-1β/μg protein | pg IL-4/μg protein | pg IL-10/μg protein |
|---|---|---|---|---|---|
| Cav-1+/+ control | 35.06 ± 14.40 | 8.06 ± 1.74 | 299.8 ± 60.62 | 30.83 ± 6.852 | 346.90 ± 21.27 |
| Cav-1+/+ DSS | 136.7 ± 31.88a | 20.96 ± 2.46a | 646.0 ± 229.5 | 25.40 ± 4.90 | 383.30 ± 66.67 |
| Cav-1−/− control | 24.11 ± 11.96 | 6.85 ± 3.04 | 69.02 ± 48.49 | 31.35 ± 5.73 | 215.60 ± 55.29 |
| Cav-1−/− DSS | 91.35 ± 18.67a | 19.35 ± 1.61a | 851.5 ± 378.1 | 23.01 ± 4.28 | 403.10 ± 26.70a |
NOTE. Values are shown in pg cytokine/μg total protein as measured by enzyme-linked immunosorbent assay of mouse colonic tissue lysates.
TNF, tumor necrosis factor.
P < .05, n = 4.
Pharmacologic Inhibition of Cav-1 Function Attenuates DSS Colitis
Studies have shown previously that interference of Cav-1 scaffolding domain function, through the use of cell-permeable peptide antagonists, suppresses Cav-1 inhibitory activity of eNOS and vascular leakage.7,13 Here we used the AP-Cav or AP control peptides during DSS colitis to determine whether therapeutic intervention targeting Cav-1 function could alter DSS colitis as seen in Cav-1−/− mice. AP-Cav peptide treatment significantly decreased DSS-induced disease activity vs mice treated with AP control peptide (Figure 3A). Likewise, AP-Cav treatment significantly prevented DSS-induced colon shortening compared with AP control peptide (Figure 3A, inset) as well as blunted DSS-induced weight loss (supplementary Figure 1C; see supplementary material online at www.gastrojournal.org). Analysis of colon histopathology was performed as described earlier for AP control (top row) and AP-Cav (bottom row) as illustrated in Figure 3B. Figure 3C shows that AP-Cav treatment conferred significant protection against DSS-induced histopathologic tissue damage compared with AP control treatment. Neutrophil and monocyte infiltrates also were measured between the peptide-treated mice that showed that AP-Cav peptide treatment significantly decreased the number of neutrophil infiltrates without a significant difference in the number of monocytic infiltrates (Figure 3D and E, respectively). Taken together these data verify that pharmacologic interruption of Cav-1 function attenuates DSS-induced disease in a manner similar to that seen in the mutant mouse studies.
Figure 3.

Pharmacologic inhibition of Cav-1 attenuates experimental colitis. (A) The disease activity index for mice treated with 3 mg/kg of AP control and AP-Cav delivered intra-peritoneally daily during the DSS model are shown. Top: colon lengths for AP control and AP-Cav–treated mice. (B) Sample H&E images of more severe (left) and less severe (right) colitis for both groups. (C) The histopathology scores are shown. (D and E) Numbers of neutrophils and monocytes counted per 40× field for both groups. (A) Statistics performed with the Student t test and comparisons for the disease activity index were made at each time point. n = 8, *P < .05. (E) P = .11.
Leukocyte and Platelet Recruitment Are Dependent on Cav-1
Leukocyte recruitment is increased in response to inflammation during DSS colitis.10 Our observation of decreased infiltrates in the Cav-1−/− mice prompted studies into leukocyte recruitment using the various Cav-1 models. Intravital microscopy was performed at day 7 before takedown to determine the effects of Cav-1 on recruitment of platelets and leukocytes during DSS colitis. Figure 4A reports a low number of basally adherent platelets in Cav-1+/+ control mice in contrast to Cav-1+/+ DSS-treated colons showing increased adherence of platelets. Conversely, DSS-induced platelet adhesion was decreased significantly in Cav-1−/− mice. Pharmacologic inhibition of Cav-1 function with AP-Cav also significantly decreased DSS-induced platelet adhesion compared with AP control peptide (Figure 4A). Figure 4B also shows that Cav-1+/+ DSS treatment leads to increased adherence of leukocytes to the endothelium, which is diminished significantly in Cav-1−/− mice or with AP-Cav peptide treatment compared with controls (Figure 4B). Lastly, both platelet and leukocyte rolling was increased significantly in response to DSS treatment in Cav-1+/+ mice, although it was decreased in Cav-1−/− mice or by treatment with AP-Cav peptide (Figure 4C and D). These data are consistent with measurements of tissue histopathology scores and leukocyte counts in DSS-treated Cav-1−/− mice and AP-Cav–treated DSS mice (Figures 2D and E, and 3D), thus providing strong evidence for the involvement of Cav-1 in mediating experimental colitis through regulation of leukocyte recruitment.
Figure 4.

Disruption of Cav-1 expression and function attenuates in vivo leukocyte recruitment. Intravital microscopy of the Cav-1+/+ control and DSS, Cav-1−/− DSS, AP-Cav DSS, and AP control DSS groups. (A and B) Numbers of firmly adherent platelets and leukocytes per mm2 observed in each group, respectively. (C and D) Numbers of slow rolling platelets and leukocytes, respectively. Statistics were performed by 1-way analysis of variance with a Bonferroni posttest among all groups. n = 8, *P < .05 vs control, #P < .05 vs Cav-1−/− DSS.
Colitis Is Dependent on Endothelial Cell Cav-1 Expression
Bone marrow chimeras were used to determine which compartment, local tissue, or immune system was responsible for the attenuation of disease seen in Cav-1−/− mice. Bone marrow from wild-type mice was transferred into irradiated wild-type mice as a control (WT/WT), these mice developed colitis normally as shown by the disease activity index in Figure 5A. Interestingly, when bone marrow taken from Cav-1−/− mice was transferred into wild-type mice (Cav-1−/−/WT), the incidence and severity of disease was not affected; however, transfer of wild-type bone marrow into Cav-1−/− mice (WT/Cav-1−/−) resulted in significant attenuation of disease (Figure 5A). DSS induced significant colon shortening in Cav-1−/−/WT but not WT/Cav-1−/− chimeras as shown in Figure 5A (inset). Likewise, DSS-induced weight loss also was increased significantly in Cav-1−/−/WT but not WT/Cav-1−/− chimeras (supplementary Figure 1D; see supplementary material online at www.gastrojournal.org). Figure 5B shows photomicrographs of the worst (left) and best (right) cases of histology in the WT/WT and WT/Cav-1−/− chimeras. A comparison of the histopathology scores from the WT/WT chimeras vs the WT/Cav-1−/− chimeras revealed significantly decreased histopathology in the WT/Cav-1−/− chimeras (Figure 5C). Measurement of the number of neutrophil infiltrates showed a significant reduction in the WT/Cav-1−/− chimeras when compared with that of the WT/WT chimeras (Figure 5D). Conversely, examination of the number of monocyte infiltrates showed no statistical significance (Figure 5E). Histopathology scores and leukocyte infiltrate data from Cav-1−/−/WT chimeras were similar to WT/WT chimeras (data not shown).
Figure 5.

Endothelial Cav-1 expression governs experimental colitis. (A) The disease activity index for WT/WT, Cav-1−/−/WT, WT/Cav-1−/− chimeras, and Cav-1 RC mice treated with DSS. Top: colon lengths for WT/Cav-1−/− and Cav-1−/−/WT chimera mice. (B) Sample images of H&E sections with less severe (left) and more severe (right) colitis for the WT/WT and the Cav-1−/−/WT groups. (C) The histology scores for these groups are shown. (D and E) Numbers of neutrophils and monocytes counted per 40× field for the WT/WT and the Cav-1−/−/WT groups. (A) Statistics performed with the Student t test vs WT/WT chimera controls and comparisons for the disease activity index were made at each time point. n = 7 for chimeras, n = 8 for Cav-1 RC, *P < .05, **P < .01. (E) P = .07.
To specifically address the importance of endothelial Cav-1 we used Cav-1 RC mice, Cav-1−/− mice that are reconstituted specifically with Cav-1 only in the endothelium. In these mice, endothelial expression of Cav-1 is driven by the preproendothelin promoter with re-expressed Cav-1 correcting cardiovascular defects present in the Cav-1−/− mice.9 Cav-1 RC mice treated with DSS showed a significant increase in disease activity similar to Cav-1+/+ mice treated with DSS (Figure 5A). Histopathology scores for Cav-1 RC DSS-treated mice were similar to those of WT/WT and Cav-1−/−/WT (data not shown). Sample images of Cav-1 RC control and DSS-treated colon histopathology are shown in supplementary Figure 2C and D (see supplementary material online at www.gastrojournal.org). These data clearly show that endothelial Cav-1 expression regulates colitis development and progression, highlighting the importance of the microvasculature during colitis. This is shown by the fact that Cav-1−/− bone marrow did not attenuate disease in wild-type mice, which express endothelial Cav-1, whereas WT/Cav-1−/− chimera mice showed attenuation, and by the fact that restoration of Cav-1 to the endothelium resulted in normal disease progression.
Loss of Cav-1 Ablates Pathologic Angiogenesis During DSS Colitis
We have shown previously that DSS colitis involves a pathologic angiogenic response in which increases in vascular density consisting of immature and torturous vessels regulate disease activity simultaneously with tissue pathology.2 This increased vascular density results in increased leukocyte recruitment contributing to the inflammatory response.2 Cav-1 regulation of angiogenesis likely is linked to this leukocyte recruitment through the ability of Cav-1 to govern angiogenesis.14 To determine what effect loss of Cav-1 has on disease-associated angiogenesis, we performed an angiogenic index analysis to determine blood vessel density in the various experimental groups. Figure 6A–C show sample images of control, Cav-1+/+ DSS-, and Cav-1−/− DSS-treated colons, respectively. In these images red staining is CD31 (PECAM-1), a marker of endothelial cells, and blue staining is DAPI nuclear counterstain (Figure 6A–C). Figure 6D reports quantitative measurement of the angiogenic index for control non–DSS-treated Cav-1+/+ and Cav-1−/− tissue, as well as angiogenic index measurements for Cav-1+/+ DSS, Cav-1−/− DSS, WT/WT chimera, WT/Cav-1−/− chimera, AP control, and AP-Cav groups. Genetic deletion or pharmacologic inhibition of endothelial Cav-1 resulted in significant decreases in vascular density to basal levels compared with control mice (Figure 6D). These data clearly show the importance of Cav-1 regulation of angiogenesis during DSS colitis and indicate that inhibition of this angiogenic response attenuates disease pathology.
Figure 6.

Loss of Cav-1 expression blunts pathologic angiogenesis during experimental colitis. (A–C) Sample images showing vascular density. Red represents CD31 staining and blue represents 4′,6-diamidino-2-phenylindole nuclear counter-stain. (A) Normal colon tissue from an untreated mouse. (B) Colitis in a Cav-1+/+ DSS-treated mouse. (C) Colitis in a Cav-1−/− DSS-treated mouse. (D) Comparison of the angiogenic indexes for all groups Cav-1−/− and Cav-1+/+ control, Cav-1−/− and Cav-1+/+ DSS, WT/WT and WT/Cav-1−/− chimera DSS, and AP control and AP-Cav peptide–treated DSS animals. Statistics were performed with the Student t test vs respective controls. n = 7 for chimeras, n = 8 for peptides, n = 10 for Cav-1 mutants, *P < .0001.
Loss of Cav-1 Attenuates Colitis-Induced Angiogenesis via Disk Assay
We next used the Millipore angiogenesis disk assay to confirm our observations that loss of Cav-1 diminishes disease activity through impairment of the pathologic angiogenic response. Disks were filled with lysate from DSS-treated C57BL/6J mice colons, which then were implanted into Cav-1+/+ or Cav-1−/− mice. Figure 7A and B show sample images of DSS lysate–treated Cav-1+/+ skin and Figure 7C and D show those of Cav-1−/− mice. The angiogenesis score and total number of branch points were measured and are shown in Figure 7E and F, respectively. These results show that in response to the DSS lysate, Cav- 1−/− mice had greatly reduced angiogenesis compared with Cav-1+/+ mice. These data show that loss of Cav-1 confers protection against experimental colitis by the role caveolae serve during the regulation of angiogenesis.
Figure 7.
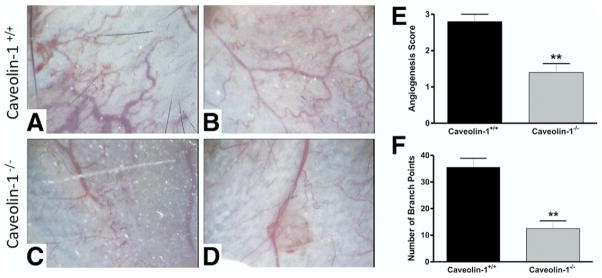
In vivo angiogenesis disk assay showing reduced angiogenesis in response to DSS colonic milieu in Cav-1−/− mice. (A and B) Sample images of skin from DSS lysate–treated Cav-1+/+ mice upon removal of the disk. (C and D) Sample images of skin from DSS lysate–treated Cav-1−/− mice upon removal of the disk. (E) The angiogenesis score and (F) the average total number of branch points for both groups. Statistics were performed by the Student t test. n = 6, **P < .01.
Discussion
The earlier experiments show that disruption of Cav-1, either by genetic deletion (Cav-1−/− mice) or pharmacologic inhibition (AP-Cav peptide), during DSS colitis resulted in attenuation of disease, suggesting that endothelial caveolae are crucial points for microvascular regulation of experimental colitis. Specifically, disruption of Cav-1 function significantly prevented weight loss and decreased tissue histopathology score, leukocyte recruitment, and angiogenic activity during disease. Importantly, these findings show endothelial Cav-1 to be necessary for the development of DSS colitis through the bone marrow chimera and Cav-1 RC experiments. This is likely owing to several primary functions of endothelial Cav-1, chief among them is the role of caveolae as a signaling platform that regulates angiogenic activity as seen during experimental colitis. In fact, disruption of Cav-1 has been shown to enhance vascular nitrosative stress and diminish NO bioavailability as a result of relocation of eNOS, as well as to disrupt cytokine stimulation of NO production and angiogenesis.6,15,16 In addition, we and others have suggested that microvascular dysfunction is a key aspect of experimental colitis and our data identify endothelial Cav-1 as a critical control point for microvascular regulation of colitis.1,17,18
We found that loss of endothelial Cav-1 reduces both the angiogenic and inflammatory responses known to occur in experimental colitis. It is possible that this loss of leukocyte recruitment, as a result of attenuated angiogenesis, decreases important signaling molecules necessary for the inflammatory response. For example, it has been shown that when Cav-1 is lost there is reduced colocalization of endothelial cell adhesion molecules and leukocyte-associated transmigration.14,16 Our intravital data showing decreased leukocyte and platelet adhesion are consistent with these previous reports and reinforce the importance of leukocyte recruitment for perpetuating pathologic angiogenesis during DSS colitis as we have reported previously.2 Importantly, the angiogenic response to DSS colitis was ablated completely in the Cav-1−/− mice although some degree of histopathology still occurred, suggesting that loss of Cav-1 interferes primarily with angiogenesis and vascular remodeling during experimental colitis.19 Inflammatory cytokine enzyme-linked immunosorbent assay data also showed that classic inflammatory cytokine production remained intact in DSS-treated Cav-1−/− mice. Interestingly, tissue levels of the immunomodulatory and anti-angiogenic cytokine IL-10 were increased significantly in Cav-1−/− mice treated with DSS compared with null mice given regular water. These data are intriguing and could indicate Cav-1 regulation of negative angiogenic mediators; however, further studies are required to substantiate this hypothesis because basal tissue levels of IL-10 were decreased with genetic deletion of Cav-1. Lastly, the angiogenesis disk assay data strongly indicate that the protection seen with the loss of Cav-1 is, in fact, owing to an impaired angiogenic response and not the result of inflammatory defects. These findings are crucial to the idea that pathologic angiogenesis drives chronic inflammation during IBD through positive feedback with the inflammatory response.
Cav-1 has been indicated in many aspects of the angiogenic response including endothelial transcytosis, vascular permeability, vascular reactivity, and tube formation. It is likely that Cav-1 regulation of angiogenesis in our model is complex and involves multiple vascular responses. One possible explanation of how Cav-1 could mediate angiogenesis in this model is through impairment of calcium signaling in Cav-1−/− endothelial cells as has been reported previously.20 Caveolae also may dictate endothelial membrane composition and surface expansion while being polarized in migrating endothelium, affecting signaling associated with cell migration.21 It has been shown that rear polarization of caveolae is specific to 2-dimensional cell movement and that during 3-dimensional endothelial transmigration caveolae are polarized to the front of the cell with differential polarization being dependent on Cav-1 binding to intermediate filaments.22,23 Importantly, the loss of Cav-1 polarity in activated endothelium prevents cell polarization and directional motility.24 Cav-1 also has been shown to enhance tube formation in Matrigel through regulation of endothelial cell differentiation and to be important for the maturation of newly formed blood vessels, indicating it as a marker of tumor vascular maturity.25,26 In addition, caveolae are crucial to the regulation of angiogenesis signaling and it has been shown that caveolins affect this signaling both within caveolae and in noncaveolar regions.27 One example of caveolae regulation of angiogenesis is transforming growth factor-β signaling, which occur in caveolae where transforming growth factor-β receptor activin receptor-like kinase 1 (ALK1) interacts with the Cav-1 scaffolding domain, thereby controlling receptor activation, which is necessary for angiogenic activity.28 Together, these data provide strong evidence for Cav-1 and the signaling platform it constructs being linked to the angiogenesis response and suggests that loss of Cav-1 is protective in colitis owing to diminished angiogenesis.
In summary, our findings make a compelling case for endothelial Cav-1 as a regulator of vascularity and inflammation during the progression of acute colitis. Because of the complexity of angiogenic and inflammatory responses during DSS colitis, further study will be necessary to determine how these responses influence one another. Moreover, the importance of Cav-1 function for angiogenic activity during mucosal healing after acute inflammatory flare remains unknown and is an important issue because interruption of wound healing–associated angiogenesis obviously would be undesirable. However, previous reports discussed earlier along with data shown here make a compelling argument for loss of Cav-1 affecting pathologic angiogenic responses during experimental colitis. Although the precise pathophysiologic mechanisms of endothelial Cav-1 during experimental colitis are not clear, these data reported here suggest that inhibition of Cav-1 could have therapeutic benefit in the treatment of human IBD.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants DK43785, DK65649, HL80482, HL61371, and HL57665.
Abbreviations used in this paper
- AP
antennapedia peptide
- Cav-1
caveolin-1
- DSS
dextran sodium sulfate
- eNOS
endothelial nitric oxide synthase
- IL
interleukin
- RC
Cav-1−/− mice that overexpress Cav-1 in the endothelium
- WT
wild type
Footnotes
The authors disclose no conflicts.
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2008.10.085.
References
- 1.Chidlow JH, Jr, Shukla D, Grisham MB, et al. Pathogenic angiogenesis in IBD and experimental colitis: new ideas and therapeutic avenues. Am J Physiol Gastrointest Liver Physiol. 2007;293:G5–G18. doi: 10.1152/ajpgi.00107.2007. [DOI] [PubMed] [Google Scholar]
- 2.Chidlow JH, Jr, Langston W, Greer JJ, et al. Differential angiogenic regulation of experimental colitis. Am J Pathol. 2006;169:2014–2030. doi: 10.2353/ajpath.2006.051021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sbaa E, Frerart F, Feron O. The double regulation of endothelial nitric oxide synthase by caveolae and caveolin: a paradox solved through the study of angiogenesis. Trends Cardiovasc Med. 2005;15:157–162. doi: 10.1016/j.tcm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Cohen AW, Hnasko R, Schubert W, et al. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 5.Sonveaux P, Martinive P, DeWever J, et al. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res. 2004;95:154–161. doi: 10.1161/01.RES.0000136344.27825.72. [DOI] [PubMed] [Google Scholar]
- 6.Schilling K, Opitz N, Wiesenthal A, et al. Translocation of endothelial nitric-oxide synthase involves a ternary complex with caveolin-1 and NOSTRIN. Mol Biol Cell. 2006;17:3870–3880. doi: 10.1091/mbc.E05-08-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernatchez PN, Bauer PM, Yu J, et al. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc Natl Acad Sci U S A. 2005;102:761–766. doi: 10.1073/pnas.0407224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Alessio A, Al-Lamki RS, Bradley JR, et al. Caveolae participate in tumor necrosis factor receptor 1 signaling and internalization in a human endothelial cell line. Am J Pathol. 2005;166:1273–1282. doi: 10.1016/S0002-9440(10)62346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murata T, Lin MI, Huang Y, et al. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdelbaqi M, Chidlow JH, Matthews KM, et al. Regulation of dextran sodium sulfate induced colitis by leukocyte beta 2 integrins. Lab Invest. 2006;86:380–390. doi: 10.1038/labinvest.3700398. [DOI] [PubMed] [Google Scholar]
- 11.Vowinkel T, Wood KC, Stokes KY, et al. Mechanisms of platelet and leukocyte recruitment in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1054–G1060. doi: 10.1152/ajpgi.00350.2007. [DOI] [PubMed] [Google Scholar]
- 12.Langston W, Chidlow JH, Jr, Booth BA, et al. Regulation of endothelial glutathione by ICAM-1 governs VEGF-A-mediated eNOS activity and angiogenesis. Free Radic Biol Med. 2007;42:720–729. doi: 10.1016/j.freeradbiomed.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bucci M, Gratton JP, Rudic RD, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 14.Millan J, Hewlett L, Glyn M, et al. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- 15.Wunderlich C, Schober K, Lange SA, et al. Disruption of caveolin-1 leads to enhanced nitrosative stress and severe systolic and diastolic heart failure. Biochem Biophys Res Commun. 2006;340:702–708. doi: 10.1016/j.bbrc.2005.12.058. [DOI] [PubMed] [Google Scholar]
- 16.Bouzin C, Brouet A, De Vriese J, et al. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol. 2007;178:1505–1511. doi: 10.4049/jimmunol.178.3.1505. [DOI] [PubMed] [Google Scholar]
- 17.Danese S, Dejana E, Fiocchi C. Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J Immunol. 2007;178:6017–6022. doi: 10.4049/jimmunol.178.10.6017. [DOI] [PubMed] [Google Scholar]
- 18.Hatoum OA, Binion DG, Otterson MF, et al. Acquired microvascular dysfunction in inflammatory bowel disease: loss of nitric oxide-mediated vasodilation. Gastroenterology. 2003;125:58–69. doi: 10.1016/s0016-5085(03)00699-1. [DOI] [PubMed] [Google Scholar]
- 19.Yu J, Bergaya S, Murata T, et al. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest. 2006;116:1284–1291. doi: 10.1172/JCI27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murata T, Lin MI, Stan RV, et al. Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J Biol Chem. 2007;282:16631–16643. doi: 10.1074/jbc.M607948200. [DOI] [PubMed] [Google Scholar]
- 21.Navarro A, Anand-Apte B, Parat MO. A role for caveolae in cell migration. FASEB J. 2004;18:1801–1811. doi: 10.1096/fj.04-2516rev. [DOI] [PubMed] [Google Scholar]
- 22.Parat MO, Anand-Apte B, Fox PL. Differential caveolin-1 polarization in endothelial cells during migration in two and three dimensions. Mol Biol Cell. 2003;14:3156–3168. doi: 10.1091/mbc.E02-11-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santilman V, Baran J, Anand-Apte B, et al. Caveolin-1 polarization in transmigrating endothelial cells requires binding to intermediate filaments. Angiogenesis. 2007;10:297–305. doi: 10.1007/s10456-007-9083-z. [DOI] [PubMed] [Google Scholar]
- 24.Beardsley A, Fang K, Mertz H, et al. Loss of caveolin-1 polarity impedes endothelial cell polarization and directional movement. J Biol Chem. 2005;280:3541–3547. doi: 10.1074/jbc.M409040200. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Wang XB, Park DS, et al. Caveolin-1 expression enhances endothelial capillary tubule formation. J Biol Chem. 2002;277:10661–10668. doi: 10.1074/jbc.M110354200. [DOI] [PubMed] [Google Scholar]
- 26.Dewever J, Frerart F, Bouzin C, et al. Caveolin-1 is critical for the maturation of tumor blood vessels through the regulation of both endothelial tube formation and mural cell recruitment. Am J Pathol. 2007;171:1619–1628. doi: 10.2353/ajpath.2007.060968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Head BP, Insel PA. Do caveolins regulate cells by actions outside of caveolae? Trends Cell Biol. 2007;17:51–57. doi: 10.1016/j.tcb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Santibanez JF, Blanco FJ, Garrido-Martin EM, et al. Caveolin-1 interacts and cooperates with the transforming growth factor-type I receptor ALK1 in endothelial caveolae. Cardiovasc Res. 2008;77:791–799. doi: 10.1093/cvr/cvm097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.