

Abstract
Background and Objective
Photodynamic therapy (PDT) is FDA-approved anti-cancer modality for elimination of early disease and palliation in advanced disease. PDT efficacy depends in part on elicitation of a tumor-specific immune response that is dependent on cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. The cytolytic potential of CTLs and NK cells is mediated by the ability of these cells to recognize major histocompatibility complex (MHC) class I and MHC class I-related molecules. The MHC class I-related molecules MICA and MICB are induced by oxidative stress and have been reported to activate NK cells and co-stimulate CD8+ T cells. The purpose of this study was to examine the effect of PDT on tumor cell expression of MHC classes I and II-related molecules in vivo and in vitro.
Study Design/Materials and Methods
Human colon carcinoma Colo205 cells and murine CT26 tumors were treated with 2-[1-hexyloxyethyl]-2-devinyl pyropheophor-bide-a (HPPH)-PDT at various doses. MHC classes I and I-related molecule expression following treatment of Colo205 cells was temporally examined by flow cytometry using antibodies specific for components of MHC class I molecules and by quantitative PCR using specific primers. Expression of MHC class I-related molecules following HPPH-based PDT (HPPH-PDT) of murine tumors was monitored using a chimeric NKG2D receptor.
Results
In vitro HPPH-PDT significantly induces MICA in Colo205 cells, but had no effect on MHC class I molecule expression. PDT also induced expression of NKG2D ligands (NKG2DL) following in vivo HPPH-PDT of a murine tumor. Induction of MICA corresponded to increased NK killing of PDT-treated tumor cells.
Conclusions
PDT induction of MICA on human tumor cells and increased expression of NKG2DL by murine tumors following PDT may play a role in PDT induction of anti-tumor immunity. This conclusion is supported by our results demonstrating that tumor cells have increased sensitivity to NK cell lysis following PDT.
Keywords: PDT, NKG2D, MICA, MHC class I
INTRODUCTION
Photodynamic therapy (PDT) causes tumor destruction through the generation of reactive oxygen species (ROS), vascular disruption, induction of inflammation, and enhancement of anti-tumor immunity [1]. ROS are produced upon illumination of tumors with a specific wavelength of light following administration of a photoreactive drug or photosensitizer. PDT is approved for clinical use in a number of countries, including the United States, for the elimination of early stage malignancies, palliation of symptoms, and reduction of obstruction in patients with late stage tumors [2,3].
PDT was initially considered to be primarily a local treatment; however, several studies have shown that local PDT treatment can lead to the induction of systemic anti-tumor immunity, resistance to subsequent tumor challenge [4,5], and an ability to combat distant disease [6–11]. Induction of anti-tumor immunity following PDT is dependent upon the presence of CD8+ T cells [4,5] and in some instances, natural killer (NK) cells [6]. While a majority of the studies demonstrating enhancement of anti-tumor immunity by PDT have been done in pre-clinical models by Abdel-Hady et al. [12] demonstrated that PDT outcome in patients can be linked to CD8+ T-cell infiltration. In addition, Kabingu et al. [13] showed that PDT of basal cell carcinoma (BCC) leads to enhanced immune recognition of a peptide derived from the BCC-associated antigen, HIP-1, when the peptide is presented in the context of the major histocompatibility complex (MHC) class I molecule HLA-A2.
Effector CD8+ T lymphocytes (CTLs) recognize target cells via engagement of T-cell receptor (TcR) with peptide-bearing classical MHC class I molecules on the surface of target cells. TcR:peptide:MHC class I binding results in activation of CTLs and killing of target cells [14]. NK cells express MHC class I-specific receptors that when engaged can inhibit NK cell killing of target cells [15]. Thus, recognition of tumor cells by both CD8+ T and NK cells depends upon expression of classical MHC class I molecules.
In addition to classical MHC class I molecules, both CD8+ T cells and NK cells are activated by recognition of MHC class I-like proteins on target cells [16]. These molecules are recognized by the NKG2D receptor and are globally denoted as NKG2D ligands (NKG2DL) [17,18]. Human NKG2DL include MHC-class I polypeptide-related chain (MIC) A and B and the cytomegalovirus UL16-binding proteins (ULBP) 1–4 and REATIG. Mice do not express MIC or ULBP proteins; ligands of murine NKG2D include the five retinoic acid early transcript 1 (RAE1) proteins, H60 and MULT-1.
NKG2DL recognition has been shown to play a major role in immune recognition and elimination of tumors [16,19–22]. NKG2DL expression is induced by many insults, including oxidative stress [23]. The dependence of PDT on CD8+ T cells and NK cells as well as the induction of NKG2DL by oxidative stress prompted us to examine the effect of 2-[1-hexyloxyethyl]-2-devinyl pyropheophor-bide-a (HPPH)-based PDT (HPPH-PDT) on the expression of classical and non-classical MHC class I molecules. HPPH is a second generation photosensitizer that was developed to overcome the side effects associated with the first generation sensitizer, sodium Porfimer [24]. HPPH-PDT has shown excellent efficacy in both pre-clinical and clinical studies [24,25] and was chosen for this research based on its ability to stimulate anti-tumor immunity [26].
We demonstrate that HPPH-PDT of colon carcinoma cells of human and murine origin results in increased expression of the NKG2DL, MICA. This increase in MICA expression is correlated with an increase in NK cytotoxicity against PDT-treated tumor cells.
MATERIALS AND METHODS
Cell Lines and Reagents
The human colon carcinoma cell line Colo205, murine colon carcinoma cell line CT26, and human NK cell line NK-92MI were purchased from ATCC (Manassas, VA). Colon carcinoma cell lines were grown in RPMI-1640 medium purchased from Life Technologies (Grand Island, NY), supplemented with 10% bovine calf serum purchased from Hyclone Laboratories (Logan, UT), and 10 mM HEPES. NK-92MI is a subclone of NK-92, which is an IL-2-dependent NK cell line derived from a patient with non-Hodgkin's lymphoma. NK-92MI was generated by transfection of the parental line with human IL-2 cDNA [27] and grows independent of IL-2. NK-92MI were propagated in alpha minimum essential medium supplemented with 2 mM l-glutamine, 1.5 g/L sodium bicarbonate, 0.2 mM inositol, 0.1 mM 2-mercaptoethanol; 0.02 mM folic acid, 12.5% horse serum, and 12.5% fetal bovine serum. All cells were grown in a Heraeus incubator (Fisher Scientific, Pittsburgh, PA) at 37°C and 5% CO2.
The transporter associated with antigen processing (TAP1)-specific antibody NOB-1, TAP-associated protein (tapasin)-specific antibody TO-3, and the MHC class I heavy chain-specific antibody TP25.99 have been previously described [28]. The mAb specific for human tubulin was purchased from Sigma–Aldrich (St. Louis, MO). Antibodies specific for STAT3 and the pan-specific antibodies for human classical MHC class I molecules were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). MICA antibody, Amo-1, and MICB antibody BMO-2 were purchased from Axxora (San Diego, CA). Antibodies to ULBPs and the murine NKG2D-Fc were purchased from R&D Systems (Minneapolis, MN). The CD45-specific antibody was purchased from BD Biosciences (Bedford, MA).
Animals
BALB/cJ mice purchased from Jackson Labs (Bar Harbour, ME) and housed in the Roswell Park Cancer Institute Animal Facility. Following tumor inoculation, animals were monitored daily. The Roswell Park Cancer Institute Institutional Animal Care and Use Committee (IACUC) approved all procedures carried out in this study.
Photodynamic Therapy
The clinical grade HPPH was obtained from the Roswell Park Pharmacy and reconstituted to 0.4 mmol/L in pyrogen-free 5% dextrose in water (D5W; Baxter Corp). Unless otherwise specified, for in vitro treatment with PDT, cells were incubated at 37°C and 5% CO2 with HPPH for 21 hours in the dark. Following the 21-hour incubation the excess photosensitizer was removed by a single wash in PBS. Cells were then incubated under the same conditions for an additional 3 hours. After the 3-hour incubation in the dark, cells were illuminated with 665 nm light given at 600–630 mW/cm2 using an argon laser-pumped dye laser (Spectra Physics, Mountain View, CA).
For in vivo PDT, animals were inoculated subcutaneously (s.c.) on their left shoulder with 3 × 105 CT26 a murine colon carcinoma cell line and allowed to grow for approximately 6 days or until they reached a size of 5 mm × 5 mm. Tumor growth was monitored every other day using HPPH was injected into the tail vein at a dose of 0.4 μmoles/kg. After 18–24 hours, the primary s.c. tumors were exposed to 665 nm light until a total fluence of 100 J/cm2 was achieved at a fluence rate of 75 mW/cm2. Animals treated with HPPH alone, were used as controls.
MTT Assay
An aliquot of cells was removed after the 21-hour incubation in photosensitizer or media (control cells) and plated in multiple wells of a 96-well plate at various concentrations. Plated cells that had not been incubated with the photosensitizer were incubated at 37°C and 5% CO2 for 3 hours and then either treated with light or not (light only control and untreated control, respectively) and so were the plates containing the cells incubated with HPPH (HPPH-PDT-treated and HPPH only control, respectively). Twenty-four hours after the treatment with PDT, 10 μl of a 4 mg/ml stock of MTT (Sigma–Aldrich) was added to all the wells and incubated in the dark for 3 hours. Following the 3-hour incubation during which the viable cells metabolized the MTT reagent to generate purple crystals, all the plates cells were spun down to preserve semi-adherent cells to the bottom of the plate and the MTT excess present in the media was removed. DMSO (100 μl) was added to all the wells and the plates were rocked at room temperature in the dark for 30 minutes. The optical density (OD) at 560 nm was determined. The photosensitizer only control was set as background given the dark toxicity observed at the HPPH concentration used during our studies.
Western Blotting
Quantitative fluorescent-based Western blotting was performed as described previously [29]. Briefly, cells were lysed using 1% Triton X-100 in 10 mM Tris, 150 mM NaCl, pH 7.4 supplemented with protease inhibitors PMSF (0.5 mM), and N-ethylmaleimide (5 mM), and incubated at 4°C for 30 minutes. Post-nuclear protein concentrations were determined by the Bradford assay (Bio Rad, Hercules, CA) and separated by SDS–PAGE. Following transfer of proteins to Immobilon-P membrane (Millipore, Bedford, MA), membranes were incubated in 3% bovine serum albumin (BSA) to reduce background signal. Proteins of interest were detected using the indicated primary antibodies (incubated overnight in an orbital shaker at 4°C). Primary antibodies were decanted and membranes were washed a minimum of three times in PBS/0.02% Tween 20. Secondary antibodies coupled to alkaline phosphatase were then added in 3% BSA and incubated at room temperature for 1 hour. After three washes in PBS/0.02% Tween 20, VistraECF substrate (Amersham Pharmacia, Piscataway, NJ) was applied and fluorescence was measured using a STORM instrument (Molecular Dynamics, Palo Alto, CA). Fluorescence measurements were acquired and analyzed using ImageQuant software.
Flow Cytometric Staining
Cells were washed and incubated for 60 minutes at 4°C with saturating concentrations of the indicated primary antibodies in FACS buffer. After washing three times to remove the excess primary antibody, 5 × 105 cells were resuspended in FACS buffer in saturating concentrations (10 μg/ml) of fluorescein isothiocyanate (FITC) or phycoerythrin (PE) conjugated, goat anti-mouse IgG antibodies for 30 minutes at 4°C in the dark. Cells were then washed three times to remove the excess secondary antibody and fixed in 2% paraformaldehyde. Isotype and experimental samples were analyzed on a FACScan instrument (Becton Dickinson, San Jose, CA). Data were analyzed with WINMDI software (Joe Trotter, The Scripps Research Institute, La Jolla, CA) or FlowJo (Ashland, OR).
RNA Extraction, RT-PCR, Q-PCR
RNA from cells was isolated using TRIZOL (Invitrogen, Carlsbad, CA) as per the manufacturer's recommendations. Two micrograms of RNA was reverse transcribed to generate cDNA using the Superscript II system (Invitrogen) and genes of interest were amplified using Accuprime taq (Invitrogen). For quantitative-PCR (Q-PCR), 2 μl of the cDNA reaction was used to amplify the indicated genes with the aid of SYBR green technology. Samples were set in triplicate wells and normalized to GAPDH and β-actin. The master mix used for the Q-PCR was purchased from Promega (Madison, WI) and the reactions were performed by Applied Biosystems HT7900 machine (Foster City, CA). The primers were designed at the 3′-end of the gene and always spanning an intron to easily detect genomic contamination if at all present. The 5′ to 3′ sequence of the primer sets were: β2-microglobulin (FP: GGCTATCCAGCGTACTCCAAA and RP: CGGCAGGCATACTCATCTTTTT), tapasin (FP: TATGCCTGTCGAATTCACCATCC and RP: TATGCCTGTCGAATTCACCATCC), heavy chain (FP: GATTACATCGCCTTGAACGAGG and RP: AGAGACAGCGTGGTGAGTCAT), GAPDH (FP: GAAGGTGAAGGTCGGAGTC and RP: GAAGATGGTGATGGGATTTC), β-actin (FP: TCCTGTGGCATCCACGAAA and RP: GCTGATCCACATCTGCTGGAA). Following either RT-PCR or Q-PCR, the amplicons size was confirmed by running the products in a 1.5–2.0% aga-rose gel in TAE. The sequence of the primer sets used to determine the induction of the indicated stress inducible genes were for MICA (FP: CCTTGGCCATGAACGTCAGG and RP: CGCAGCGAGGCCTCAGAGG), MICB (FP: ACC TTGGCTATGAACGTCACA and RP: CCCTCTGAGACCTCGCTGCA), ULBP-1 (FP: GTACTGGGAACAAATGCTGGAT and RP:AACTCTCCTCATCTGCCAGCT), ULBP-2 (FP: TTACTTCTCAATGGGAGACTGT and RP: TGTGCCTGAGGACATGGCGA), and ULBP-3 (FP: CCTGATGCACAGGAAGAAGAG and RP: TATGGCTTTGGGTTGAGCTAAG). All primer sets were purchased from Integrated DNA Technologies (San Diego, CA).
NK Cytotoxicity Assay
NK cytotoxicity was measured using a flow cytometry-based assay as previously described [30]. Colo205 cells were treated with HPPH-PDT (0.3 J/cm2) and incubated overnight at 37°C, 5% CO2. Following incubation, PDT-treated cells were washed once and labeled with a cyanine membrane dye 3,3′-dioctadecyloxacarbocyanine (DiO, 30 μM) by a 20-minute incubation at 37°C. NK92MI cells and the target cell lines were then incubated for 2 hours at 37°C in the presence of 75 μM propidium iodide (PI; Molecular Probes, Eugene, OR). After the incubation, the samples were analyzed by flow cytometry. A minimum of 2,000 target cells was analyzed per sample and dead target cells were identified as DiO+PI+. Results are presented as percent-specific lysis which was calculated as follows: percent-specific lysis = [(observed experimental lysis – the spontaneous lysis observed when target cells were incubated with medium alone)/(maximal lysis observed when target cells alone were incubated with 0.3% Triton X-100 – the spontaneous lysis)] × 100.
Statistical Analysis
Statistical analyses were performed using a standardized Student's t-test with Welch's correction, where equal variances were not assumed, to compare experimental groups. Differences were considered significant when P-values were ≤0.05.
RESULTS
Determination of PDT Efficacy on Colo205 Cells
Colo205 cells were subjected to HPPH-PDT to establish a dose–response curve. A summary of the in vitro treatment strategy can be found in Figure 1A; Colo205 cells incubated with HPPH at 0.8 μM were exposed to increasing fluences (0.05–1 J/cm2 administered at 600–630 mW/cm2) and their viability was determined 24 hours post-treatment by MTT assay (Fig. 1B). Subsequent studies were performed at LD30 (0.2–0.3 J/cm2) or over a dose range of LD30–LD99 (>1 J/cm2).
Fig. 1.

Determination of HPPH-PDT dose–response in Colo205 cells. A: Strategy for the in vitro treatment of Colo205 cells with HPPH-PDT. B: Colo205 cells were treated at several fluences and 24 hours post-HPPH-PDT their viability was determined by MTT. C: Colo205 cells were harvested immediately following treatment with HPPH-PDT at different fluences, lysed, and examined for the presence of STAT-3 cross-linking by Western blot.
Previous studies have shown that the signal transducer and activator of transcription 3 (STAT-3) is cross-linked following PDT treatment and that the magnitude of the cross-linking directly correlates with the combination of the photosensitizer and fluence [31]. To confirm the response to HPPH-PDT seen in Colo205 cells, the amount of STAT-3 cross-linking was determined by Western blot (Fig. 1C). As expected, the amount of cross-linked STAT-3 increased with treatments of higher fluence.
PDT Does Not Affect the Regulation, Protein Level or Surface Expression of Classical MHC Class I Molecules
Long-term control of PDT-treated tumors relies on CD8+ T cells and NK cells [4,5,32]. CD8+ T cells recognize antigens by ligation of TcR to MHC class I molecules, which are composed of MHC class I heavy chain, β2-microglobulin, and cognate peptide. It has been shown in several models that expression levels of MHC class I surface molecules regulates the CD8+ T cell and NK cell response to tumors [15,33]. To evaluate whether HPPH-PDT modulates the expression of classical MHC class I molecules, we examined the mRNA induction of MHC class I heavy chain and β2-microglobulin by Q-PCR on control (HPPH alone) and HPPH-PDT-treated Colo205 cells. RNA was isolated at different time points following treatment at 0.3 J/cm2 (LD30) to determine the effect on the indicated genes. Samples were analyzed by Q-PCR using the △△Ct method and normalized to housekeeping genes GAPDH and β-actin. Q-PCR analysis revealed the lack of an induction of the examined genes immediately, 1, 3, 6, 12, or 24 hours following treatment (Fig. 2A).
Fig. 2.

HPPH-PDT does not affect classical MHC class I molecule expression. A: Colo205 cells treated with HPPH-PDT at 0.3 J/cm2 were harvested immediately following, or 1, 3, 6, 12, and 24 hours post-treatment. cDNA generated from each of the time points was used in Q-PCR reactions using primers specific to β2-microglobulin (upper panel) or MHC class I heavy chain (lower panel). Induction was normalized to both GAPDH and β-actin. B: Total protein levels for the indicated proteins were examined 1, 6, 12, and 24 hours post-HPPH-PDT at 0.3 J/cm2. C: The surface expression of classical MHC class I molecules was determined as a function of fluence 24 hours post-HPPH-PDT. D: Surface levels of classical MHC class I molecules were examined as a function of time following HPPH-PDT at 0.8 J/cm2.
Surface expression of classical MHC class I molecules is controlled by several proteins in the endoplasmic reticulum (ER) [34]. Among the many proteins involved in assembly and loading of classical MHC class I molecules, the TAP and tapasin have been shown to be necessary for maximal expression at the cell surface [35,36]. Control or HPPH-PDT-treated (0.3 J/cm2) Colo205 cells were isolated 1, 6, 12, or 24 hours post-treatment and analyzed for the indicated proteins by Western blot. Analysis of total cell lysates revealed that HPPH-PDT did not affect the protein level of any of the tested proteins (Fig. 2B).
To rule out the possibility that the distribution of MHC class I molecules and not their total level is altered, cell surface expression of classical MHC class I molecules was determined pre-PDT and 24 hours post-PDT as a function of fluence. Cell surface expression of MHC class I molecules was not altered 24 hours after post-PDT at any of the tested fluences (Fig. 2C). Expression was also unchanged when cells treated with LD60 dose of HPPH-PDT were examined 48 hours post-treatment (Fig. 2D). In total these results suggest that factors other than MHC class I expression are responsible for increased NK and CD8+ T-cell cytolytic activity following PDT.
The NKG2DL MICA Is Induced Following PDT
To examine the effect of HPPH-PDT on NKG2DL expression, Colo205 cells were treated with HPPH alone or HPPH-PDT (0.3 J/cm2; LD30). Cells were examined by flow cytometric analysis using a MICA-specific antibody 24 hours following treatment. HPPH-PDT significantly enhanced MICA expression at the cell surface when compared to untreated or HPPH-treated cells (Fig. 3A). HPPH treatment alone also significantly increased MICA expression over that observed in untreated cells. The effect of HPPH-PDT on MICB was determined by RT-PCR. MICB mRNA is expressed constitutively by Colo205 cells and was unaltered by HPPH-PDT (Fig. 3B). The gene promoters of the NKG2DL contain heat shock responsive elements [18,23]; therefore hyperthermia was used as a positive control for gene expression. MICA expression was induced by treatment of Colo205 cells with hyperthermia (Fig. 3D); hyperthermia had no effect on MICB expression (data not shown).
Fig. 3.

The human NKG2D ligand MICA is induced following HPPH-PDT. A: Untreated Colo205 cells, HPPH only or treated with HPPH-PDT at 0.3 J/cm2 were examined 24 hours after treatment by flow cytometry for the expression of MICA at the cell surface. B: Changes in MICB mRNA levels were determined following HPPH-PDT 24 hours post-treatment. C: Colo205 cells treated with HPPH-PDT at 0.2, 0.8, and 1.6 J/cm2 were analyzed for the expression of ULBP family members at the cell surface by flow cytometry 24 hours following treatment. D: Colo205 cells were either left untreated or treated with hyperthermia, 6-hour incubation at 39.5°C followed by a 16-hour rest period prior analysis with the indicated antibodies.
Treatment of Colo205 cells with HPPH-PDT failed to induce cell surface expression of ULBP family members at any fluence tested (Fig. 3C). ULBP-2 and -3 expression was increased by treatment with hyperthermia (Fig. 3D), indicating that the lack of induction of ULBP family members was not due to an inability of Colo205 cells to express these ligands.
MICA expression can also be increased on colon carcinoma cells by hydrogen peroxide (H2O2) treatment [37]; therefore PDT induction of MICA on Colo205 cells was also compared to induction by H2O2. Colo205 cells were treated with HPPH-PDT at 0.3 J/cm2 or H2O2 at 50 and 250 μM. MICA surface expression was determined 24 hours post-treatment by flow cytometric analysis. H2O2 treatment resulted in a significantly higher induction of MICA when compared to HPPH-PDT (Fig. 4).
Fig. 4.
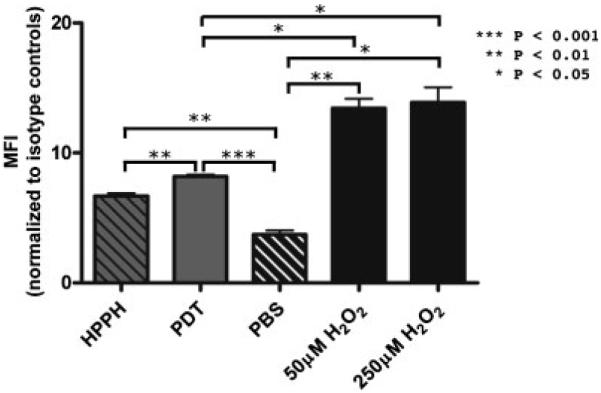
Comparison of MICA induction by HPPH-PDT and hydrogen peroxide. Colo205 cells treated with HPPH only or HPPH-PDT at 0.3 J/cm2 were examined 24 hours after treatment by flow cytometry for the expression of MICA at the cell surface. MICA levels were also examined in cells incubated with hydrogen peroxide. Results are shown as average mean fluorescence intensity (MFI); error bars represent SEM.
PDT Results in the Induction of NKG2D Ligands In Vivo
To determine if our findings translate to the in vivo setting, CT26 tumor-bearing animals were treated with HPPH-PDT at a tumor LD50 and the surviving cells were examined for the expression of MHC class I and NKG2DL expression. PDT-treated and control tumors were harvested 24 hours post-treatment and a single cell suspension was generated [38]. Tumor cells were distinguished from host immune cells as those cells lacking CD45. A chimeric NKG2D receptor moiety was used to detect NKG2DL expression by flow cytometry. NKG2DL expression was increased on tumor cells following HPPH-PDT treatment of CT26 tumors (Fig. 5); MHC class I expression was unchanged by PDT (data not shown).
Fig. 5.
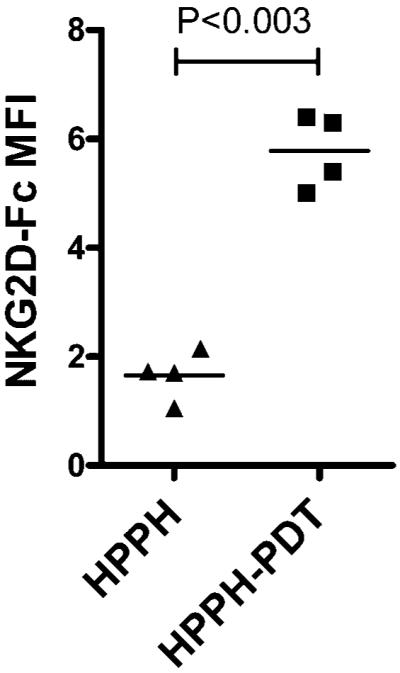
In vivo HPPH-PDT results in the significant induction of murine NKG2D ligands. BALB/c mice were inoculated with CT26 tumor cells in the left shoulder. Once the tumor reached a size of 5 × 5 mm2, mice were injected with HPPH, and 24 hours later half of the animals were illuminated with light of a specific wavelength, while the other were used as controls (HPPH alone). Tumor tissue was isolated 24 hours post-PDT and a single cell suspension was generated. Tumor cells (CD45−) were analyzed by flow cytometry for the expression of NKG2D ligands using an NKG2D-Fc chimera. Results are presented as NKG2D-Fc MFI; each symbol represents an individual tumor.
PDT Treatment Enhances NK Cytotoxicity
Previous studies have shown that increased expression of NKG2DL leads to enhanced NK cytotoxicity [16,19–22,39]. Therefore, we tested whether PDT-treated Colo205 cells showed increased sensitivity to NK lysis. Colo205 cells were treated with HPPH-PDT (0.3 J/cm2) or H2O2 (50 μM) and incubated for 24 hours to allow expression of MICA. Treated cells were then cultured in the presence of a human NK cell line (NK-92MI) and specific cytotoxicity was determined by flow cytometric analysis. Treatment with either HPPH-PDT or H2O2 significantly increased tumor cell susceptibility to NK lysis at effector to target ratios of 5:1 and 10:1 (P 0.01 when compared to cells treated with PBS alone); H2O2-treated target cells were also significantly more sensitive to lysis by NK-92MI cells at ratios of 25:1 and 50:1. These findings correspond to the degree of MICA induction found following treatment with HPPH-PDT and H2O2 (Fig. 6).
Fig. 6.
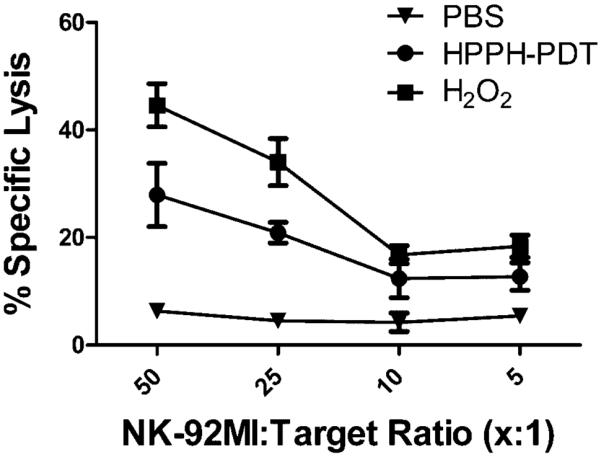
HPPH-PDT enhances NK killing of Colo205 cells. Colo205 cells were treated with PBS, HPPH-PDT (0.3 J/cm2) or H2O2 (50 μM). Following a 24-hour incubation cells were used as target cells in a NK cytotoxicity assay as described in Materials and Methods Section. Results are shown as mean-specific lysis ± SEM.
DISCUSSION
We show that PDT of human colon carcinoma cells in vitro and murine colon tumors in vivo leads to increased expression of non-classical MHC class I molecules. These molecules are recognized by the cytotoxicity receptor NKG2D expressed on CD8+ T cells and NK cells. The induction of NKG2DL expression in human Colo205 colon carcinoma cells by PDT appears to be limited to induction of MICA. We were unable to detect induction of either MICB or any of the ULBP family members. In contrast to the induction of MICA by PDT, treatment does not result in increased expression of either classical MHC class I molecules or molecules associated with antigen presentation by MHC class I molecules. Induction of MICA by HPPH-PDT corresponded to an increase in NK killing of the PDT-treated tumor cells.
NKG2DL expression can be induced by oxidative stress [23,40]. Interestingly, induction of MICA by PDT was significantly less than that observed following treatment with H2O2. It is possible that the half-life of PDT-induced ROS is too short to allow for gene induction or that PDT induction of MICA is independent of oxidative stress. This possibility is supported by the results showing that treatment with HPPH alone leads to induction of MICA. HPPH localizes to the mitochondrial membrane [41]; mitochondrial damage or stress can lead to NF-κB activation and cytokine secretion [42], which may lead to increases in MICA expression [16,23].
The differential expression of MICA and MICB in response to HPPH-PDT is somewhat surprising given that these two molecules share significant homology and are encoded by evolutionarily conserved genes [43]. However, MICA and MICB are highly polymorphic and several studies have shown that they can be differentially regulated [23,40]. It is also apparent that NKG2DL expression and regulation varies between tumor cell lines [16]. Critical to our study are the findings showing that transcriptional regulation of NKG2DL is both cell type and environment dependent [16]. Importantly, previous studies have shown that hematoporphyrin-based PDT induces ULBP-1 and -2 genes in a gastric tumor cell line and MICA/B, ULBP-1, -2, and -3 in a lung cancer cell line [39].
Other studies have also examined the effects of PDT on MHC class I expression with mixed results. Verteporfin-based PDT lead to a downregulation of classical MHC class I surface expression on murine dendritic cells but had no effect on B cell expression of these molecules [44]. Blom et al. [45] demonstrated that hematoporphyrin-based PDT of human ocular melanoma cells led to rapid changes in expression that returned to pre-treatment levels within 6 hours. Unfortunately, our study does little to clarify these discrepancies other to further suggest that tumor cell expression of MHC class I appears to be unaffected by PDT. This conclusion is supported by the study of Abdel-Hady et al. [12], which shows that 5-aminolevulinic acid-based PDT is unable to induce MHC class I expression on tumor cells lacking MHC class I.
Our laboratory has previously shown that PDT enhanced anti-tumor immunity is dependent on CD8+ T cells and NK cells. In the current study, we show that both in vitro treatment of human tumor cells and in vivo treatment of murine tumors with HPPH-PDT results in increased NKG2DL expression. Our studies and those of Park et al. [39] showed that PDT-induced NKG2DL expression was accompanied by increased sensitivity to NK cell killing. Therefore, it is likely that induction of NKG2DL following PDT contributes to PDT enhancement of anti-tumor immunity. Understanding the regulation of NKG2D by different types of PDT treatments may provide answers to the differing ability of various PDT photosensitizers to enhance anti-tumor immunity. Furthermore, this understanding is fundamental for the improvement and design of new treatment strategies that will elicit the long-term immune response mediated by PDT in the clinic.
ACKNOWLEDGMENTS
We thank to Ms. Patricia Maier for her assistance and technical expertise. This work was supported by NIH Grants CA55791 and CA98156 and in part by the Roswell Park Cancer Center Support Grant CA16056.
Contract grant sponsor: NIH; Contract grant numbers: CA55791; CA98156; Contract grant sponsor: Roswell Park Cancer Center Support; Contract grant number: CA16056.
Footnotes
Conflict of interest: none.
REFERENCES
- 1.Henderson BW, Gollnick SO. Mechanistic principles of photodynamic therapy. In: Vo-Dinh T, editor. Biomedical photonics handbook. CRC Press; Boca Raton, FL: 2003. pp. 36-1–36-27. [Google Scholar]
- 2.Brown SB, Brown EA, Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5:497–508. doi: 10.1016/S1470-2045(04)01529-3. [DOI] [PubMed] [Google Scholar]
- 3.Dougherty TJ. An update on photodynamic therapy applications. J Clin Laser Med Surg. 2002;20:3–7. doi: 10.1089/104454702753474931. [DOI] [PubMed] [Google Scholar]
- 4.Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer. 2006;6:535–545. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gollnick SO, Brackett CM. Enhancement of anti-tumor immunity by photodynamic therapy. Immunol Res. 2010;46:216–226. doi: 10.1007/s12026-009-8119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kabingu E, Vaughan L, Owczarczak B, Ramsey KD, Gollnick SO. CD8+ T cell-mediated control of distant tumours following local photodynamic therapy is independent of CD4+ T cells and dependent on natural killer cells. Br J Cancer. 2007;96:1839–1848. doi: 10.1038/sj.bjc.6603792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blank M, Lavie G, Mandel M, Keisari Y. Effects of photodynamic therapy with hypericin in mice bearing highly invasive solid tumors. Oncol Res. 2001;12:409–418. doi: 10.3727/096504001108747864. [DOI] [PubMed] [Google Scholar]
- 8.Castano AP, Gad R, Zahra T, Hamblin MR. Specific anti-tumor immune response with photodynamic therapy mediated by benzoporphyrin derivative chlorin(e6). In: Jacques SL, Duncan DD, Kirkpatrick SJ, Kriete A, editors. The International Society for Optical Engineering, Proceedings of SPIE. Laser-Tissue Interaction XIV:Photochemical, Phototheramal and Photomechanical; 2003. pp. 1–9. Ref Type: Conference Proceeding. [Google Scholar]
- 9.Friedberg JS, Mick R, Stevenson JP, Zhu T, Busch TM, Shin D, Smith D, Culligan M, Dimofte A, Glatstein E, Hahn SM. Phase II trial of pleural photodynamic therapy and surgery for patients with non-small-cell lung cancer with pleural spread. J Clin Oncol. 2004;22:2192–2201. doi: 10.1200/JCO.2004.07.097. [DOI] [PubMed] [Google Scholar]
- 10.Thong PS, Ong KW, Goh NS, Kho KW, Manivasager V, Bhuvaneswari R, Olivo M, Soo KC. Photodynamic-therapy-activated immune response against distant untreated tumours in recurrent angiosarcoma. Lancet Oncol. 2007;8:950–952. doi: 10.1016/S1470-2045(07)70318-2. [DOI] [PubMed] [Google Scholar]
- 11.Thong PS, Olivo M, Kho KW, Bhuvaneswari R, Chin WW, Ong KW, Soo KC. Immune response against angiosarcoma following lower fluence rate clinical photodynamic therapy. J Environ Pathol Toxicol Oncol. 2008;27:35–42. doi: 10.1615/jenvironpatholtoxicoloncol.v27.i1.40. [DOI] [PubMed] [Google Scholar]
- 12.Abdel-Hady ES, Martin-Hirsch P, Duggan-Keen M, Stern PL, Moore JV, Corbitt G, Kitchener HC, Hampson IN. Immunological and viral factors associated with the response of vulval intraepithelial neoplasia to photodynamic therapy. Cancer Res. 2001;61:192–196. [PubMed] [Google Scholar]
- 13.Kabingu E, Oseroff AR, Wilding GE, Gollnick SO. Enhanced systemic immune reactivity to a basal cell carcinoma associated antigen following photodynamic therapy. Clin Cancer Res. 2009;15:4460–4466. doi: 10.1158/1078-0432.CCR-09-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cronin SJ, Penninger JM. From T-cell activation signals to signaling control of anti-cancer immunity. Immunol Rev. 2007;220:151–168. doi: 10.1111/j.1600-065X.2007.00570.x. [DOI] [PubMed] [Google Scholar]
- 15.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 16.Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev. 2010;235:267–285. doi: 10.1111/j.0105-2896.2010.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol. 2007;7:737–744. doi: 10.1038/nri2144. [DOI] [PubMed] [Google Scholar]
- 18.Stern-Ginossar N, Mandelboim O. An integrated view of the regulation of NKG2D ligands. Immunology. 2009;128:1–6. doi: 10.1111/j.1365-2567.2009.03147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diefenbach A, Raulet DH. The innate immune response to tumors and its role in the induction of T-cell immunity. Immunol Rev. 2002;188:9–21. doi: 10.1034/j.1600-065x.2002.18802.x. [DOI] [PubMed] [Google Scholar]
- 21.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA. 2001;98:11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28:571–580. doi: 10.1016/j.immuni.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene. 2008;27:5944–5958. doi: 10.1038/onc.2008.272. [DOI] [PubMed] [Google Scholar]
- 24.Henderson BW, Bellnier DA, Greco WR, Sharma A, Pandey RK, Vaughan LA, Weishaupt KR, Dougherty TJ. An in vivo quantitative structure–activity relationship for a congeneric series of pyropheophorbide derivatives as photosensitizers for photodynamic therapy. Cancer Res. 1997;57:4000–4007. [PubMed] [Google Scholar]
- 25.Nava HR, Allamaneni SS, Dougherty TJ, Cooper MT, Tan W, Wilding G, Henderson BW. Photodynamic therapy (PDT) using HPPH for the treatment of precancerous lesions associated with Barrett's esophagus. Lasers Surg Med. 2011;43:705–712. doi: 10.1002/lsm.21112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kousis PC, Henderson BW, Maier PG, Gollnick SO. Photodynamic therapy (PDT) enhancement of anti-tumor immunity is regulated by neutrophils. Can Res. 2007;67:10501–10510. doi: 10.1158/0008-5472.CAN-07-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther. 1999;10:1359–1373. doi: 10.1089/10430349950018030. [DOI] [PubMed] [Google Scholar]
- 28.Tanabe M, Sekimata M, Ferrone S, Takiguchi M. Structural and functional analysis of monomorphic determinants recognized by monoclonal antibodies reacting with the HLA class I alpha 3 domain. J Immunol. 1992;148:3202–3209. [PubMed] [Google Scholar]
- 29.Stolina M, Sharma S, Huang M, Lin Y, Zhu L, Chen K, Lim K, Mao JT, Miller PW, Dubinett SM. Tumor cyclooxygenase-dependent modulation of lymphocyte IL-10 production in murine lung cancer. Am Assoc Cancer Res. 1997;38:121. [Google Scholar]
- 30.Korbelik M, Sun J. Cancer treatment by photodynamic therapy combined with adoptive immunotherapy using genetically altered natural killer cell line. Int J Cancer. 2001;93:269–274. doi: 10.1002/ijc.1326. [DOI] [PubMed] [Google Scholar]
- 31.Henderson BW, Daroqui C, Tracy E, Vaughan LA, Loewen GM, Cooper MT, Baumann H. Cross-linking of signal transducer and activator of transcription 3—A molecular marker for the photodynamic reaction in cells and tumors. Clin Cancer Res. 2007;13:3156–3163. doi: 10.1158/1078-0432.CCR-06-2950. [DOI] [PubMed] [Google Scholar]
- 32.Korbelik M. PDT-associated host response and its role in the therapy outcome. Lasers Surg Med. 2006;38:500–508. doi: 10.1002/lsm.20337. [DOI] [PubMed] [Google Scholar]
- 33.Campoli M, Ferrone S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Onco-gene. 2008;27:5869–5885. doi: 10.1038/onc.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peaper DR, Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu Rev Cell Dev Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 35.Belicha-Villanueva A, McEvoy S, Cycon K, Ferrone S, Gollnick SO, Bangia N. Differential contribution of TAP and tapasin to HLA class I antigen expression. Immunology. 2008;124:112–120. doi: 10.1111/j.1365-2567.2007.02746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grandea AG, III, Van KL. Tapasin: An ER chaperone that controls MHC class I assembly with peptide. Trends Immunol. 2001;22:194–199. doi: 10.1016/s1471-4906(01)01861-0. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto K, Fujiyama Y, Andoh A, Bamba T, Okabe H. Oxidative stress increases MICA and MICB gene expression in the human colon carcinoma cell line (CaCo-2) Biochim Biophys Acta. 2001;1526:10–12. doi: 10.1016/s0304-4165(01)00099-x. [DOI] [PubMed] [Google Scholar]
- 38.Gollnick SO, Liu X, Owczarczak B, Musser DA, Henderson BW. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res. 1997;57:3904–3909. [PubMed] [Google Scholar]
- 39.Park MJ, Bae JH, Chung JS, Kim SH, Kang CD. Induction of NKG2D ligands and increased sensitivity of tumor cells to NK cell-mediated cytotoxicity by hematoporphyrin-based photodynamic therapy. Immunol Invest. 2011;40:367–382. doi: 10.3109/08820139.2010.551435. [DOI] [PubMed] [Google Scholar]
- 40.Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol. 2007;178:961–969. doi: 10.4049/jimmunol.178.2.961. [DOI] [PubMed] [Google Scholar]
- 41.MacDonald IJ, Morgan J, Bellnier DA, Paszkiewicz G, Whitaker JE, Litchfield DJ, Dougherty TJ. Subcellular localization patterns and their relationship to photodynamic activity of pyropheophorbide-a derivatives. Photochem Photobiol. 1999;70:789–797. [PubMed] [Google Scholar]
- 42.Kepp O, Galluzzi L, Kroemer G. Mitochondrial control of the NLRP3 inflammasome. Nat Immunol. 2011;12:199–200. doi: 10.1038/ni0311-199. [DOI] [PubMed] [Google Scholar]
- 43.Bahram S, Bresnahan M, Geraghty DE, Spies T. A second lineage of mammalian major histocompatibility complex class I genes. Proc Natl Acad Sci USA. 1994;91:6259–6263. doi: 10.1073/pnas.91.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King DE, Jiang H, Simkin G, Obochi M, Levy JG, Hunt DWC. Photodynamic alteration of the surface receptor expression pattern of murine splenic dendritic cells. Scand J Immunol. 1999;49:184–192. doi: 10.1046/j.1365-3083.1999.00498.x. [DOI] [PubMed] [Google Scholar]
- 45.Blom DJ, Schuitmaker HJ, De Waard-Siebinga I, Dubbelman TM, Jager MJ. Decreased expression of HLA class I on ocular melanoma cells following in vitro photodynamic therapy. Cancer Lett. 1997;112:239–243. doi: 10.1016/s0304-3835(96)04578-8. [DOI] [PubMed] [Google Scholar]