

Abstract
The Z-selective ethenolysis activity of chelated ruthenium metathesis catalysts was investigated with experiment and theory. A five-membered chelated catalyst that was successfully employed in Z-selective cross metathesis reactions has now been found to be highly active for Z-selective ethenolysis at low ethylene pressures, while tolerating a wide variety of functional groups. This phenomenon also affects its activity in cross metathesis reactions and prohibits crossover reactions of internal olefins via trisubstituted ruthenacyclobutane intermediates. In contrast, a related catalyst containing a six-membered chelated architecture is not active for ethenolysis and seems to react through different pathways more reminiscent of previous generations of ruthenium catalysts. Computational investigations of the effects of substitution on relevant transition states and ruthenacyclobutane intermediates revealed that the differences of activities are attributed to the steric repulsions of the anionic ligand with the chelating groups.
INTRODUCTION
The discovery of transition metal alkylidene catalysts has allowed olefin metathesis to permeate the literature in a wide variety of fields including green chemistry,1 natural product synthesis,2 and polymer chemistry,3 since its discovery in the 1950’s. The development of catalysts that exhibit preference for kinetically versus thermodynamically controlled products, such as the generation of terminal olefins from internal olefins, termed ethenolysis, and the formation of Z-olefins in cross metathesis (CM) reactions, is a particular challenge in the field.
In order for a metathesis catalyst to be active in ethenolysis reactions, it must exhibit high activity and stability as a propagating methylidene. However, many known metathesis catalysts are unstable as methylidene complexes and undergo rapid decomposition, thus exhibiting poor ethenolysis reactivity.4 The desired ethenolysis catalytic cycle is depicted in Scheme 1. Initial reaction of an internal olefin with a metal methylidene proceeds via a 1,2-metallacycle and produces a terminal olefin and the corresponding substituted metal alkylidene. Further reaction with ethylene forms a second equivalent of terminal olefin and regenerates the catalytically active methylidene.
Scheme 1.

Ethenolysis and related side reactions.
For an ethenolysis catalyst to show high selectivity at appropriate ethylene pressures, formation of terminal olefin products must be favored over back reactions and side reactions that produce internal olefins (Scheme 1). Side reactions that reduce selectivity for the desired terminal olefin products include self-metathesis and secondary metathesis. Self-metathesis is when a metathesis reaction occurs between two substrate molecules instead of between a substrate molecule and ethylene, and secondary metathesis involves the CM of two terminal olefins to generate an internal olefin and ethylene (Scheme 1). Industrially, the ethenolysis of seed oil derivatives affords chemically desirable products with applications in cosmetics, detergents, polymer additives, and renewable biofuels.5
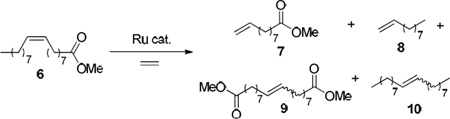
The ability to selectively form the kinetically preferred Z-olefin products in CM reactions is another significant challenge in metathesis research, as catalysts have generally been observed to favor formation of the thermodynamic E-isomer. The Z-olefin motif is prevalent in a variety of small molecules, including many natural products and pharmaceutical targets. The first example of a catalyst-controlled system capable of predominantly forming the Z-isomer in CM reactions was reported by the Hoveyda and Schrock laboratories. The Z-selectivity of the reported tungsten and molybdenum catalysts was attributed to the difference in the size of the two axial ligands. This size difference influences the orientation of the substituents on the forming metallacyclobutane intermediate and leads to productive formation of Z-olefins.6 These catalysts have shown great utility in the synthesis of complicated natural products and stereoregular polymers.7 A particular Z-selective molybdenum catalyst (1) was shown to be effective for the Z-selective ethenolysis of internal olefins.8 In this process, the corresponding molybdenum methylidene reacts preferentially with cis-olefins to produce two terminal olefins, while trans-olefins react to a significantly smaller extent (Scheme 2). Since ethenolysis is the reverse of cross metathesis, the same 1,2-disubstituted metallacyclobutane complex must be formed as an intermediate in both reactions. Hence, if a catalyst is highly Z-selective when forming cross products, it is expected to also be able to selectively degrade cis-olefins by ethenolysis, assuming that the corresponding metal methylidene complex is stable.
Scheme 2.

Z-selective ethenolysis reaction using molybdenum catalyst 1.
A family of functional group tolerant Z-selective ruthenium-based catalysts has recently been reported that contain a chelating N-heterocyclic carbene (NHC) ligand derived from an intramolecular carboxylate-driven C-H bond insertion of an N-bound substituent (Figure 1).9 Catalyst 3 is derived from C–H activation of the benzylic position of the N-mesityl substituent of complex 2 and thus contains a six-membered chelated structure that imparts slightly improved Z-selectivity compared to previous generations of ruthenium catalysts. Catalysts 4 and 5 contain five-membered chelates derived from C–H activation of an N-adamantyl substituent, and exhibit activity and Z-selectivity rivaling the aforementioned group IV systems. The identity of the anionic ligand has been found to have great consequence on reactivity and selectivity, as replacement of a carboxylate on 4 for a nitrato-type ligand (5) results in greater stability, Z-selectivity, and close to 1000 turnovers for homodimerization reactions. Z-selective CM, macrocyclic ring closing metathesis (RCM), and ring opening metathesis polymerization (ROMP) reactions have been reported using this family of chelated catalysts.10
Figure 1.

Prominent ruthenium metathesis catalysts (Mes = 2,4,6-trimethylphenyl).
Our groups and others have used density functional theory (DFT) calculations to elucidate important information about the mechanism of action, origin of Z-selectivity, and stability of chelated ruthenium catalysts 4 and 5.11 The metathesis reaction occurs via a side-bound mechanism, different from that with non-chelated ruthenium catalysts. The olefin approaches cis to the NHC ligand on the catalyst.12 The N-adamantyl chelating group positions the N-mesityl substituent directly over the forming metallacyclobutane, thus causing its substituents to be oriented away to avoid steric repulsions and leading to high Z-selectivity of the metathesis products.
In order to design better catalysts for Z-selective metathesis, a more thorough understanding of this family of chelated ruthenium catalysts is required. The goal of this study is to explore the Z-selectivity of these catalysts for ethenolysis reactions and concurrently investigate how this can help us better understand their CM reactivity. The stability and structure of metallacyclobutane intermediates greatly influences metathesis reactivity and selectivity, thus we sought to study the effects of substitution on relevant ruthenacyclobutane intermediates using experimental and theoretical techniques. Both catalysts 3 and 5 were tested so that the effects of chelate size could be investigated.13 Herein, we report a method for the functional group tolerant Z-selective ethenolysis of internal olefins and explore other unique reactivity of chelated ruthenium catalysts, providing hypotheses of observed behavior based on ruthenacyclobutane stability and structure.
RESULTS AND DISCUSSION
Ethenolysis Experimental Investigations
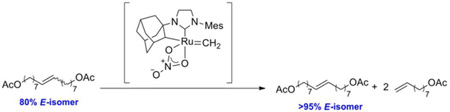
The ethenolysis activity of chelated ruthenium complexes 3 and 5 was initially investigated in order to directly compare our chelated catalysts to previously reported catalysts.14 We first explored their activity and selectivity for the ethenolysis of the completely cis-olefin substrate, methyl oleate (Table 1). The ethenolysis of methyl oleate is a standard assay used to compare ethenolysis reactivity and selectivity of metathesis catalysts. It should be noted that selectivity here refers to the formation of the desired ethenolysis products, terminal olefins 7 and 8, and not to the catalyst’s E/Z selectivity. Although catalyst 3 showed no reactivity at the catalyst loadings tested, catalyst 5 was able to catalyze the transformation with high turnovers and high selectivity at low loadings.15 The fact that 5 is highly active as an ethenolysis catalyst and previously exhibited high Z-selectivity in CM reactions strongly suggests that it would exhibit high selectivity for Z-olefins in ethenolysis reactions.
Table 1.
Ethenolysis reactions of methyl oleate catalyzed by catalysts 3 and 5.
 | |||||
|---|---|---|---|---|---|
| entrya | catalyst | mol % | yieldb | selectivityc | TONd |
| 1 | 3 | 0.1 | 0% | - | 0 |
| 2 | 3 | 0.01 | 0% | - | 0 |
| 3 | 5 | 0.1 | 80% | >95% | 800 |
| 4 | 5 | 0.01 | 12% | >95% | 1160 |
The reactions were run in a minimal amount of CH2Cl2 for 1 h at 40 °C and 10.2 atm of ethylene.
Yield = (moles of ethenolysis products 7 + 8)*100%/(initial moles of 6).
Selectivity = (moles of ethenolysis products 7 + 8) *100%/(moles of total products 7 + 8 + 9 + 10).
TON = yield*[(moles of 6)/(moles of catalyst)]
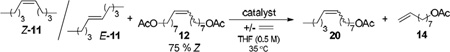
We tested chelated catalysts 3 and 5 in the ethenolysis of ~ 4:1 mixtures of the trans- and cis- isomers of two internal olefins, 5-decene and the acetate-substituted substrate 12, to determine if these catalysts exhibited any selectivity for Z-olefins. We were pleased to find that under the optimized conditions depicted in Scheme 3, catalyst 5 was able to enrich both internal olefin mixtures (~80% E) to >95% of the E-isomer at 5 atm of ethylene and 0.5 mol % catalyst loading for the two substrates (Scheme 3); the products of both reactions were recovered by flash column chromatography.16 For 5-decene, the purely E- isomer (>95% E) was isolated in 90% yield based upon initial E-content.17 For 12, the purely E-internal olefin (>95% E) and 8-nonenyl acetate produced by ethenolysis of the Z-olefins were both quantitatively recovered (Scheme 3). Exposure of catalyst 3 (0.5 mol %) to a ~ 4:1 mixture of the trans- and cis- isomers of 12 at 5 atm of ethylene led to a very small amount of ethenolysis (<3% conversion) and no observable selectivity. Additionally, only olefin migration of the starting material 12 was observed when the reaction was carried out at 1 atm of ethylene at higher loadings of catalyst 3 (5 mol %).
Scheme 3.
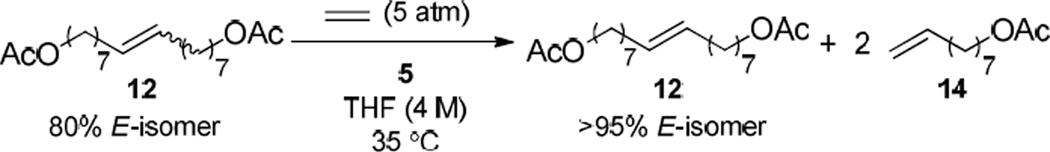
Z-selective ethenolysis reaction of substrate 12.
With the knowledge that 5 was an effective Z-selective ethenolysis catalyst, we sought to investigate its functional group compatibility in E-isomer enrichment reactions.18 A variety of functional groups were tolerated under the reaction conditions, including acetates, alcohols, esters, amines, and ketones (Table 2). Reaction performed with 5 atm of ethylene for all E-dominant substrates (79–82% E) led to enrichment of the internal olefins with >95% of the E-isomer as monitored by 1H NMR. Although the same reactions performed under 1 atm of ethylene proceeded with high selectivity, reaction at 5 atm was necessary to push the ~80% mixtures to >95% of the E-isomer.
Table 2.
E-isomer enrichment by Z-selective ethenolysis of various functionalized symmetrical internal olefins with the formula R(CH2)nCH=CH-(CH2)nR using catalyst 5.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | compound | R; n | initial %E |
mol % 5 |
pressure (atm) |
time (h) |
final %E |
| 1 | 11 | CH3; 3 | 79 | 0.5 | 1 | 4 | 90 |
| 2 | 79 | 0.5 | 5 | 4 | >95 | ||
| 3 | 52 | 0.5 | 5 | 4 | 90 | ||
| 4 | 12 | OAc; 7 | 78 | 0.5 | 1 | 4 | 93 |
| 5 | 78 | 0.5 | 5 | 4 | >95 | ||
| 6 | 15 | OH; 4 | 82 | 0.5 | 1 | 4 | 92 |
| 7 | 82 | 0.5 | 5 | 4 | >95 | ||
| 8 | 68 | 0.5 | 5 | 4 | 90 | ||
| 9 | 16 | CO2Me; 6 | 80 | 0.5 | 1 | 6 | 88 |
| 10 | 80 | 0.5 | 5 | 6 | >95 | ||
| 11 | 17 | NHPh; 3 | 80 | 0.5 | 1 | 4 | 92 |
| 12 | 80 | 1.0 | 5 | 4 | >95 | ||
| 13 | 60 | 1.0 | 5 | 6 | 86 | ||
| 14 | 18 | C(O)Me; 2 | 72 | 0.5 | 1 | 4 | 90 |
| 15 | 72 | 0.5 | 5 | 4 | >95 | ||
We next attempted to quantify the ethenolysis selectivity of catalyst 5 by investigating the relative rates of degradation of E and Z internal olefins using 1H NMR spectroscopy. 5-Decene was chosen as a substrate since the stereopure E- and Z-isomers are commercially available. The rate of ethenolysis was found to be first-order in substrate8 and the relative rates of 5-decene ethenolysis were determined by 1H NMR spectroscopy under 1 atm of ethylene (see Supporting Information). Neither the ethenolysis of Z-5-decene nor E-5-decene proceeded to completion under the reaction conditions.19 Nevertheless, log plots of substrate concentration versus time at early reaction times were found to be linear (E-5-decene, R2 = 0.93, Z-5-decene, R2 = 0.98). From the slopes of these plots, the ratio of the rate constants for ethenolysis of Z-5-decene and E-5-decene (kZ/kE) was found to be ca. 4.5. The corresponding kZ/kE value reported for molybdenum catalyst 1, 30 ± 5, is significantly higher, implying that catalyst 1 is inherently more selective than 5. However, all reactions catalyzed by 1 were conducted under high ethylene pressures not suitable for benchtop reactions (4–20 atm).20 The functional group tolerance of catalyst 5 and the Z-selectivity at ethylene pressures as low as 1 atm highlights advantages of this particular ruthenium-based system for the preparation of terminal olefins from internal olefins and for the purification of Z/E mixtures.21 The further development of chelated catalysts with increased Z-selectivity will lead to ruthenium catalysts with increased kZ/kE values.22
Ethenolysis Computational Investigations
In order to understand the mechanism of ethenolysis, and the origin of Z-selectivity with catalyst 5, we computed the ethenolysis reaction pathways and the Z/E-selectivity with density functional theory (DFT). The calculations were performed using Gaussian 0923 with a theoretical level found to be satisfactory in our previous computational studies of chelated ruthenium catalysts.11 Geometries were optimized in the gas phase with B3LYP24/LANL2DZ–6-31G(d). Single point calculations were performed with M0625/SDD–6-311+G(d,p) and the SMD26 solvation model with THF solvent.
Reaction pathways initiated from both ruthenium methylidene and alkylidene complexes were investigated, since these interconvert during the ethenolysis reaction. The most favorable pathway of the ethenolysis of cis-2-butene with catalyst 5 involves the side-bound approach of the internal olefin to the ruthenium methylidene complex 21 (Figure 2). Formation of the ruthenacyclobutane intermediate 23 requires an activation free energy of 8.8 kcal/mol (TS22). In 21, TS22, and 23, the nitrate is syn to the α-H on the chelating adamantyl group. Ruthenacyclobutane 23 isomerizes to form a less stable ruthenacycle 24, in which the nitrate is anti to the adamantyl α-H.27 Cleavage of the ruthenacycle 24 via TS25 requires a comparable activation energy as TS22 (ΔG‡ = 8.6 kcal/mol), and generates a ruthenium–propene πcomplex 26. In contrast, productive cleavage of 23 without isomerization to 24 requires a much higher barrier (ΔG‡ = 20.9 kcal/mol) and forms an unstable ruthenium ethylidene complex in which the ethylidene is trans to the chelating Ru–C bond. Thus, the isomerization to 24 is necessary before cleaving the ruthenacyclobutane. Decoordination of propene from 26 yields ruthenium ethylidene 27, which then binds to an ethylene molecule to form πcomplex 28. Subsequent steps involve the formation and cleavage of monosubstituted ruthenacyclobutane intermediates via TS29 and TS32, respectively, and eventually regeneration of the ruthenium methylidene complex 21. The monosubstituted transition states in the second half of the catalytic cycle (TS29 and TS32) are both 3~4 kcal/mol more stable than the disubstituted transition states TS22 and TS25. Thus, the reaction of ruthenium methylidene with the internal olefin is the rate-limiting step in the catalytic cycle (TS22), while the productive cleavage of the disubstituted metallacycle (TS25) requires essentially identical activation energy.
Figure 2.

The most favorable pathway of ethenolysis of cis-2-butene with catalyst 5. Gibbs free energies and enthalpies (in parenthesis) are in kcal/mol and with respect to the most stable ruthenium ethylidene complex 47 (an isomer of 27, see Figure 4). For clarity, the chelating adamantyl group is not shown in the 3D transition state structures.
The anionic nitrate ligand binds bidentate to the ruthenium in all four transition states in the catalytic cycle, although the Ru–O bond trans to the alkylidene is significantly longer than the Ru–O bond trans to the NHC (~2.4 Å versus ~2.2 Å). The transition states with monodentate nitrate are five-coordinated with trigonal bipyramidal geometries and 1~4 kcal/mol less stable than the corresponding bidentate transition states (see Supporting Information). The small energy differences between mono- and bidentate nitrate complexes suggest that the monodentate transition structures might become favorable with bulkier olefin substituents and/or bulkier anionic ligands. Structures containing both mono- and bidentate binding modes are considered in the following computations and only the most favorable structures are shown.
In the analogous reaction with trans-2-butene, both transition states TS33 and TS36 are less stable than the corresponding transition states TS22 and TS25 in the reaction with cis-2-butene (Figure 3). In TS33 and TS36, one of the olefin substituents is pointing towards the N-mesityl group and leads to significant steric repulsions. The overall activation barrier is 5.2 kcal/mol higher than the ethenolysis of cis-2-butene. This explains the observed Z-selectivity in ethenolysis reactions.28 Interestingly, the ruthenacyclobutane intermediates 34 and 35 in the reaction with trans-2-butene are slightly more stable than the corresponding cis-substituted metallacycles 23 and 24. Unlike the metathesis transition states, in which the olefin and the Ru-alkylidene are almost in the same plane, the four-membered metallacycle intermediates are puckered. The methyl substituents on the metallacycles in 34 and 35 are not directly pointing towards the N-mesityl group on the ligand (See SI for the 3D structures of the metallacycle intermediates). The ligand-metallacycle repulsions in the metallacycle are smaller than in the transition states.
Figure 3.

The most favorable pathway of ethenolysis of trans-2-butene with catalyst 5. Gibbs free energies and enthalpies (in parenthesis) are in kcal/mol and with respect to the most stable ruthenium ethylidene complex 47 (an isomer of 27, see Figure 4). For clarity, the chelating adamantyl group is not shown in the 3D transition state structures. In subsequent steps, 27 reacts with ethylene to regenerate 21. This is identical to the second half of the catalytic cycle in the reaction with cis-2-butene (see Figure 2).
Crossover Experimental Studies
Research presented in a previous report led us to believe that ethenolysis plays a major role in CM reactions catalyzed by 5.10c The CM reaction between a cis-internal olefin and a terminal olefin was monitored over time and revealed that internal olefins must be broken down by ethenolysis before a hetero-cross product can be generated; it was proposed that the required methylidene complex was generated by homodimerization of the terminal olefin substrate. In addition to this, no crossover was observed when two cis-internal olefins were reacted in the presence of catalyst 5, and it was suggested that this was due to high steric demands associated with forming trisubstituted ruthenacycles using this particular catalyst.29 Since CM between two internal olefins is a common occurrence for previous generations of metathesis catalysts including molybdenum-based Z-selective catalysts,6 we desired to further probe this unique reactivity of catalyst 5. Previously, species 20 was synthesized by reacting the two terminal olefins 1-hexene and 8-nonenyl acetate in the presence of catalyst 5 (0.5 mol %) and proceeded with high yield (67%) and cis-selectivity (91% Z-olefin). We explored whether catalysts 5 and 3 were able to form substrate 20 from (1) an internal olefin and a terminal olefin, or (2) two internal olefins. Catalysts 5 and 3 were both investigated in order to elucidate differences in reactivity, activity, and selectivity between the complexes with different chelate sizes.
The reaction of 5-decene (11) and 8-nonenyl acetate (14) to form compound 20 was initially probed (Table 3). When catalyst 5 was employed, use of the cis- and trans- isomers of 5-decene greatly affected its metathesis activity (entries 1 and 2). Reaction with cis-5-decene led to formation of 20 with 57% yield and 91% Z-isomer at 0.2 mol % of 5. The analogous reaction under the same conditions with trans-5-decene led to only trace amounts of 20. In both cases, the undesired homodimer of 8-nonenyl acetate (compound 12) was also formed in similar quantities and Z-selectivities, regardless of the isomer of 5-decene used. Conversely, catalyst 3 was able to form compound 20 with both isomers of 5-decene (entries 3 and 4). The % Z of 20 and 12 were notably low compared to the reactions catalyzed by 5, however this is attributed to extensive Z/E isomerization by secondary metathesis processes at the long reaction times.
Table 3.
Internal-terminal cross metathesis reaction of 5-decene (11) and 8-nonenyl acetate (14) catalyzed by 3 or 5.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | catalyst | mol % | substrate | time (h) |
yield of 20a |
yield of 12a |
% Z of 20b |
% Z of 12b |
| 1 | 5 | 0.2 | Z-11 | 6 | 57% | 21% | 91% | 83% |
| 2 | 5 | 0.2 | E-11 | 24 | <1% | 19% | - | 87% |
| 3 | 3 | 2.5 | Z-11 | 2 | 69% | 14% | 23% | 22% |
| 4 | 3 | 2.5 | E-11 | 2 | 53% | 17% | 25% | 33% |
Determined by gas chromatography.
Determined by 1H NMR.
The unique behavior of catalyst 5 gives important insight into the reactivity of this chelated catalyst.30 Since 5 is effective for the Z-selective ethenolysis of internal olefins, its inability to react with trans-olefins in CM reactions further suggests that all internal olefins must undergo ethenolysis first to generate terminal olefins, and the productive CM reaction occurs between two terminal olefin molecules. In the reaction shown in Scheme 4 catalyzed by 5, it is proposed that a methylidene is initially formed by homodimerization of 8-nonenyl acetate (14) to form 12. This methylidene can then react with cis-5-decene to form 1-hexene and the corresponding substituted alkylidene, both of which can react further with the terminal olefin 8-nonenyl acetate to generate cross product 20 (Scheme 4). Since 8-nonenyl acetate must initially be homodimerized for productive CM to occur, larger amounts of 12 will be generated compared to other catalysts, as was observed. It is also interesting to note that because catalyst 3 is not particularly active as an ethenolysis catalyst, CM reactions catalyzed by 3 seems to proceed through different pathways that are more similar to previous generations of ruthenium catalysts, like 2. In order to further test these hypotheses, the reactions of two internal olefins in the presence and absence of ethylene were attempted.
Scheme 4.
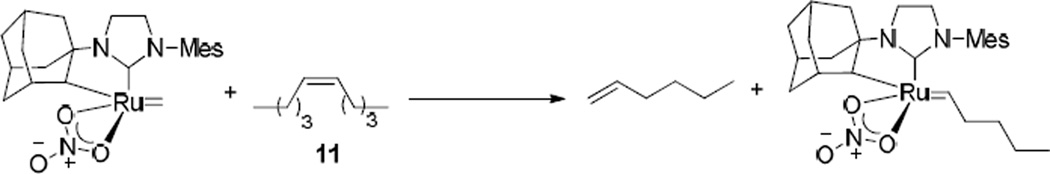
Reaction of a methylidene with cis-5-decene to produce 1-hexene and the corresponding substituted alkylidene.
Exposure of catalyst 5 to a mixture of the internal olefins 5-decene (11) and 12 (75 % Z) under the conditions shown in Table 4 led to no formation of cross product 20 regardless of which isomer of 5-decene was employed (entries 1 and 2).31,32 However, addition of 1 atm of ethylene into the headspace of the reaction vessel for 2 hours followed by stirring for 4.5 hours did lead to formation of product 20 with cis-5-decene (entry 3). No crossover was observed under these conditions when the trans-isomer was used (entry 4). This again supports the hypothesis that productive CM reactions involving internal olefins first proceed via Z-selective ethenolysis. In contrast, under the same conditions depicted in Table 4, catalyst 3 catalyzes the CM of two internal olefins in the absence of ethylene regardless of which isomer of 5-decene is employed (entries 5 and 6). Thus, CM with catalyst 3 proceeds through a completely different pathway and with this catalyst, trisubstituted ruthenacyclobutane intermediates are accessible. The low E/Z ratio is again attributed to extensive Z/E isomerization by secondary metathesis processes as evidenced by the degradation of 12 from 75% to 46% of the Z-isomer.
Table 4.
Internal- internal cross metathesis reaction of 5-decene (11) with 12 catalyzed by 3 or 5.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | catalyst | mol % |
substrate | ethylene exposurea |
time (h) |
yield of 20b |
yield of 14b |
% Z of 20c |
% Z of 12c |
| 1 | 5 | 1.0 | Z-11 | - | 24 | <1% | <1% | - | 76% |
| 2 | 5 | 1.0 | E-11 | - | 24 | <1% | <1% | - | 75% |
| 3 | 5 | 1.0 | Z-11 | + | 4.5 | 21% | 8% | 95% | 70% |
| 4 | 5 | 1.0 | E-11 | + | 24 | <1% | 2% | - | 76% |
| 5 | 3 | 2.5 | Z-11 | - | 2 | 37% | <1% | 31% | 46% |
| 6 | 3 | 2.5 | E-11 | - | 2 | 30% | <1% | 31% | 60% |
+ = 1 atm of ethylene was introduced into the headspace of the reaction vessel for 2 hours prior to reaction.
Determined by gas chromatography.
Determined by 1H NMR.
Crossover Computational Studies
We employed computations to determine the activation energies to form and cleave the di- and trisubstituted ruthenacycle intermediates involved in reactions catalyzed by chelated catalysts 5 and 3, and the non-chelated catalyst 2. We first investigated the metathesis reactions of two internal cis-olefins with catalysts 2, 3, and 5.33 To simplify the calculations, we used trimethyl substituted ruthenacyclobutanes (i.e. the reaction of ruthenium ethylidene with cis-2-butenes) in the calculations as a model of the long conformationally mobile substrates used experimentally. Figure 4 shows the reactions of alkylidenes formed from catalysts 2, 3, and 5 with cis-2-butene. In these reactions, trans-2-butene is formed preferentially with catalyst 2, and catalysts 3 and 5 give cis-2-butene product.
Figure 4.

Reactions of ruthenium ethylidene complexes with cis-2-butene. These are the rate-determining steps in the metathesis of two cis internal olefins in the absence of ethylene. Free energies and enthalpies (in parenthesis) are given in kcal/mol with respect to the ruthenium alkylidene complexes (37, 41, and 47, respectively). For clarity, the chelating adamantyl group is not shown in the 3D structures of TS48 and TS51.
Reaction with the unchelated catalyst 2 (Figure 4a) forms trans-olefin product via the bottom-bound mechanism, i.e. the olefin approaches trans to the NHC ligand.34,12 In the reactions with chelated catalysts 3 and 5, the most favorable pathway involves the side-bound mechanism (Figure 4b and 4c).11 The activation barrier of the reaction catalyzed by 5 is 5.6 and 3.7 kcal/mol higher than that with catalysts 2 and 3, respectively. The activation energy of the rate-determining transition state TS51 is 14.1 kcal/mol with respect to the ruthenium alkylidene complex 47. In the reactions with catalysts 2 and 3, the activation energies are 8.5 and 10.4 kcal/mol, respectively (TS40 and TS45). The overall barrier of the reaction with catalyst 5 is likely to be higher than 14.1 kcal/mol, since the catalyst resting state may be more stable than the energy zero in the calculations (47). This suggests that formation of trisubstituted ruthenacyclobutanes with catalyst 5 is much more difficult than with catalysts 2 and 3, in agreement with the observed low crossover reactivities of 5 (Table 4).
The low crossover reactivity of catalyst 5 is attributed to one particular trisubstituted transition state, TS51, which is 4.4 kcal/mol less stable than the other trisubstituted transition state TS48. In TS51, the ethylidene is syn to the α-hydrogen on the chelating adamantyl group, while in TS48 the ethylidene is anti to the α-hydrogen. Interestingly, TS51 is the only transition state involving a nitrate ligand bound monodentate among all the transition states investigated in this study. Its bidentate isomer TS51’ is 0.8 kcal/mol less stable, which is in contrast to other bidentate nitrate transition states that are typically 3 kcal/mol more stable than corresponding monodentate nitrate TS. To better illustrate the steric interactions with the nitrate ligand, “side-view” of the di- and trisubstituted transition states with catalyst 5 are shown in Figure 5. In the high energy trisubstituted transition state TS51’, the nitrate is located between the bulky chelating adamantyl group and one of the methyl substituents on the olefin. The distances between the nitrate and the olefin and between the nitrate and the adamantyl group are both significantly shorter than the sum of the van der Waals radii (the N–H distances are 2.57 and 2.51 Å, respectively, compared to the sum of van der Waals radii of N and H, 2.75 Å). The steric repulsions of the anionic nitrate ligand with the chelating adamantyl group and the substituent on the olefin clearly destabilize the bidentate transition state TS51’ and force the trisubstituted reaction to proceed via a generally less favorable monodentate transition state (TS51). As described earlier, the isomerization of the metallacyclobutane intermediate is necessary for a productive turnover. In the other trisubstituted metathesis transition state TS48, the nitrate is located on the less crowded side that is syn to the α-adamantyl hydrogen. In TS48, the nitrate–adamantyl and nitrate–olefin distances are both longer than corresponding distances in TS51’. With the diminished steric repulsions with the nitrate, TS48 is 5.4 kcal/mol more stable than TS51’. Thus, the rate-limiting step in the trisubstituted catalytic cycle is TS51.
Figure 5.

Side view of the dimethyl substituted transition states (TS22 and TS25) and the trimethyl substituted TS48 and TS51’. TS51’ is destabilized due to steric repulsions of the nitrate with the chelating adamantyl group and the methyl group on the olefin.
In the presence of ethylene, ruthenium methylidene complexes are formed by the reaction of ethylene and ruthenium alkylidenes. The reaction of ruthenium methylidene 21 with cis-2-butene proceeds through disubstituted transition states TS22 and TS25 (Figure 2). TS22 and TS25 are both more stable than the corresponding trisubstituted transition states: TS22 is only 0.9 kcal/mol more stable than TS48 because of smaller steric repulsions of the internal olefin with the methylidene than with the ethylidene (Figure 5). Replacing the methyl substituent with hydrogen, TS25 is dramatically stabilized by 6.3 kcal/mol compared to TS51’, due to alleviation of steric repulsions with the nitrate (see Figure 5 for direct comparison of the structures of di- and trisubstituted TS). The activation energy of the reaction of cis-2-butene with this methylidene is 5.3 kcal/mol lower than the corresponding pathway to form the trisubstituted ruthenacycle (ΔG‡ = 8.8 kcal/mol, TS22 compared to 14.1 kcal/mol, TS51). Thus, in the presence of ethylene, crossover products are formed, as was observed experimentally, from the reaction of internal olefins with the methylidene complex of catalyst 5 rather than with corresponding alkylidene complexes.
The trisubstituted metathesis pathways with catalysts 2 and 3 were also calculated and shown in Figure 4a and 4b. In contrast to the high activation barrier of the trisubstituted pathway with catalyst 5, catalysts 2 and 3 both have lower activation barrier in the trisubstituted reaction (ΔG‡ = 8.5 and 10.4 kcal/mol, respectively, compared to ΔG‡ = 14.1 kcal/mol with 5). This is attributed to less steric demand in the trisubstituted transition states with these catalysts; the chelating mesityl group in catalyst 3 is less bulky than the chelating adamantyl in 5. With the unchelated catalyst 2, the olefin approaches from the bottom, trans to the NHC ligand and thus there are no unfavorable ligand–substrate steric repulsions in the transition states.
As a comparison with the disubstituted pathway of catalyst 5 (Figure 2), we also computed the activation barriers of the reactions of cis-2-butene and the ruthenium methylidenes derived from catalysts 2 and 3 (Figure 6a and 6b, respectively). For the unchelated catalyst 2, the reaction of cis-2-butene with methylidene 52 requires an activation energy of 8.9 kcal/mol. This is slightly higher than the barrier in the reaction with corresponding ethylidene 37 (ΔG‡ = 8.5 kcal/mol, Figure 4a). This is attributed to fact that ruthenium methylidene 52 is 5.8 kcal/mol less stable than corresponding ethylidene 37 as well as the absence of unfavorable ligand-substrate steric repulsions in the trisubstituted transition states. The reaction of cis-2-butene with the methylidene complex of catalyst 3 (56) requires a slightly lower activation energy than corresponding ethylidene (41) (ΔG‡ = 8.7 kcal/mol compared to 10.4 kcal/mol). With both catalysts 2 and 3, the differences between di- and trisubstituted activation barriers are within 1~2 kcal/mol. This suggests that the rate of crossover of internal olefins will not be significantly affected by the exposure to ethylene.
Figure 6.

Free energies and enthalpies (in parenthesis) of the reaction of ruthenium methylidene complexes with cis-2-butene catalyzed by (a) catalyst 2 and (b) catalyst 3. These are the rate-determining steps in the ethenolysis of internal olefins. All energies are with respect to the ruthenium ethylidene complexes (37 and 41, see Figure 4) and are given in kcal/mol. See Figure 2 for the reaction catalyzed by catalyst 5.
To complete the computational investigations, another two scenarios involving disubstituted ruthenacyclobutanes, homodimerization and non-productive metathesis of terminal olefins, were also computed and the detailed results are provided in the Supporting Information. The homodimerization pathways of propene to form E- or Z-2-butene with catalysts 2, 3, and 5 are shown in Figure S1, and the competing non-productive reactions of propene and ruthenium ethylidenes are shown in Figure S2.35 The computations predicted that the non-productive equilibration with catalysts 2 and 3 both requires lower activation barrier than the corresponding productive homodimerization pathway. In contrast, with catalyst 5, the non-productive pathway requires 1.5 kcal/mol higher activation energy than homodimerization (12.7 kcal/mol compared to 11.2 kcal/mol, see Supporting Information for details). Similar to the trisubstituted transition state, the 1,3-disubstituted non-productive transition state is destabilized by steric repulsions with the nitrate, which is also located between the adamantyl and the methyl substituent on the olefin.
CONCLUSION
In summary, we have investigated the ethenolysis behavior of a new class of ruthenium metathesis catalysts, 3 and 5, containing chelating NHC ligands. Catalyst 5 was found to catalyze Z-selective ethenolysis reactions at low ethylene pressures (1–5 atm) with substrates containing a wide variety of functional groups. DFT calculations showed that the Z-selectivity in ethenolysis reactions catalyzed by 5 is a result of steric effects that prohibit E-olefins from productively reacting with the corresponding methylidene. Two internal olefins could not undergo CM in the presence of 5 to form cross products but must first react with ethylene to form terminal olefins. In addition to this, no crossover was observed when trans-internal olefins were employed as substrates in CM reactions. This implies that the Z-selective ethenolysis behavior of 5 plays a large role not only in its ethenolysis reactivity, but also in its CM reactivity. In contrast, catalyst 3 containing a six-membered chelated exhibited poor ethenolysis reactivity and was capable of catalyzing the crossover of two internal olefins in the absence of ethylene, and thus reacts by a different pathway compared to 5. Density functional theory calculations revealed the origins of the different reactivities of catalysts 3 and 5 in the crossover of internal olefins. The low crossover reactivity of two internal olefins with catalyst 5 is attributed to the steric repulsions of the nitrate anionic ligand with the chelating adamantyl group and the olefin substituent in the trisubstituted metathesis transition state. In ethenolysis reactions with catalyst 5, similar steric control also prevents the ruthenium alkylidene from reacting with internal olefins. In contrast, the most favorable ethenolysis pathway catalyzed by 5 involves the reaction of internal olefin with ruthenium methylidene to avoid trisubstituted metathesis transition states. Catalyst 3 has a smaller mesityl chelating group, and thus the steric repulsions with the anionic ligand are diminished, making it capable to productively form and cleave trisubstituted metallacycles.
The elucidation of a functional-group-tolerant ruthenium metathesis catalyst capable of performing Z-selective ethenolysis at ethylene pressures as low as 1 atm should enable the widespread use of this technology in academic and industrial settings. In addition to providing important insight into the ethenolysis behavior of catalyst 5, a better understanding of its CM reactivity has been gained and should provide important information for researchers planning on using this catalyst for a variety of applications. It is envisioned that the further development of new Z-selective catalysts should make this Zselective ethenolysis methodology even more selective and efficient.
Supplementary Material
ACKOWLEDGMENT
Dr. David VanderVelde is thanked for his assistance with NMR characterization and experiments. This work was financially supported by the NIH (NIH 5R01GM031332-27, R.H.G.), the NSF (CHE-1048404, R.H.G. and CHE-0548209, K.N.H.), the NDSEG (fellowship to B.K.K.), and Mitsubishi Tanabe Pharma Corporation (H.M.). Materia, Inc. is acknowledged for its generous donation of metathesis catalysts. Calculations were performed on the Hoffman2 cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details, NMR spectra, optimized Cartesian coordinates and energies, details of computational methods, and complete reference of Gaussian 09 are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Schrodi Y, Ung T, Vargas A, Mkrtumyan G, Lee CW, Champagne TM, Pederson RL, Hong SH. CLEAN-Soil, Air, Water. 2008;36:669. [Google Scholar]
- 2.Cossy J, Arseniyadis S, Meyer C. Metathesis in Natural Product Synthesis: Strategies, Substrates, and Catalysts. 1st ed. Weinheim: Wiley-VCH; 2010. [Google Scholar]
- 3.a) Leitgeb A, Wappel J, Slugovc C. Polymer. 2010;51:2927. [Google Scholar]; b) Liu X, Basu A. J. Organomet. Chem. 2006;691:5148. [Google Scholar]; c) Sveinbjornsson BR, Weitekamp RA, Miyake GM, Xia Y, Atwater HA, Grubbs RH. Proc. Natl. Acad. Sci. USA. 2012;109:14332. doi: 10.1073/pnas.1213055109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong SH, Wenzel AG, Salguero TT, Day MW, Grubbs RH. J. Am. Chem Soc. 2007;129:7961. doi: 10.1021/ja0713577. [DOI] [PubMed] [Google Scholar]
- 5.a) Burdett KA, Harris LD, Margl P, Aughon BR, Mokhtar-Zadeh T, Saucier PC, Wasserman EP. Organometallics. 2004;23:2027. [Google Scholar]; b) Lysenko Z, Maughon BR, Bicerano J, Burdett KA, Christenson CP, Cummins CH, Dettloff ML, Maher JM, Schrock AK, Thomas PJ, Varjian RD, White JE. WO 2003/ 093215 A1. priority date of November 13, 2003.; c) Olson ES. US 2010/0191008 A1. priority date of July 29, 2010.; d) DuBois J-L, Sauvageot O. WO 2010/103223 A1. priority date of September 16, 2010.; e) Herbinet O, Pitz WJ, Westbrook CK. Combust. Flame. 2010;157:893. [Google Scholar]
- 6.Flook MM, Jiang AJ, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2009;131:7962. doi: 10.1021/ja902738u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Yu M, Wang C, Kyle AF, Jukubec P, Dixon DJ, Schrock RR, Hoveyda AH. Nature. 2011;479:88. doi: 10.1038/nature10563. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461. doi: 10.1038/nature09957. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Flook MM, Ng VWL, Schrock RR. J. Am. Chem. Soc. 2011;133:1784. doi: 10.1021/ja110949f. [DOI] [PubMed] [Google Scholar]; d) Jiang AJ, Zhao Y, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:16630. doi: 10.1021/ja908098t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marinescu SC, Levine DS, Zhao Y, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2011;133:11512. doi: 10.1021/ja205002v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Endo K, Grubbs RH. J. Am. Chem. Soc. 2011;133:8525. doi: 10.1021/ja202818v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Keitz BK, Endo K, Patel PR, Herbert MB, Grubbs RH. J. Am. Chem. Soc. 2012;134:693. doi: 10.1021/ja210225e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Keitz BK, Endo K, Herbert MB, Grubbs RH. J. Am. Chem. Soc. 2011;133:9686. doi: 10.1021/ja203488e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Keitz BK, Fedorov A, Grubbs RH. J. Am. Chem. Soc. 2012;134:2040. doi: 10.1021/ja211676y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Herbert MB, Marx VM, Pederson RL, Grubbs RH. Angew. Chem. Int. Ed. 2013;52:310. doi: 10.1002/anie.201206079. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Marx VM, Herbert MB, Keitz BK, Grubbs RH. J. Am. Chem. Soc. 2013;135:94. doi: 10.1021/ja311241q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Liu P, Xu X, Dong X, Keitz BK, Herbert MB, Grubbs RH, Houk KN. J. Am. Chem. Soc. 2012;134:1464. doi: 10.1021/ja2108728. [DOI] [PubMed] [Google Scholar]; b) Herbert MB, Lan Y, Keitz BK, Liu P, Endo K, Day MW, Houk KN, Grubbs RH. J. Am. Chem. Soc. 2012;134:7861. doi: 10.1021/ja301108m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dang Y, Wang Z-X, Wang X. Organometallics. 2012;31:7222. [Google Scholar]; d) Dang Y, Wang Z-X, Wang X. Organometallics. 2012;31:8654. [Google Scholar]
- 12.In contrast, the bottom-bound mechanism is favored with previous unchelated ruthenium catalysts. For examples of recent computational studies of olefin metathesis with unchelated ruthenium catalysts: Adlhart C, Hinderling C, Baumann H, Chen P. J. Am. Chem. Soc. 2000;122:8204. Adlhart C, Chen P. Angew. Chem. Int. Ed. 2002;41:4484. doi: 10.1002/1521-3773(20021202)41:23<4484::AID-ANIE4484>3.0.CO;2-Q. Cavallo L. J. Am. Chem. Soc. 2002;124:8965. doi: 10.1021/ja016772s. Vyboishchikov SE, Buhl M, Thiel W. Chem.-Eur. J. 2002;8:3962. doi: 10.1002/1521-3765(20020902)8:17<3962::AID-CHEM3962>3.0.CO;2-X. Bernardi F, Bottoni A, Miscione GP. Organometallics. 2003;22:940. Fomine S, Martinez Vargas S, Tlenkopatchev MA. Organometallics. 2003;22:93. Adlhart C, Chen P. J. Am. Chem. Soc. 2004;126:3496. doi: 10.1021/ja0305757. Cavallo L, Costabile C. J. Am. Chem. Soc. 2004;126:9592. doi: 10.1021/ja0484303. Suresh CH, Koga N. Organometallics. 2004;23:76. Benitez D, Goddard WA. J. Am. Chem. Soc. 2005;127:12218. doi: 10.1021/ja051796a. Suresh CH, Baik MH. Dalton Trans. 2005:2982. doi: 10.1039/b508192k. Tsipis AC, Orpen AG, Harvey JN. Dalton Trans. 2005:2849. doi: 10.1039/b506929g. Straub BF. Angew. Chem. Int. Ed. 2005;44:5974. doi: 10.1002/anie.200501114. Cavallo L, Correa A. J. Am. Chem. Soc. 2006;128:13352. doi: 10.1021/ja064924j. Occhipinti G, Bjorsvik HR, Jensen VR. J. Am. Chem. Soc. 2006;128:6952. doi: 10.1021/ja060832i. Straub BF. Adv. Synth. Catal. 2007;349:204. Zhao Y, Truhlar DG. Org. Lett. 2007;9:1967. doi: 10.1021/ol0705548. Torker S, Merki D, Chen P. J. Am. Chem. Soc. 2008;130:4808. doi: 10.1021/ja078149z. Benitez D, Tkatchouk E, Goddard WA. Chem. Commun. 2008:6194. doi: 10.1039/b815665d. Cavallo L, Bahri-Laleh N, Credendino R. Beilstein J. Org. Chem. 2011;7:40. doi: 10.3762/bjoc.7.7. Yang HC, Huang YC, Lan YK, Luh TY, Zhao Y, Truhlar DG. Organometallics. 2011;30:4196. Hillier IH, Pandian S, Percy JM, Vincent MA. Dalton Trans. 2011;40:1061. doi: 10.1039/c0dt01314e. Martinez H, Miró P, Charbonneau P, Hillmyer MA, Cramer CJ. ACS Catalysis. 2012:2547.
- 13.Previous experiments have shown that catalyst 4 is unstable to excess ethylene and thus was not investigated in this study.
- 14.Basic olefin metathesis reactions are near thermoneutral and at equilibrium produce a statistical mix of products. Fortunately the pre-equlibrium mixture of products can be controlled by the catalysts and by removal of one of the products. In RCM the ethylene product can be removed and the structure of the catalyst can be used to control the initial product ration and the rate of approach to equilibrium. In the present paper, the standard metathesis reaction is driven backward by the addition of ethylene and the structure of the catalyst controls the rate of reaction of the components of the reaction mixture.
- 15.Catalyst 5 exhibits smaller turnover numbers for ethenolysis when compared to the state of the art ruthenium, molybdenum, and tungsten catalysts: Anderson DR, Ung T, Mkrtumyan G, Bertrand G, Grubbs RH, Schrodi Y. Organometallics. 2008;27:563. doi: 10.1021/om7008028. Thomas RM, Keitz BK, Champagne TM, Grubbs RH. J. Am. Chem. Soc. 2011;133:7490. doi: 10.1021/ja200246e. Marinescu SC, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2009;131:10840. doi: 10.1021/ja904786y.
- 16.The yields reported herein were calculated based on the assumption that only the Z-internal olefin isomer underwent ethenolysis and that it reacted completely.
- 17.It should be noted that the volatility of the generated 1-hexene prevented it from being recovered and it was thus removed in vacuo.
- 18.The reactions depicted in table 2 were performed to showcase the functional group tolerance of this method and the final %E. Isolated yields of highly %E products were obtained for two of the substrates (see Supporting Information), 12 (96% yield, >95%E) and the relatively volatile 11 (90% yield, >95%E).
- 19.The catalyst loading for E-5-decene was five times higher than for Z-5-decene.
- 20.The reactivity differences between 5 and 1 can be partially explained by the fact that 5 is not soluble in the 5-decene substrate, meaning that neat reactions could not be performed as with catalyst 5. The necessary addition of solvent to these reactions seemingly reduced the activity of catalyst 5.
- 21.It is envisioned that this Z-selective ethenolysis method can be used to purify products from reactions other than metathesis that produce E-olefins but that are not perfectly selective for their formation.
- 22.An improved ruthenium-based Z-selective catalyst was recently reported, and its ethenolysis reactivity will be investigated in a subsequent report: Rosebrugh LE, Herbert MB, Marx VM, Keitz BK, Grubbs RH. J. Am. Chem. Soc. 2013;135:1276. doi: 10.1021/ja311916m.
- 23.Gaussian 09, Revision B.01: Frisch MJ, et al. Wallingford CT: Gaussian, Inc.; 2010.
- 24.a) Becke AD. J. Chem. Phys. 1993;98:5648. [Google Scholar]; b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 25.a) Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215. [Google Scholar]; b) Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 26.Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2009;113:6378. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 27.Isomerization of 23 to 24 occurs via monodentate nitrate complexes. See Supporting Information for details.
- 28.Theoretical calculations predicted higher Z-selectivity than what the observed relative rates of E and Z olefins would suggest.
- 29.It should be noted that catalyst 4 was also not able to catalyze the CM of two internal olefins and, as such, we believe that trisubstituted metallacycle intermediates are also unfavorable.
- 30.For catalyst 4, it is thought that formation of a methylidene is possible and that the reaction proceeds via the same mechanism as with 5, but reaction with a large excess of ethylene leads to decomposition.
- 31.Raising the temperature from 35 °C to 80 °C did not affect reactivity as formation of 20 was still not observed.
- 32.This suggests that the self-metathesis processes outlined in Scheme 1 are prevented, helping explain the high selectivity observed in the ethenolysis of methyl oleate by catalyst 5.
- 33.In order to simplify calculations, an analog of catalyst 3 where the pivalate ligand is replaced by an acetate ligand was used for modeling.
- 34.The competing pathway to form cis-2-butene requires a barrier of 8.7 kcal/mol, only 0.2 kcal/mol higher than the trans pathway, indicating poor Z/E-selectivity from kinetic control.
-
35.Computational studies of olefin homodimerization with catalysts 2, 4, and 5 have already been reported see ref. 11). Here we repeated some of the computations to make direct comparisons between different catalysts and between productive and non-productive metathesis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.