

Abstract
Genome-wide protein structure prediction and structure-based function annotation have been a long-term goal in molecular biology but not yet become possible due to difficulties in modeling distant-homology targets. We developed a hybrid pipeline combining ab initio folding and template-based modeling for genome-wide structure prediction applied to the Escherichia coli genome. The pipeline was tested on 43 known sequences, where QUARK-based ab initio folding simulation generated models with TM-score 17% higher than that by traditional comparative modeling methods. For 495 unknown hard sequences, 72 are predicted to have a correct fold (TM-score > 0.5) and 321 have a substantial portion of structure correctly modeled (TM-score > 0.35). 317 sequences can be reliably assigned to a SCOP fold family based on structural analogy to existing proteins in PDB. The presented results, as a case study of E. coli, represent promising progress towards genome-wide structure modeling and fold family assignment using state-of-the-art ab initio folding algorithms.
With the tremendous success of genome sequencing in past decades, it becomes an increasingly urgent goal to determine structure of protein molecules as expressed by all genes in organisms, which is essential to a systematical understanding of the functional roles that individual molecules play in the interaction network of involved cellular procedures. However, experimental approaches, e.g. X-ray crystallography and nuclear magnetic resonance (NMR), are far too slow and expensive for genome-wide protein structural determinations. In human genome, for example, there are 6,054 proteins with an experimental structure in the Protein Data Bank (PDB)1, which counts only for ~ 29% of nearly 21 k open reading frames (ORFs); if counting the variations from alternative splicing and post-translational processing2, this fraction reduces to < 0.6%. For the best-studied E. coli genome, there are only 23.8% proteins having experimental structure.
Thanks to the significant efforts made by the community in last four decades3,4,5,6,7,8, the 3D structure models of an increasing portion of genes in organisms can be built by computational programs9. Among the earliest attempts of genome-wide protein structure modeling, for example, Fischer and Eisenberg10 successfully assigned structural fold to 22% of 468 protein ORFs in M. genitalium by sequence profile alignments. Sanchez and Sali11 applied MODELLER to S. cerevisiae which generated atomic models for substantial segments of 17% of all yeast proteins. Later, Zhang and Skolnick12 generated full-length models by TASSER for all ORFs in E. coli with 68% proteins having high confidence scores. Baker and coworkers13 identified homologous templates for 47% of ORFs in S. cerevisiae and had 12% of small proteins below 150 residues built by Rosetta ab initio modeling. Despite the impressive success, however, a major obstacle of the genome-wide protein structure prediction is on the modeling of a substantial portion of hard proteins that have no close homologous templates in the PDB and therefore request efficient ab initio folding algorithms to construct model predictions from scratch14.
Recent CASP experiments have witnessed considerable progress in ab initio protein folding15,16. Of note, QUARK was recently developed to construct low-to-medium resolution structures by assembling continuously sized fragments (1–20 residues) excised from unrelated protein structures17,18. In CASP9, for example, QUARK successfully predicted models of correct folds (TM-score > 0.5) for 8 out of 18 Free Modeling (FM) target proteins with length below 150 residues that have no analogous templates in the PDB. In CASP10, QUARK had models of TM-score > 0.5 for two FM targets (R0006 and R0007) with length > 150 residues, which represents probably the largest size range of successful FM modeling in the history of CASP experiments.
In this work, we intend to re-examine the capacity of genome-wide protein structure prediction using the state of the art ab initio protein structure modeling algorithms. Because the proteins of close homologous templates are relatively easy to model using comparative modeling tools7,8,19, our focus is on the protein targets which have no strong alignments by threading programs. We select E. coli genome for case study, partially because it is the best-studied species at the molecule level which has the highest number of solve proteins (except for human genome) to help testify the validity of our approach. To quantitatively assess the quality of the ab initio modeling, we developed a confidence score based on the quality of fragment collection and the convergence of the assembly simulations. As an application of the low-resolution ab initio modeling, we assign the modeled proteins with standard fold families as defined in the SCOP database20. All the prediction and fold assignment data are publicly available at http://zhanglab.ccmb.med.umich.edu/QUARK/ecoli/.
Results
Classification of the E. coli genome sequences
Escherichia coli is a Gram-negative, rod-shaped bacterium that is commonly found in the lower intestine of warm-blooded organisms. Since it can be grown easily and inexpensively in laboratory setting, E. coli has been intensively investigated for over 60 years as the most widely studied prokaryotic model organism.
To analyze the genome data, we first download the list of all E. coli sequences from UniProt database (http://www.uniprot.org/), which contains 4,279 non-redundant entries. To identify targets of experimentally solved structures, we obtained the PDB IDs by scanning the UniProt annotation text. In case that one sequence matches with multiple PDB chains, we choose the experimental structure which has the highest sequence identity to the target sequence, where the pair-wise sequence identity is calculated by NW-align (http://zhanglab.ccmb.med.umich.edu/NW-align). In total, 1,019 E. coli sequences have full or partial structures solved by experiments. Proteins which have structures covering less than half of the sequence are not considered here.
For the remaining 3,260 proteins, we use LOMETS21, a meta-threading method of nine fold-recognition programs, to thread the sequences through the PDB library. In 2,764 cases (64.6%), there is at least one threading program that can identify a strong homologous template with a high confidence Z-score. These proteins are categorized as Easy/Medium targets in LOMETS (see Methods). The remaining 495 proteins are Hard targets since no close templates could be identified by any threading programs. A pie chart of the sequence distribution is showed in Figure 1a.
Figure 1. Distribution of E. coli genome sequences.
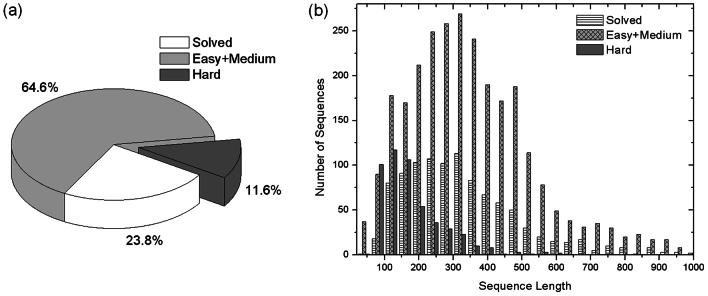
(a) Classification of sequences based on their homology to the PDB structures. (b) Histogram of sequence length in different categories.
The distributions of the targets in terms of the sequence length are presented in Figure 1b. The average length of the sequences with solved structures is largely similar to that of the unsolved ones, which indicates that there is no obvious bias of experimentally determined structures towards protein size. Most of the Hard proteins, however, have a relatively small size (< 400 residues). In particular, for proteins with around 100 residues, 50% of cases belong to the Hard targets. This is partially due to the attribution of the threading algorithms, since the larger proteins tend to have better constructed sequence profiles which can easily identify significant template hits at least for part of the sequence domains.
Since the targets in Easy/Medium category have strong homologous templates, we use the Iterative Threading Assembly Refinement algorithm, I-TASSER19, to generate all model predictions with both template alignment and full-length models deposited at http://zhanglab.ccmb.med.umich.edu/QUARK/ecoli2. In the following, our analyses are mainly focused on the modeling of the Hard proteins which are generated by QUARK ab initio assembly simulations17,18.
Identification of transmembrane proteins
Membrane proteins are the molecules embedded in the cell surface, which fold with a highly regular scaffold, i.e. a hydrophobic domain associated with the bilayer lipid membrane capped by two intra- and extra-cellular hydrophilic domains.
We use two transmembrane helix prediction programs to label the membrane proteins in E. coli since most membrane proteins are alpha-helical proteins. In TMHMM2.022, a protein is considered as transmembrane protein if the predicted number of amino acids in transmembrane helices is larger than 18. In MEMSAT323, it is regarded as a transmembrane protein, if the number of residues in transmembrane segments is larger than 7. Due to the smaller cutoff, MEMSAT3 has a slightly more number of predicted transmembrane proteins than TMHMM2.0. Nevertheless, most of the predictions by the two programs are consistent, with a Pearson's correlation coefficient 0.98 based on the 4,279 target sequences in E. coli. Hence, we use the average number of amino acids in transmembrane helices predicted by the two programs to assign the member proteins, i.e. if the average number of transmembrane residues is > 13, this target is counted as a transmembrane protein.
In total, 1,076 out of the 4,279 sequences (25%) are predicted to be transmembrane proteins where 74 of them have the solved structure in the PDB. This ratio of transmembrane proteins is consistent with the data in the former analysis on E. coli (23–26%)12,24. For most of the E. coli sequences, UniProtKB has a record of ontology which specifies if the targets contain transmembrane helix. 883 E. coli targets are assigned as transmembrane proteins from the UniProt annotation, 97.6% of which are consistent with our membrane prediction here. The remaining 21 proteins from the UniProt annotation turn out to be false positives by our manual check since these sequences contains too few hydrophobic transmembrane residues to span the membranes.
Benchmark test of ab initio folding on known E. coli proteins
Before the application of QUARK to the entire E. coli genome, we firstly test the algorithm on the Hard proteins that have known experimental structures. For this purpose, we use LOMETS to thread all the 1,019 sequences of known structure through the PDB where all homologous templates that have a sequence identity > 30% to the query are excluded. This procedure results in 43 Hard proteins, where 21 have a length < 150 residues and 22 have length > 150 residues.
Summary of QUARK modeling on 21 small proteins
In Table I, we present the folding results of QUARK on the 21 smaller size proteins, where all homologous proteins with a sequence identity > 30% or detectable to PSI-BLAST with E-value < 0.1 are excluded when the fragments were generated for QUARK. In control, we also list the modeling result by the widely-used comparative modeling tool, Modeller7, which constructs models based on the LOMETS tempaltes. As expected, most of the targets have incorrect threading templates with TM-score < 0.5; the exception is from ZAPB_ECOLI that has a trivial single-long helix topology (TM-score = 0.711). The best in top five models by Modeller has an average TM-score = 0.318, which is marginally higher than that of the threading templates (0.295). This TM-score increase is mainly due to length elongation of the full-length models by filling the alignment gaps. There is no target that has a Modeller model with TM-score > 0.5 in Table I.
Table 1. Summary of template-based modeling and ab initio modeling on 21 known E. coli proteins with length < 150 residues.
| TM-score of the best in top 5 models | ||||||||
|---|---|---|---|---|---|---|---|---|
| UniProt Entry | PDB ID | Compact | Length | Template | Modeller | QUARK | ModRefiner | Estimated |
| ARIR_ECOLI | 2oxlA | YES | 88 | 0.415 | 0.439 | 0.614 | 0.614 | 0.614 |
| EMRE_ECOLI | 3b61A | NO | 110 | 0.360 | 0.362 | 0.314 | 0.319 | 0.605 |
| EUTN_ECOLI | 2z9hA | YES | 95 | 0.196 | 0.203 | 0.255 | 0.259 | 0.409 |
| FLHD_ECOLI | 2avuB | NO | 116 | 0.279 | 0.280 | 0.295 | 0.302 | 0.427 |
| GCSH_ECOLI | 3a7lA | YES | 129 | 0.406 | 0.439 | 0.280 | 0.269 | 0.351 |
| ISCX_ECOLI | 1uj8A | YES | 66 | 0.331 | 0.348 | 0.438 | 0.464 | 0.461 |
| METJ_ECOLI | 1cmaA | NO | 105 | 0.185 | 0.220 | 0.254 | 0.247 | 0.397 |
| MQSR_ECOLI | 3hi2B | YES | 98 | 0.243 | 0.250 | 0.424 | 0.449 | 0.381 |
| PTHP_ECOLI | 1pohA | YES | 85 | 0.349 | 0.437 | 0.568 | 0.570 | 0.440 |
| RL14_ECOLI | 1vt2K | YES | 123 | 0.203 | 0.215 | 0.380 | 0.392 | 0.388 |
| RL17_ECOLI | 1vs6N | NO | 127 | 0.198 | 0.236 | 0.256 | 0.255 | 0.468 |
| RL20_ECOLI | 1vs6Q | YES | 118 | 0.236 | 0.363 | 0.420 | 0.412 | 0.435 |
| RL21_ECOLI | 1vs6R | NO | 103 | 0.184 | 0.187 | 0.204 | 0.209 | 0.389 |
| RL27_ECOLI | 1vs6W | NO | 85 | 0.138 | 0.179 | 0.170 | 0.177 | 0.348 |
| RS16_ECOLI | 1vs5P | YES | 82 | 0.210 | 0.241 | 0.336 | 0.336 | 0.377 |
| RUSA_ECOLI | 2h8eA | YES | 120 | 0.330 | 0.374 | 0.306 | 0.307 | 0.378 |
| YBCO_ECOLI | 3g27A | YES | 96 | 0.330 | 0.328 | 0.416 | 0.407 | 0.356 |
| YEEU_ECOLI | 2h28A | YES | 122 | 0.327 | 0.329 | 0.363 | 0.378 | 0.344 |
| YFJZ_ECOLI | 2ea9A | YES | 105 | 0.313 | 0.320 | 0.393 | 0.398 | 0.359 |
| YHBY_ECOLI | 1ln4A | YES | 97 | 0.260 | 0.436 | 0.488 | 0.515 | 0.453 |
| ZAPB_ECOLI | 2jeeA | NO | 81 | 0.711 | 0.490 | 0.512 | 0.523 | 0.786 |
| Average | 102 | 0.295 | 0.318 | 0.366 | 0.371 | 0.436 | ||
Although without using global templates, QUARK generates backbone model predictions with an average TM-score = 0.366, where three targets (ARIR_ECOLI, PTHP_ECOLI and ZAPB_ECOLI) have a TM-score > 0.5. After the atomic-level refinement of ModRefiner25, the number of targets with correct fold increase to 4 (with YHBY_ECOLI added) and the average TM-score increases to 0.371, which is 17% higher than that by Modeller.
In the last column of Table I, we also present the estimated TM-score based on the confidence score (Eqs. 1 and 2 in Methods), which is on average 19% higher than the actual TM-score. This difference is mainly caused by proteins with the irregular shape (including trivial super-long tail and open helices etc, as listed in Column 3 in Table I), where QUARK tends to over-predict the quality due to the well-contracted packing for these proteins in the assembly simulations. If we exclude these irregular targets, the estimated TM-score of the best in top 5 models is very close to the actual TM-score (0.409 vs. 0.405).
Illustrative example from E. coli YmgB
In Figures 2a–d, we show two successful examples of QUARK assembly simulations in this set. Figure 2a is an α-protein of three-helix bundle from E. coli YmgB; this protein is critical for biofilm formation and acid-resistance26. QUARK folds the target with a high accuracy (RMSD = 2.91 Å, TM-score = 0.61). Although the target was solved in the dimmer form (PDB ID: 2oxl), each monomer has a hydrophobic core on its own, which is constituted by the leucine (Leu), isoleucine (Ile) and valine (Val) residues.
Figure 2. Examples of successful QUARK modeling results on known Hard E. coli proteins.

In the structural superposition, QUARK models and experimental structures are shown in green and red cartoons respectively. (a) Superposition of the QUARK model and the experimental structure for ARIR_ECOLI with side-chains of the seven hydrophobic residues highlighted. (b) Solvent accessibility distributions for ARIR_ECOLI with data from sequence-based prediction, QUARK model and experimental structure, respectively. (c) Superposition of the QUARK model and experimental structure for PTHP_ECOLI, where the beta-turns are highlighted in blue in the experimental structure. (d) The four-state secondary structure distribution of PTHP_ECOLI shown for sequence-based prediction, the QUARK model and the experimental structure. Coil, helix, strand and turn are marked in green, red, yellow and blue, respectively. (e) Superposition of the QUARK model and the experimental structure for RSD_ECOLI, which contains 158 residues.
In Figure 2b, we plot the solvent accessibilities of the native structure and the model as calculated by EDTSurf27, in comparison with the solvent accessibility prediction by neural network (NN) (Residues 1–24 missed in experimental structure are not shown). The average difference of solvent accessibility between the model and the native structure is 7.4%, while the difference between the sequence-based NN prediction and the native structure is 10.6%. Although QUARK starts from the sequence-based solvent accessibility predictions, the QUARK assembly simulations improve the packing of helices by the incorporation of the inherent knowledge-based force field.
Seven hydrophobic residues are highlighted in Figure 2b whose actual solvent accessibility values are below 0.1. Except for Val72, all the residues in the QUARK model have the same solvent accessibility as that of the native structure. As shown in Figure 2a, the hydrophobic interactions between the six residues in the second and third helices, as well as the rotamer orientations, in the QUARK model are highly close to that in the native structure. The modeling of these interactions is the key for QUARK to correctly pack the orientation of the two helices.
Illustrative example from E. coli HPr
Target in Figure 2c is an αβ-protein from E. coli histidine-containing phosphocarrier protein HPr, containing three short helices paired with four β-strands. In the QUARK model, the three helices and three strands have the same orientation as the native structure, which results in a reasonably high TM-score = 0.57. Two long-range β-strands, which have a sequence separation of 28 residues, are successfully drawn together to form an antiparallel β-sheet. However, the N-terminal β-strand is misplaced in the model.
In the native structure, there are 7 β-turns as defined by the PROMOTIF program28. These β-turns (shown in blue in Figure 2c) determine the relative orientation of the three helices and four β-strands, since each β-turn joins two secondary structure elements together. In our sequence-based NN prediction, 7 β-turns were initially predicted from sequence where 6 of them agree with the PROMOTIF assignments. The QUARK simulations eventually construct 8 β-turns in the predicted model, which cover all the 7 predicted β-turns.
In Figure 2d, we show the distribution of standard secondary structure (SS) elements (coil, helix, strand) along the sequence, where the SS in native structure and QUARK model is defined by DSSP29 and that in target sequence is predicted by PSSpred (http://zhanglab.ccmb.med.umich.edu/PSSpred). Compared with the DSSP assignment, the Q3 accuracy of the PSSpred is 86%. This high accuracy of SS and β-turn predictions is essential to the successful QUARK modeling for this target.
Summary of QUARK modeling on 22 large proteins
In Table II, we summarize the QUARK folding results on 22 big hard proteins which have the length longer than 150 residues. The average TM-score of this set of proteins (0.323) is lower than that of the smaller proteins (0.371), which is however 16% higher than that of template-based modeling by Modeller (TM-score = 0.279). There are five proteins (CHEZ_ECOLI, MOAC_ECOLI, RSD_ECOLI, YFBM_ECOLI, NCPP_ECOLI) which have TM-score > 0.4, and one protein with TM-score > 0.5 (RSD_ECOLI). Again, after excluding the proteins with irregular topology, the estimated TM-score of the QUARK models (0.337) is close to the actual TM-score to the native structure (0.338).
Table 2. Summary of template-based modeling and ab initio modeling on 22 known E. coli proteins with length > 150 residues.
| TM-score of the best in top 5 models | ||||||||
|---|---|---|---|---|---|---|---|---|
| UniProt Entry | PDB ID | Compact | Length | Template | Modeller | QUARK | ModRefiner | Estimated |
| CHEZ_ECOLI | 1kmiZ | NO | 214 | 0.349 | 0.358 | 0.441 | 0.430 | 0.515 |
| PTGA_ECOLI | 1glaF | YES | 169 | 0.172 | 0.232 | 0.241 | 0.248 | 0.319 |
| ECOT_ECOLI | 1ecyA | NO | 162 | 0.175 | 0.265 | 0.194 | 0.194 | 0.325 |
| GCH1_ECOLI | 1fbxA | NO | 222 | 0.192 | 0.195 | 0.250 | 0.245 | 0.364 |
| GFCB_ECOLI | 2in5A | YES | 214 | 0.310 | 0.342 | 0.321 | 0.326 | 0.354 |
| HFLD_ECOLI | 1qz4A | YES | 213 | 0.240 | 0.258 | 0.350 | 0.354 | 0.392 |
| ISPF_ECOLI | 3esjA | YES | 159 | 0.232 | 0.261 | 0.349 | 0.342 | 0.352 |
| MOAC_ECOLI | 1ekrA | YES | 161 | 0.277 | 0.330 | 0.399 | 0.405 | 0.364 |
| MUKE_ECOLI | 3euhC | NO | 234 | 0.225 | 0.245 | 0.269 | 0.274 | 0.342 |
| PAGP_ECOLI | 3gp6A | YES | 186 | 0.212 | 0.244 | 0.223 | 0.222 | 0.280 |
| PHNH_ECOLI | 2fsuA | YES | 194 | 0.216 | 0.338 | 0.261 | 0.260 | 0.324 |
| IPYR_ECOLI | 1inoA | YES | 176 | 0.221 | 0.236 | 0.260 | 0.267 | 0.334 |
| RIBA_ECOLI | 2bz0B | YES | 196 | 0.243 | 0.316 | 0.350 | 0.346 | 0.356 |
| RSD_ECOLI | 2p7vA | YES | 158 | 0.205 | 0.297 | 0.587 | 0.585 | 0.455 |
| SEQA_ECOLI | 3fmtA | NO | 181 | 0.227 | 0.246 | 0.340 | 0.342 | 0.372 |
| YAEQ_ECOLI | 3c0uB | YES | 181 | 0.256 | 0.299 | 0.348 | 0.348 | 0.326 |
| YCEB_ECOLI | 3l6iA | NO | 186 | 0.209 | 0.259 | 0.235 | 0.233 | 0.354 |
| YECM_ECOLI | 1k4nA | YES | 188 | 0.274 | 0.303 | 0.260 | 0.259 | 0.330 |
| YFBM_ECOLI | 1rylA | YES | 167 | 0.192 | 0.255 | 0.399 | 0.401 | 0.364 |
| YFEY_ECOLI | 2qzbB | YES | 191 | 0.185 | 0.189 | 0.313 | 0.317 | 0.296 |
| NCPP_ECOLI | 1u5wB | YES | 170 | 0.285 | 0.469 | 0.453 | 0.453 | 0.369 |
| ZINT_ECOLI | 1txlA | YES | 216 | 0.187 | 0.208 | 0.257 | 0.254 | 0.310 |
| Average | 188 | 0.231 | 0.279 | 0.323 | 0.323 | 0.354 | ||
Figure 2e presents the superposition of the QUARK model and the target structure from RSD_ECOLI. The experimental structure contains 4 super-long helices and one short helix in the N-terminal. The QUARK modeling correctly assembles the topology of the helix bundle with a minor error in the orientation in the short N-terminal helix.
Overall, the average TM-score of the QUARK models is 16.5% higher than that by Modeller using LOMETS templates in the 43 Hard proteins, which corresponds to a p-value = 0.00019 in the paired Student's t-test. These modeling results are consistent with the performance of QUARK in blind CASP experiments, in terms of the folding rate of FM targets for both small and large size proteins.
Summary of ab initio structure predictions for unknown E. coli proteins
QUARK ab initio folding algorithm is used to generate 3D structures for all 495 Hard protein targets in the E. coli genome which have no reliable templates identified by LOMETS. Since the experimental structures are unsolved, to guide the use of the QUARK predictions, we provide an estimated TM-score for each target based on the confidence score calculations, which are benchmarked mainly for single-domain proteins (see Eqs. 1 and 2). For multiple domain proteins which all have domains modeled separately (see Methods), the estimated TM-score is calculated as a sum of TM-score of individual domains, with weight proportional to the length of the domains.
In Figure 3, we present the histogram of the estimated TM-score for the unknown E. coli protein set, in comparison with that for an independent benchmark set of 145 non-redundant globular proteins randomly selected from the PDB library with length in [70, 150] residues (see Methods). The two histograms generally agree with each other. However, the unknown E. coli protein set has a slightly higher percentage of proteins in the low TM-score regions than the benchmark set, which is mainly due to the fact that the average length of the unknown E. coli proteins is longer than that in the benchmark test set (155 vs. 107 residues). We also show the distribution of the actual TM-score to the native for the benchmark set proteins in the figure (curve with triangle), which has a slightly flatter shape than that of the estimation although the average values of them are almost the same. For the proteins with TM-score ≥ 0.45, however, the distributions of the estimated and the actual TM-scores become very close. Hence, we can infer that the estimated TM-score for the unknown E. coli proteins should be most trustable for the proteins with a TM-score ≥ 0.45.
Figure 3. Histograms of estimated and actual TM-scores.

The blind set is from 495 unknown E. coli hard sequences and the benchmark set consists of 145 non-redundant proteins from the PDB.
In total, there are 72 out of 495 targets whose estimated TM-score is higher than 0.5, which are supposed to have the same folds as their native structures30. Among the 72 successfully folded targets, 67 are small proteins with length shorter than 150, and 62 are α-proteins. This data highlights the fact that QUARK works better for small proteins and α-proteins, which partially because these proteins generally have a smaller conformational space (i.e. with simpler topology) than the big proteins and β-proteins, and therefore relatively easier to fold by ab initio folding approaches. Nevertheless, the majority of the hard targets (64.8%), including big proteins and β-proteins, have a prediction with a significant TM-score estimation > 0.35.
Modeling of transmembrane proteins
The modeling of membrane protein structures has been considered as a major challenge to computational structure predictions, because there are too few membrane proteins in the PDB which can be used as templates. Structurally, the strong hydrogen bonding in the membrane causes the backbone to form regular secondary structures, with the major conformational variances from the arrangements of secondary structure elements and various loop connections. Such structural characteristics are consistent with the QUARK methodology, where regular secondary structure elements are excised from other proteins and used to rearrange the global topology (see Methods).
Among the 72 successfully folded targets, 28 are from membrane proteins. Figure 4 shows one example of the high confidence prediction for YQJK_ECOLI, where the first QUARK model has an estimated TM-score = 0.727. Following the ab initio solvent accessibility prediction, most residues in the region of [35T, 86R] are hydrophobic except for the loop region that connects the two helices. QUARK therefore folded the target into 2 domains, i.e. a C-terminal transmembrane domain with two helix bundle embedded in the lipid bilayer plus a single-helix extracellular domain. This topology is consistent with the transmembrane prediction from MEMSAT331 although the latter was not exploited in the QUARK assembly simulations.
Figure 4. QUARK modeling result for transmembraine protein YQJK_ECOLI in E. coli.

(a) Cartoon representation of the model. Side-chains of residues 35T, 62I and 71L, which mark the location of lipid bilayer, are highlighted in sticks. (b) Predicted secondary structure type and solvent accessibility for the target.
SCOP fold family assignments of E. coli proteins
As an application of the genome-wide structure prediction, we assign the E. coli proteins with standard fold families by matching the ab intio models with known structures in the SCOP family database20. We first compare the top QUARK models with the proteins in the PDB using the structural alignment algorithm TM-align32. If the QUARK model includes multiple domains, DomainParser33 will be used to split the chain to domains. The PDB structures are then listed in descending order based on their TM-score value to the QUARK models. The nearest neighbor classification method34 is then used to classify the predicted models based on the TM-score list. In case that the top PDB structure has no SCOP code in the SCOP database, the code of the protein that is closest to the QUARK model is used. Here, we note that the TM-score is calculated as the average of the two TM-scores which are normalized by the target length and the analogy length separately. We found that the TM-score normalized by the target length may pick up some big proteins with artificial alignments while the use of average TM-score from both target and analog proteins help recognize the closest analogs with the similar size.
Benchmark of fold assignment strategy
To examine the accuracy of this fold assignment strategy, we apply the procedure on the 688 E. coli sequences that have both known structure and SCOP ID. If we use the native structure as a probe to identify the closest PDB structure, 97% of targets can be assigned to a SCOP code which is correct in the most detailed family level (the remaining 3% were mis-assigned due to the close structural similarity between proteins in the same families). If we use the predicted models as probe, the targets are correctly assigned in the family, superfamily and fold levels for 96%, 97% and 97% of cases, respectively. These results confirm the feasibility of the structural analogy-based approach in fold family assignments for the E. coli proteins.
Moreover, the TM-score between predicted models and the most similar analogs in the PDB is found highly correlated with the actual TM-score of the predicted model to the native. Figure 5 show the actual TM-score of predicted QUARK models versus the TM-score between model and its closest analog identified by TM-align from the PDB, which has a Pearson's correlation coefficient 0.71 for the first model or 0.72 for the best in top five models. Accordingly, a positive correlation between the accuracy of the SCOP family assignment and the TM-score between QUARK model and the analogy protein was observed, i.e. the targets with a closer analogy in the PDB have generally a higher successful rate of fold assignment than that without close analogy (data not shown).
Figure 5. TM-score of the QUARK models versus TM-score between model and its closest analogy for the benchmark set proteins.

Examples of SCOP family assignments on known E. coli proteins
In Figure 6, we show two typical examples on the successful fold family assignments at different level of modeling accuracy. Figure 6a is an example from the histidine-containing phosphocarrier protein HPr (PTHP_ECOLI), the experimental structure of which has an open-faced beta-sandwich fold, consisting of four antiparallel beta-strands and three alpha-helices. The QUARK model has the global topology correctly modeled but with the first strand shifted and the last helix tilted, which results in a medium TM-score ( = 0.503) to the native. The TM-align search identified an analog from another HPr protein from Bacillus subtilis. Interestingly, this analogy protein has a structure much closer to the target than the QUARK model that was used as a probe for the structure search. This high structural analogy helps to correctly assign PTHP_ECOLI to the SCOP HPr-like family (d.94.1.1) at the most detailed family level.
Figure 6. SCOP fold family assignment results.

(a) PTHP_ECOLI; (b) NCPP_ECOLI. The QUARK models, the closest analogy structures in the PDB, and the experimental structure of targets are shown in Columns 1, 2 and 3, respectively. Blue to red runs from N- to C-terminals.
NCPP_ECOLI in Figure 6b is a hypothetical protein of hitherto unknown function. The QUARK model has a quite low estimated TM-score ( = 0.369). Nevertheless, the two beta-hairpins and paired helices in the core region are correctly assembled in the QUARK simulations, which enable the TM-align structure comparison to pick up an analogous ITPase-like protein from T. brucei (PDB ID: 2amhA). Similar to PTHP_ECOLI, the analogy protein by TM-align for NCPP_ECOLI has a higher TM-score to the native (0.523) which has all four beta-hairpins and the helix domain on the top much better packed compared to the QUARK model. This allows the TM-align structure comparison correctly transfer the SCOP ID at up to the superfamily level (i.e. c.51.4: ITPase-like); but at the family level, 2amhA is Maf-like (c.51.4.2) while NCPP_ECOLI is YjjX-like (c.51.4.3).
Here, although the analogy proteins closer to the target exist in the PDB in both examples, we note that all the analogous proteins have been excluded from the QUARK fragment library. These data demonstrate the advantage of ab initio modeling in fold family assignment. Since the ab inito models are constructed from scratch, a substructure similarity to real PDB structures with a modest TM-score are usually significant and sufficient to recognize the correct global topology for fold family assignment as shown in the above examples.
SCOP family assignment for unknown proteins
When we scan the QUARK predictions of the 495 Hard proteins through the SCOP database, we find 317 targets which have a TM-score between model and analogy above 0.5. Based on correlations observed on the known proteins, these targets should have the highest reliability for the fold family assignments. In Figure 7, we show four illustrative examples of the QUARK predictions and the SCOP family assignment from the 495 hard unknown proteins, which cover different topology of structures and in different range of model qualities.
Figure 7. QUARK models and the closest analogies for four representative E. coli hard sequences.

(a) YFCL_ECOLI; (b) YIJD_ECOLI; (c) YDAF_ECOLI; (d) YDBJ_ECOLI.
First, YFCL_ECOLI in Figure 7a is an uncharacterized protein in the UniProt database. QUARK generated a model of bromodomain-like four-helix bundle, which has a high estimated TM-score = 0.806 because of the high normalized cluster size (0.645) of the first cluster, i.e. 64.5% of the decoy structures converge to this fold although the replica-exchange Monte Carlo simulations start from different random conformations. The closest analogy protein (PDB ID: 1ng6A) has a slightly more tilted helix-hairpin structure which results in a slightly lower TM-score to the model (0.608). Nevertheless, both the high TM-score of estimation and the high TM-score in the analogy protein comparison guarantee that the SCOP family assignment (a.182.1.1: GatB/YqeY domain) is in a range of high confidence prediction.
Figure 7b is an example of transmembrane protein. Most residues in the putative transmembrane region are hydrophobic with low solvent accessibility value according to the QUARK prediction. The QUARK model has a relatively low TM-score estimated (0.448) due to the low decoy converge rate (0.134). However, the TM-score between the model and the closest analogy is high ( = 0.775); such similarity is highly significant considering that the model was constructed from scratch by ab intio folding. The target is eventually assigned into the IVS-encoded protein-like family (SCOP ID: a.29.16.1), following the analogy structure comparison.
Figure 7c is for the target YDAF_ECOLI. The QUARK simulations generate a model prediction of 4 antiparallel beta-strands sandwiched with an alpha-helix at the N-terminal. The prediction has a confidently estimated TM-score ( = 0.502), which is structurally closest to Disulfide bond isomerase, DsbC, N-terminal domain. The target is therefore assigned to the family of the N-terminal domain of Thiol:disulfide interchange protein DsbG (SCOP ID: d.17.3.1), which has a high confidence based on the estimated TM-score of the modeling and the close analogy to the template.
Finally, the target YDBJ_ECOLI in Figure 7d is also an αβ-protein which has a relatively low estimated TM-score (0.416). The sequence contains six cysteines, which form two well-packed disulfide bonds in the QUARK full-atomic model as highlighted in the left of Fig. 7d. The TM-align search picks up a close analog structure from the periplasmic protein in Bacteroides vulgatus ATCC 8482 with a TM-score = 0.510, which categorizes the target in the BT0923-like family (SCOP ID: d.98.2.1).
Discussion
We developed a hybrid pipeline to predict 3D structure of all sequences in the E. coli genome, where targets with close homologous templates are generated by threading-based fragment assembly method (I-TASSER)19 and those without homologous templates built by ab initio folding algorithm (QUARK)17. The emphasis of the study is on the ab initio folding of the hard proteins which has been a major obstacle in the genome-wide structure prediction studies.
We first benchmarked the algorithms on the 43 Hard E. coli proteins with structures experimentally solved, which demonstrate that the quality of the models by ab initio folding is significantly higher than that by traditional comparative modeling approaches. Although no templates are used, QUARK built models of correct fold (TM-score > 0.5) for five targets (ARIR_ECOLI, PTHP_ECOLI, ZAPB_ECOLI and RSD_ECOLI) where none of them can be generated by the template-based modeling approaches. On average, the TM-score of the QUARK ab initio models is 16-17% higher than that of the template-based modeling for both small and large-size proteins, corresponding to a statistically significant p-value (< 2 × 10−4) in paired Student's t-test.
The QUARK ab initio folding algorithm is applied to model all 495 Hard proteins in the E. coli genome, where 72 protein targets, including 28 transmembrane proteins, are predicted to have correct folds with an estimated TM-score > 0.5. In addition, 321 (64.8%) of the targets have a substantial fraction of structures correctly modeled with an estimated TM-score > 0.35. To assign the fold family of the E. coli proteins, we match the ab initio models to the known structures in the SCOP database. 317 targets have a close analogy with a TM-score > 0.5 where a reliable SCOP family assignment can be obtained for the target sequences.
In conclusion, despite the rapid accumulation of the experimental structures in the PDB library, there are still a substantial number of proteins in genomes which lack close homologous templates that can be detected by current fold-recognition approaches. The data analysis in this study shows that the current state of the art ab initio folding procedure is ready to generate useful structural and function predictions for the hard proteins of small-to-medium size. Further development of ab initio fold algorithms, including the hybrid approaches combining sparse spatial restraints from NMR and mutagenesis experiments, should significantly enlarge the scope of the genome-wide structure prediction and structure-based function annotations.
Methods
Outline of genome-wide structure prediction procedure
The procedure for modeling the tertiary structures of the entire E. coli genome is illustrated in Figure 8. For a given sequence, the multiple-threading program, LOMETS21, is used to detect homologous templates from the PDB library. For each threading program, the significance of the target-template alignments is measured by Z-score which is defined by the difference of raw alignment score and the mean in the unit of derivation. A target sequence is defined as “Hard” if none of the threading programs in LOMETS detects a template with Z-score higher than the specific cutoffs; a target is defined as “Easy” if on average at least one template per threading program has the Z-score higher than the cutoff; otherwise, the target is classified as a “Medium” target.
Figure 8. Flowchart of structure modeling and fold family assignment for E. coli genome sequences.

QUARK17 is developed to model the structure of the Hard sequences, where a set of 200 structural segments with length from 1 to 20 residues are first generated at each position of the sequence by gapless threading. Full-length models are then assembled from the segments by replica-exchanged Monte Carlo (REMC) simulations35, which are accommodated by a composite knowledge- and physics-based force field. Meanwhile, non-covalent contact and distance profiles are extracted from the segments that come from the same PDB structures, which are used to guide the REMC structural assembly simulation18. To facilitate the movements, protein conformations are specified in two sets of Cartesian and torsion-angle systems, both being at a reduce-level (i.e. each residue is represented by the backbone heavy atoms and the side-chain center of mass).
For each target, 10 parallel QUARK simulations are implemented, each starting from a different random number. In each simulation, 200 cycles of REMC sweeps are conducted. Following the simulations, structural decoys from the last 150 cycles in the 10 low-temperature replicas are submitted to SPICKER36 for structure clustering. Finally, the full-atomic models are built from the top cluster models by ModRefiner which refines the hydrogen-bonding network and physical realism through a two-step Monte Carlo based energy minimization25.
For the proteins of multiple domains, we first split the sequences into individual domains based on the LOMETS threading alignments and the secondary structure prediction from PSSpred. Full-length models are then constructed by QUARK for each domain. In the second step, the domain models are assembled together by 1,000 short Monte Carlo simulation runs, which start from random connection of the domain structures on different orientations. During the simulations, the domain structures are kept rigid, while regions in the domain linkers are flexible which keeps the domain orientation updated. The domain assembly is guided by the same knowledge-based potential as used by QUARK. Finally, the conformation with the lowest energy is selected as the final full-length model. For the 495 Hard E. coli proteins, we found that 61 proteins are putative multi-domain targets which are modeled using this procedure. Table S1 lists the domain parsing results of the targets which all have more than 250 residues.
For the Easy/Medium targets, we use the standard I-TASSER pipeline to generate the full-length models, which was designed to reassemble the continuous fragments from the threading alignments19. I-TASSER has been extensively tested in both benchmarking and blind experiments, and consistently ranked as the best method for template-based protein structure predictions in recently CASP experiments37,38,39,40,41.
Estimation of model accuracy
It is important to estimate the accuracy of the predicted models without knowing their experimental structures, since this estimation will essentially determine how the biologists use our model predictions.
Here, we find that two factors are highly correlated with the actual quality of the final QUARK models, which may be used as quantitative estimators of the modeling accuracy. First, in general Metropolis Monte Carlo simulations, the number of decoys at each conformational cluster nc is proportional to the partition function Zc, i.e. 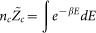 . The logarithm of normalized cluster size is then related to the free-energy of the simulation, i.e.
. The logarithm of normalized cluster size is then related to the free-energy of the simulation, i.e. 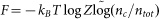 , where ntot is the total number of decoys submitted for clustering. In other word, the conformation with the larger cluster size should correspond to the state of lower free-energy. As a test, in Figure S1a we present the TM-score of the final models versus the normalized cluster size (nc/ntot) on a set of 145 non-redundant benchmark proteins with known structures, which indeed shows a strong correlation with a Pearson's correlation coefficient (PCC) equal to 0.73 for the first model and 0.76 for the best in top five models.
, where ntot is the total number of decoys submitted for clustering. In other word, the conformation with the larger cluster size should correspond to the state of lower free-energy. As a test, in Figure S1a we present the TM-score of the final models versus the normalized cluster size (nc/ntot) on a set of 145 non-redundant benchmark proteins with known structures, which indeed shows a strong correlation with a Pearson's correlation coefficient (PCC) equal to 0.73 for the first model and 0.76 for the best in top five models.
Second, the quality of the final models is strongly influenced by the quality of initial fragments that are used to assemble the final models. In Figure S1b, we show the correlation between the average alignment score of the top 6 fragments with a length = 20 and TM-score of the first and the best in top 5 models, which have PCC = −0.59 and −0.65, respectively.
Thus, we define a confidence score, C-score, as the linear combination of the normalized cluster size of the first cluster and the average gapless threading score of the top fragments (f-score):
where the weight w = −0.03 is used to balance the two terms. Figure S1c shows the C-score versus the TM-score of the final QUARK models. The correlation coefficients increase to 0.75 and 0.79 for the first and the best in top 5 models, respectively. If we define a model with TM-score > 0.5 to native as of correct fold and use C-score = −0.258 as the cutoff of correct predictions, the false positive rate and false negative rate are 0.034 and 0.296 respectively for the best in top 5 models. The C-score cutoff here corresponds to the highest Matthews correlation coefficient (MCC) value 0.714.
Following the correlation in Figure S1c, we can further estimate the true TM-score of the predicted models to the native based on the C-score, i.e.
where TM-score1 and TM-score5 are TM-score for the first and best in top five models, respectively. In our benchmark test, the average errors between the estimated and the true TM-scores based on Eq. 2 are 0.0833 and 0.0778 for TM-score1 and TM-score5, respectively.
Author Contributions
D.X. and Y.Z. conceived the project; D.X. conducted the calculations and analyzed the data; D.X. and Y.Z. wrote the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
The project is supported in part by the NSF Career Award (DBI 1027394), the National Institute of General Medical Sciences (GM083107, GM084222), and the NSFC (31128004).
References
- Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N. & Bourne P. E. The Protein Data Bank. Nucleic Acids Res 28(1), 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen O. N. Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol 8(1), 33–41 (2004). [DOI] [PubMed] [Google Scholar]
- Levitt M. & Warshel A. Computer-Simulation of Protein Folding. Nature 253(5494), 694–698 (1975). [DOI] [PubMed] [Google Scholar]
- Lewis P. N., Momany F. A. & Scheraga H. A. Folding of Polypeptide Chains in Proteins - Proposed Mechanism for Folding. P Natl Acad Sci USA 68(9), 2293–& (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccammon J. A., Gelin B. R. & Karplus M. Dynamics of Folded Proteins. Nature 267(5612), 585–590 (1977). [DOI] [PubMed] [Google Scholar]
- Bowie J. U., Luthy R. & Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170 (1991). [DOI] [PubMed] [Google Scholar]
- Sali A. & Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234(3), 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W. & Lipman D. J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17), 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S., Szilagyi A., Roy A. & Zhang Y. Genome-wide protein structure prediction. In: Kolniski A, editor. Multiscale approaches to protein modeling: structure prediction, dynamics, thermodynamics and macromolecular assemblies: Springer-London; p 810–842 (2010). [Google Scholar]
- Fischer D. & Eisenberg D. Assigning folds to the proteins encoded by the genome of Mycoplasma genitalium. Proc Natl Acad Sci U S A 94(22), 11929–11934 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R. & Sali A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins Suppl. 1, 50–58 (1997). [DOI] [PubMed] [Google Scholar]
- Zhang Y. & Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc Natl Acad Sci USA 101, 7594–7599 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmstrom L., Riffle M., Strauss C. E., Chivian D., Davis T. N., Bonneau R. & Baker D. Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. Plos Biol 5(4), e76 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Progress and challenges in protein structure prediction. Curr Opin Struct Biol 18(3), 342–348 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinch L., Yong Shi S., Cong Q., Cheng H., Liao Y. & Grishin N. V. CASP9 assessment of free modeling target predictions. Proteins 79 Suppl 10, 59–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. K. Free modeling assessment in CASP10. 10th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction. Gaeta, Italy; 2012. [Google Scholar]
- Xu D. & Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80(7), 1715–1735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D. & Zhang Y. Towards optimal fragment generations for ab initio protein structure assembly. Proteins 81(2), 229–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Kucukural A. & Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5(4), 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A. G., Brenner S. E., Hubbard T. & Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247(4), 536–540 (1995). [DOI] [PubMed] [Google Scholar]
- Wu S. & Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res 35(10), 3375–3382 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A., Larsson B., von Heijne G. & Sonnhammer E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3), 567–580 (2001). [DOI] [PubMed] [Google Scholar]
- Jones D. T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23(5), 538–544 (2007). [DOI] [PubMed] [Google Scholar]
- Jones D. T. Do transmembrane protein superfolds exist? FEBS Lett 423(3), 281–285 (1998). [DOI] [PubMed] [Google Scholar]
- Xu D. & Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 101(10), 2525–2534 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Page R., Garcia-Contreras R., Palermino J. M., Zhang X. S., Doshi O., Wood T. K. & Peti W. Structure and function of the Escherichia coli protein YmgB: a protein critical for biofilm formation and acid-resistance. J Mol Biol 373(1), 11–26 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D. & Zhang Y. Generating triangulated macromolecular surfaces by Euclidean distance transform. PLoS One 4(12), e8140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson E. G. & Thornton J. M. PROMOTIF--a program to identify and analyze structural motifs in proteins. Protein Sci 5(2), 212–220 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. & Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22(12), 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- Xu J. & Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26(7), 889–895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. T., Taylor W. R. & Thornton J. M. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33(10), 3038–3049 (1994). [DOI] [PubMed] [Google Scholar]
- Zhang Y. & Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33(7), 2302–2309 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Xu D. & Gabow H. N. Protein domain decomposition using a graph-theoretic approach. Bioinformatics 16(12), 1091–1104 (2000). [DOI] [PubMed] [Google Scholar]
- Cover T. M. & Hart P. E. Nearest neighbor pattern classification. IEEE Trans Inf Theory 13(1), 21–27 (1967). [Google Scholar]
- Zhang Y., Kihara D. & Skolnick J. Local energy landscape flattening: Parallel hyperbolic Monte Carlo sampling of protein folding. Proteins 48, 192–201 (2002). [DOI] [PubMed] [Google Scholar]
- Zhang Y. & Skolnick J. SPICKER: a clustering approach to identify near-native protein folds. J Comput Chem 25(6), 865–871 (2004). [DOI] [PubMed] [Google Scholar]
- Kopp J., Bordoli L., Battey J. N., Kiefer F. & Schwede T. Assessment of CASP7 predictions for template-based modeling targets. Proteins 69(S8), 38–56 (2007). [DOI] [PubMed] [Google Scholar]
- Battey J. N., Kopp J., Bordoli L., Read R. J., Clarke N. D. & Schwede T. Automated server predictions in CASP7. Proteins 69(S8), 68–82 (2007). [DOI] [PubMed] [Google Scholar]
- Cozzetto D., Kryshtafovych A., Fidelis K., Moult J., Rost B. & Tramontano A. Evaluation of template-based models in CASP8 with standard measures. Proteins 77 Suppl 9, 18–28 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani V., Kiefer F., Schmidt T., Haas J. & Schwede T. Assessment of template based protein structure predictions in CASP9. Proteins 79 Suppl 10, 37–58 (2011). [DOI] [PubMed] [Google Scholar]
- Montelione G. T. Template based modeling assessment in CASP10. 10th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction. Gaeta, Italy; 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information