

Abstract
Although the mutated protein causing Huntington's disease (HD) is expressed throughout the body, the major pathology of HD is localized to the striatum of the brain. We previously reported that the striatal-enriched protein Rhes binds the mutated huntingtin protein and enhances its cytotoxicity. We now demonstrate that Rhes-deleted mice are dramatically protected from neurotoxicity and motor dysfunction in a striatal-specific model of HD elicited by 3-nitropropionic acid. This finding suggests that Rhes may, in part, determine the striatal selectivity of HD.
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease manifested by highly selective destruction of the brain's corpus striatum (Ross and Tabrizi, 2011). Surprisingly, the gene mutated in HD encodes a protein (Htt) uniformly expressed throughout the brain and body. Rhes, or Ras homolog enriched in striatum, is a unique small G-protein that displays striking enrichment in the striatum and can regulate signaling through G-protein-coupled receptors (Falk et al., 1999; Vargiu et al., 2004; Mealer and Snyder, 2012). We recently reported that Rhes binds physiologically to Htt and with markedly greater avidity to mutant Htt (mHtt) (Subramaniam et al., 2009). This binding of Rhes markedly augments mHtt cytotoxicity, a finding recently confirmed in a primary striatal neuron model of HD (Seredenina et al., 2011). Many post-translational modifications of mHtt have been identified (Ehrnhoefer et al., 2011). One of these, sumoylation, decreases aggregation to promote toxicity (Steffan et al., 2004). Rhes enhances sumoylation of mHtt in cell lines to account for the stimulation of cytotoxicity (Subramaniam et al., 2009). These observations imply that Rhes is responsible for the selective striatal damage and associated neurobehavioral abnormalities of HD, and that drugs inhibiting Rhes or blocking Rhes–mHtt binding may delay/prevent onset of HD.
We tested this hypothesis using Rhes-deleted mice in a rapid and striatal-selective model of HD. Genetic mouse models of HD often employ very large polyglutamine repeats, producing global brain dysfunction resembling juvenile HD to a greater extent than the striatal-specific and more common adult-onset HD (Crook and Housman, 2011). By contrast, systemic treatment with 3-nitropropionic acid (3-NP), a mitochondrial complex II inhibitor, produces a striatal-specific lesion and attendant movement disorder resembling HD (Beal et al., 1993). The lesion caused by 3-NP is characterized by extensive loss of striatal medium spiny neurons, while sparing striatal nNOS (NADPH-diaphorase) interneurons as well as neurons in the other brain regions, a hallmark of human HD. However, the precise mechanisms accounting for the selective toxicity of 3-NP remain a mystery (Brouillet et al., 2005). We report that Rhes deletion profoundly diminishes striatal destruction and associated motor defects caused by 3-NP, suggesting that Rhes is responsible for the striatal susceptibility to both 3-NP and HD.
Materials and Methods
Rhes-knockout mice.
Rhes-knockout mice were generated as described previously (Spano et al., 2004). All experiments were performed in accordance with the guidelines established by the Animal Care and Use Committee at Johns Hopkins University School of Medicine under protocol no. MO10M17. Wild-type (WT) and Rhes-deleted (knockout, KO) mice were genotyped by Transnetyx. All mice used in this study were males between 12–16 weeks of age and were from lines previously backcrossed to C57/BL6 mice (The Jackson Laboratory).
3-Nitropropionic acid.
3-Nitropropionic acid injections were based on protocols previously reported for mice (Blum et al., 2003). In brief, 3-NP (Sigma) was dissolved to 10 mg/ml in sterile 0.1 m PBS and adjusted to pH 7.4 with sodium hydroxide. For lesion studies and behavioral analysis, mice received intraperitoneal (IP) injections of 60 mg/kg 3-NP twice a day, with 2 h between injections for 3 consecutive days. For the survival study, mice received a single IP injection of 3-NP starting at 30 mg/kg, increased daily by 15%. The experimenter was blinded to genotype for both injections and data analysis.
Lesion analysis.
Lesion analysis was performed after 3 days of 3-NP injections. Transcardial perfusion employed 4% PFA in 0.1 m PB, pH 7.2. Brains were removed and postfixed in 4% PFA for 2 h and then incubated in 30% glycerol overnight. Five micrometer sections were taken every 50 μm through the entirety of the striatum and stained using H&E. Sections were examined for pyknotic nuclei and a paucity of staining representing striatal lesions. 3-NP lesions were localized to the dorsolateral striatum, consistent with previous findings in rodents (Beal et al., 1993; Blum et al., 2003). The lesion areas with the largest diameter were consistently centered between bregma +0.4 and 0. Slides with the largest lesion diameter were scanned for quantification by the Tissue MicroArray Core facility at Johns Hopkins University, and lesion area was determined using Aperio ImageScope software.
Beam crossing.
Bean crossing was measured on a 100 cm steel rod, diameter 14 mm, suspended ∼30 cm from the bench top with a safety box at one end. For 2 days mice were habituated to the safety box for 10 min and then removed. The training protocol was 4 days: 2 days traversing half the distance of the beam (50 cm), and 2 days traversing the entirety of the beam (100 cm). Training sessions consisted of 10 min habituation in the safety box, followed immediately by 4 attempts to cross the bar, each separated by 2 min to allow the mice to rest. After training, daily performance was measured after a 10 min habituation period with 4 crossing attempts per mouse. Baseline performance was measured on day 0 before injections, and testing was performed each subsequent morning before injections for 3 days. Sessions were video taped and reviewed later for time to cross the 100 cm beam, the number of foot slips below the bar, the average distance traveled on each attempt, and the percentage of failed attempts.
Open field analysis.
Open field analysis was performed using the TruScan System from Coulbourn Instruments. Data were collected each day before the 3-NP injections. Horizontal activity represents the total distance traveled in 15 min, normalized to the animal's baseline activity. Total rears represent the number of times the mouse crossed above a vertical plane during the 15 min session.
Succinate dehydrogenase activity assay.
Succinate dehydrogenase (SDH) activity assay was adapted from previous reports (Brouillet et al., 1998; Blum et al., 2003). WT and KO mice were administered a single 120 mg/kg dose of 3-NP (or an equivalent volume of PBS) and killed 6 h later. Brains were quickly removed, rinsed with cold PBS, dissected on a cold surface into various brain regions, and snap frozen with liquid nitrogen. Brains were Dounce homogenized in cold PBS, and a crude mitochondrial fraction was isolated by 2 rounds of differential centrifugation: total homogenate was spun at 1000 × g for 4 min to remove nuclei and cell debris, and then the supernatant was spun at 15,000 × g for 15 min. The resulting pellet containing the mitochondria was resuspended in fresh PBS, and protein concentration was quickly measured using Coomassie Plus (Bradford) Protein Assay (Thermo Scientific). Mitochondria (25 μg) were incubated in 400 μl PBS with 0.3 mm nitro blue tetrazolium (Sigma) and 0.05 m sodium succinate (Sigma) for 10 min at 37°C. The insoluble formazan end product was pelleted by centrifugation at 15,000 × g for 2 min and resuspended by sonication in DMSO. The absorbance at 695 nm was quickly measured, and samples were compared against controls without sodium succinate as an electron acceptor, representing the nonspecific reduction of nitro blue tetrazolium by enzymes other than SDH.
Statistical analysis.
Statistical analysis was performed using Student's t test with results being considered significant if p < 0.05. Data are expressed as means ± SEM. Sample sizes are indicated in the figure legends.
Results
We confirm previous reports that acute systemic treatment with 3-NP in wild-type mice elicits macroscopic lesions in the dorsolateral striatum (Fig. 1A). By contrast, Rhes KO mice are almost totally protected from the striatal lesions caused by 3-NP (Fig. 1B). Whereas 9 of 10 WT mice display large striatal lesions, 8 of 10 KO mice fail to manifest any lesion whatsoever, and the lesions present in the two mutant mice are generally much smaller than those of their WT counterparts (Fig. 1C) (mean lesion area ± SEM: WT 131.3 ± 32.5 vs KO 16.8 ± 10.8 μm × 104, p value <0.005). Microscopic analysis of the 3-NP-induced lesions demonstrates pyknotic nuclei and a paucity of neural staining in WT mice (Fig. 1D), while KO striata display healthy nuclei and normal neural staining (Fig. 1E).
Figure 1.
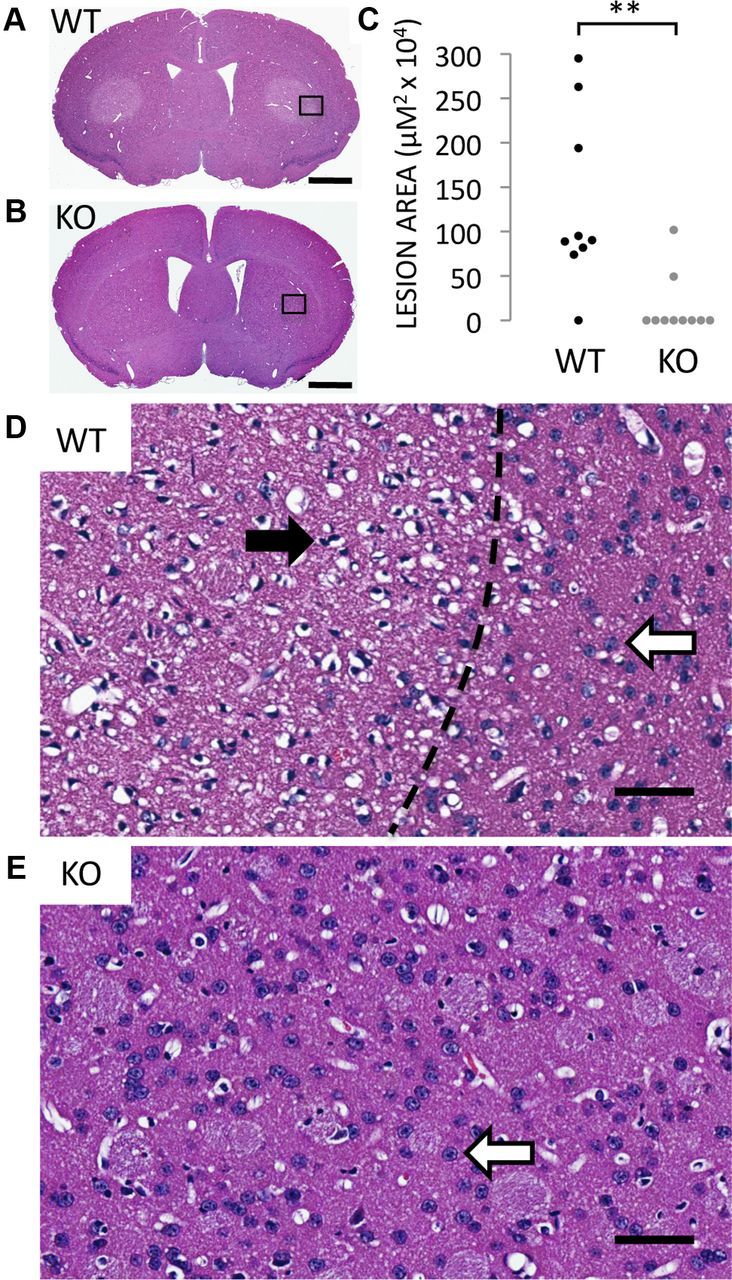
Rhes-deleted mice are protected from striatal lesions caused by systemic 3-NP intoxication. A, B, H&E stain of striatum from WT (A) and Rhes-deleted (KO) mice (B) treated with 3-NP. Scale bar, 1 mm in A and B. C, Area of striatal lesions caused by 3-NP; n = 9 for WTs, n = 10 for KOs; **p < 0.005. D, High-magnification view of region indicated in A. Dotted line indicates border between lesioned and normal striata. Closed arrowhead points to a pyknotic nucleus from an apoptotic neuron, and open arrowheads point to normal nuclei from healthy neurons. E, High-magnification view of region indicated in B. Scale bars, 50 μm for D and E.
Motoric abnormalities associated with 3-NP lesions are also markedly reduced in Rhes KO mice. Following 2 days of 3-NP, the time required to cross a 100 cm balance beam is reduced by 40% in KO mice compared to WT controls (Fig. 2A) (WT 40.0 ± 3.9 vs KO 24.5 ± 4.2 s, p value <0.005). Rhes KO mice also manifest improved performance on other measures of motor function and coordination in this task, including a reduction in foot slips (Fig. 2B) (WT 1.7 ± 0.44 vs KO 0.41 ± 0.02 foot slips/trial, p value <0.005), increased distance traveled (Fig. 2C) (WT 38.4 ± 11.0 vs KO 75.5 ± 8.6 cm, p value <0.0005), and a decrease in failed crossings (Fig. 2D) (WT 71.8 ± 12.9% vs KO 32.5 ± 11.2%, p value <0.05). Although Rhes knockout mice have been reported to have a small decrease in motor function on the rotarod test (Spano et al., 2004), this phenotype was not reproduced in a subsequent study using mice on a C57/BL6 genetic background (Errico et al., 2008). We failed to detect any difference from WT mice in the ability of Rhes KO mice to learn the balance beam task or in baseline performance.
Figure 2.

Rhes KO mice are protected from motor dysfunction after 2 days of 3-NP treatment. Time to cross a balance beam (A), number of foot-slips per trial (B), distance traveled before falling (C), and the percentage of trials ending in failure (D) are indicated. *p < 0.05, **p < 0.005, ***p < 0.0005; n = 10 mice per group. No difference in baseline performance or following 1 day of 3-NP treatment was detected between groups.
Modest changes in basal locomotor activity have also been reported in Rhes KO mice, but the differences are subtle and the direction of effect differs between studies (Spano et al., 2004; Errico et al., 2008; Quintero and Spano, 2011). To reduce variability and determine changes in locomotion caused specifically by 3-NP treatment, activity in an open field was monitored each day before 3-NP injections and normalized to each animal's baseline activity. Following 2 days of injections, WT mice display a near 75% reduction in horizontal activity, a deficit reduced by more than half in KO mice (Fig. 3A) (WT 27.1 ± 9.1% vs. KO 65.1 ± 9.8% baseline activity, p value <0.05). Rearing movements are also dramatically reduced in WT mice after 1 d of 3-NP treatment and further decreased on day 2, while Rhes KO mice show no significant decrease in rearing movements even after 2 days of injections (Fig. 3B) (day 1: WT 63.7 ± 11.9 vs KO 108.0 ± 14.5 rears, p value <0.05; day 2 WT 28.9 ± 9.65 vs KO 82.0 ± 16.2 rears, p value <0.05).
Figure 3.

Rhes deletion prevents the deterioration of basal activity monitored by open field analysis in mice treated with 3-NP. Horizontal movement as a percentage of baseline activity (A) and number of entries above the vertical plane (rears) (B) are shown. *p < 0.05, **p < 0.03; n = 10 mice per group.
Systemic 3-NP treatment inhibits succinate dehydrogenase (mitochondrial complex II, SDH) similarly in all regions of the brain, demonstrating that differences in mitochondrial inhibition cannot account for the striatal vulnerability to 3-NP (Brouillet et al., 1998). To determine whether the dramatic protection from lesions and motor dysfunction caused by deletion of Rhes might result from changes in 3-NP efficacy, we measured the decrease of SDH activity in WT and Rhes KO mice following a single dose of 3-NP. We confirm similar diminution of SDH activity in WT and Rhes KO mice in both the cortex (Fig. 4A) (WT 0.58 ± 0.03 vs WT + 3-NP 0.44 ± 0.02, p value <0.005; KO 0.57 ± 0.01 vs KO + 3-NP 0.44 ± 0.03, p value <0.0005) and striatum (Fig. 4B) (WT 0.50 ± 0.02 vs WT + 3-NP 0.37 ± 0.01, p value <0.005; KO 0.48 ± 0.02 vs KO + 3-NP 0.39 ± 0.02, p value <0.05). Thus, neither regional differences in mitochondrial inhibition nor systemic clearance of 3-NP can account for the striatal protection afforded by deletion of Rhes. Consistent with this notion, the lethality caused by high doses of 3-NP, which is due to global mitochondrial inhibition and systemic toxicity, is similar in WT and KO mice (Fig. 4C) (mean survival WT 9.0 ± 0.36 vs KO 9.6 ± 0.31 days, p value = 0.171).
Figure 4.
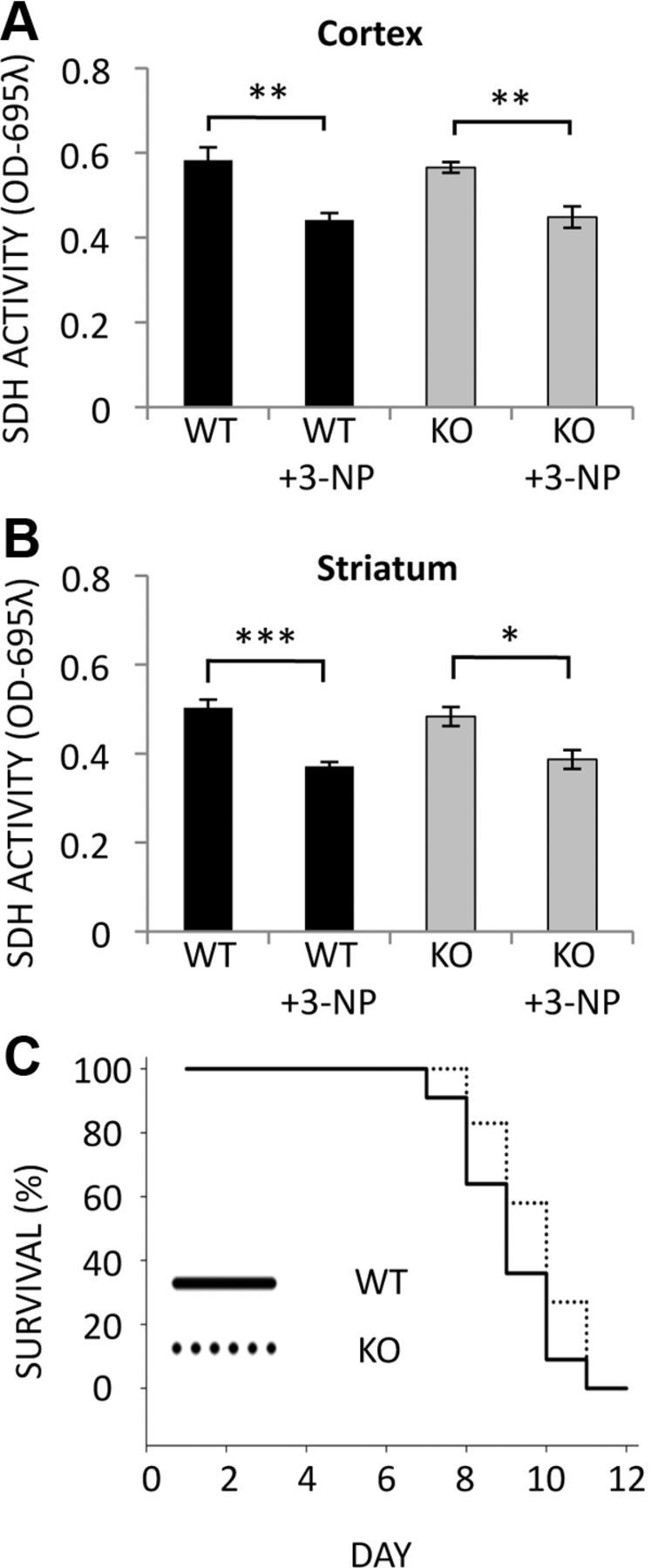
SDH inhibition and survival following 3-NP treatment are similar for WT and Rhes KO mice. A, B, SDH activity was measured in mitochondria isolated from cortex (A) and striatum (B) of mice treated with 120 mg/kg 3-NP or PBS for 6 h; n = 3 mice per group, and reactions were performed in triplicate; *p < 0.05, **p < 0.005, ***p < 0.005. C, Kaplan-Meier survival analysis of WT and Rhes KO mice treated with 3-NP starting at 30 mg/kg and increasing 15% daily until death. No significant difference was found between WTs and KOs; n = 11 for WTs, n = 12 for KOs.
Discussion
In this study we have found that deleting Rhes profoundly protects mice from striatal damage and motor dysfunction caused by 3-NP, a toxin that elicits selective striatal lesions resembling the disposition of those in HD. These findings support the hypothesis that Rhes determines the selective striatal vulnerability seen in HD. Accordingly, it is conceivable that drugs impairing Rhes function may delay or prevent the onset of HD. The small decrease in behavioral performance of Rhes KO mice following 3-NP treatment is likely unrelated to striatal dysfunction, instead reflecting toxicity from global mitochondrial inhibition, as the majority of KO mice have no striatal lesion whatsoever (Fig. 1C). Succinate dehydrogenase activity is inhibited equivalently by 3-NP in both WT and Rhes KO mice, indicating that the unique striatal vulnerability to 3-NP cannot be attributed to mitochondrial inhibition, but instead more likely involves other systems linked to Rhes. It has long been appreciated that differences in SDH inhibition by 3-NP cannot account for selective striatal toxicity, as such inhibition is similar in striatum and other brain regions (Brouillet et al., 1998). The protective effect of Rhes deletion is therefore independent of any effect on mitochondrial SDH activity.
The selective localization of 3-NP neuropathology is remarkable considering that systemically administered 3-NP is uniformly distributed throughout the brain, which parallels the paradox of the ubiquitously expressed mHtt being associated with selective striatal pathology. Nevertheless, the 3-NP model lacks the mutated form of huntingtin. It is therefore possible that Rhes binds wild-type huntingtin to mediate the cytotoxic effects of 3-NP. We have previously demonstrated that Rhes is capable of interacting with wtHtt, albeit with a lower affinity than mHtt (Subramaniam et al., 2009). Rhes did not enhance cytotoxicity or promote sumoylation of wtHtt in cells under basal conditions. Perhaps, following 3-NP administration, wtHtt is sumoylated in the presence of Rhes, but because measuring the sumoylation of any endogenous protein in an in vivo setting is nearly impossible, we have not evaluated such a possibility. Thus, we suggest that 3-NP might selectively damage the striatum via the association of Rhes with wild-type Htt, potentially through novel mechanisms that are independent from sumoylation but induced by 3-NP.
Lowering mHtt levels has been proposed as a therapeutic approach for HD, and recent studies in animal models have shown promising results (Sah and Aronin, 2011; Kordasiewicz et al., 2012). As Htt occurs throughout the brain and the rest of the body, its global depletion may elicit unforeseen adverse effects. By contrast, Rhes is brain-specific and highly localized to the striatum. Accordingly, inhibitors may have a diminished propensity for causing adventitious damage, an important feature for agents that would be administered to individuals at risk for many years.
Rhes-specific drugs may also find utility in the therapy of Parkinson's Disease, as our recent study indicates that Rhes mediates the dyskinetic influences of l-DOPA acting through mammalian target of rapamycin (mTOR) (Subramaniam et al., 2011). Rubinsztein and colleagues (Ravikumar et al., 2004) demonstrated that aggregates of mHtt sequester mTOR, leading to decreased kinase activity of mTOR and enhanced cellular autophagy. Autophagy degrades mHtt, and increasing evidence suggests that changes in autophagy may be critical in both the pathology and treatment of HD (Banerjee et al., 2010; Jimenez-Sanchez et al., 2012). Loading of autophagic vesicles is deficient in HD cells (Martinez-Vicente et al., 2010), and molecules that promote autophagy are protective in multiple models of HD (Tsvetkov et al., 2010; Harris and Rubinsztein, 2012). The huntingtin protein itself has been suggested to function in the autophagic pathway (Steffan, 2010), and changes in huntingtin polyglutamine length affect levels of neuronal autophagy (Zheng et al., 2010). Whether Rhes contributes to HD pathology through the regulation of mTOR or autophagy remains to be determined.
Footnotes
This work was supported by United States Public Health Service Grant MH18501 (S.H.S.) and a grant from the Cure Huntington's Disease Initiative (S.H.S.). We thank S. Karuppagounder and T. and V. Dawson for technical support.
The authors declare no competing financial interests.
References
- Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549. doi: 10.1016/j.tins.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum D, Galas MC, Pintor A, Brouillet E, Ledent C, Muller CE, Bantubungi K, Galluzzo M, Gall D, Cuvelier L, Rolland AS, Popoli P, Schiffmann SN. A dual role of adenosine A2A receptors in 3-nitropropionic acid-induced striatal lesions: implications for the neuroprotective potential of A2A antagonists. J Neurosci. 2003;23:5361–5369. doi: 10.1523/JNEUROSCI.23-12-05361.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Guyot MC, Mittoux V, Altairac S, Condé F, Palfi S, Hantraye P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. 1998;70:794–805. doi: 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J Neurochem. 2005;95:1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- Crook ZR, Housman D. Huntington's disease: can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Sutton L, Hayden MR. Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist. 2011;17:475–492. doi: 10.1177/1073858410390378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico F, Santini E, Migliarini S, Borgkvist A, Centonze D, Nasti V, Carta M, De Chiara V, Prosperetti C, Spano D, Herve D, Pasqualetti M, Di Lauro R, Fisone G, Usiello A. The GTP-binding protein Rhes modulates dopamine signalling in striatal medium spiny neurons. Mol Cell Neurosci. 2008;37:333–345. doi: 10.1016/j.mcn.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Falk JD, Vargiu P, Foye PE, Usui H, Perez J, Danielson PE, Lerner DL, Bernal J, Sutcliffe JG. Rhes: A striatal-specific Ras homolog related to Dexras1. J Neurosci Res. 1999;57:782–788. [PubMed] [Google Scholar]
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8:108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez M, Thomson F, Zavodszky E, Rubinsztein DC. Autophagy and polyglutamine diseases. Prog Neurobiol. 2012;97:67–82. doi: 10.1016/j.pneurobio.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW. Sustained therapeutic reversal of Huntington's disease by transient repression of Huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealer RG, Snyder SH. Rhes is a striatal-enriched protein with pathophysiologic relevance. Eur J Neurodegen Dis. 2012;1:89–100. [Google Scholar]
- Quintero GC, Spano D. Exploration of sex differences in Rhes effects in dopamine mediated behaviors. Neuropsychiatr Dis Treat. 2011;7:697–706. doi: 10.2147/NDT.S25888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- Sah DW, Aronin N. Oligonucleotide therapeutic approaches for Huntington disease. J Clin Invest. 2011;121:500–507. doi: 10.1172/JCI45130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seredenina T, Gokce O, Luthi-Carter R. Decreased striatal RGS2 expression is neuroprotective in Huntington's disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation. PLoS One. 2011;6:e22231. doi: 10.1371/journal.pone.0022231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano D, Branchi I, Rosica A, Pirro MT, Riccio A, Mithbaokar P, Affuso A, Arra C, Campolongo P, Terracciano D, Macchia V, Bernal J, Alleva E, Di Lauro R. Rhes is involved in striatal function. Mol Cell Biol. 2004;24:5788–5796. doi: 10.1128/MCB.24.13.5788-5796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS. Does Huntingtin play a role in selective macroautophagy? Cell Cycle. 2010;9:3401–3413. doi: 10.4161/cc.9.17.12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Napolitano F, Mealer RG, Kim S, Errico F, Barrow R, Shahani N, Tyagi R, Snyder SH, Usiello A. Rhes, a striatal-enriched small G protein, mediates mTOR signaling and l-DOPA-induced dyskinesia. Nat Neurosci. 2011;15:191–193. doi: 10.1038/nn.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–16987. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargiu P, De Abajo R, Garcia-Ranea JA, Valencia A, Santisteban P, Crespo P, Bernal J. The small GTP-binding protein, Rhes, regulates signal transduction from G protein-coupled receptors. Oncogene. 2004;23:559–568. doi: 10.1038/sj.onc.1207161. [DOI] [PubMed] [Google Scholar]
- Zheng S, Clabough EB, Sarkar S, Futter M, Rubinsztein DC, Zeitlin SO. Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. 2010;6:e1000838. doi: 10.1371/journal.pgen.1000838. [DOI] [PMC free article] [PubMed] [Google Scholar]