

Allogeneic hematopoietic stem cell transplantation has proven benefit in controlling sickle cell disease-related vasculopathy and organ damage. However, the difficulty in finding HLA-matched donors in the family or registry is a therapeutic limitation. Umbilical cord blood transplantation allows for more mismatching from the graft-versus-host disease perspective and this donor pool is expandable with effort and education. Successful unrelated donor cord blood transplantation (UCBT) can overcome this limitation and formal trials of UCBT merit consideration in this disorder.
Summary
Allogeneic hematopoietic stem cell transplantation has proven benefit in controlling sickle cell disease-related vasculopathy and organ damage. Myeloablative matched sibling donor cord transplants have excellent outcomes in sickle cell disease. Unrelated donor transplant options are often deferred because of a lack of suitable human leukocyte antigen-matched donors, a problem especially relevant to minority populations. Umbilical cord blood transplantation allows for more mismatching from the graft-versus-host disease perspective and the donor pool is expandable with effort and education. Drawbacks such as increased rates of graft rejection, a fixed cell dose, delayed immune reconstitution, and transplant-related mortality have deterred unrelated cord transplant efforts. However, the transplant community continues to make enormous strides in this transplant realm in areas of immunogenetics, stem cell expansion, conditioning regimens, and supportive care. This has allowed the development of new studies that are currently ongoing, exploring ways to make cord blood transplantation successful and safer. The goal is to make unrelated donor cord blood transplantation for sickle cell disease merit early consideration in patients who stand to benefit from this approach.
Introduction
Sickle cell disease (SCD) is a genetic disorder driven by a single amino acid substitution in the β-globin chain of adult hemoglobin. In the homozygous state or in conjunction with other hemoglobin disorders (such as hemoglobin S-β thalassemia), it results in red-cell deformity/polymerization, chronic vasculopathy, irreversible organ damage, and early mortality. Although supportive care has advanced by leaps and bounds over the last decade, it often does not fully control ongoing organ destruction (recurrent strokes, pulmonary hypertension) or is toxic/ineffective (red-cell alloimmunization, failed hydroxyurea, or iron overload) [1]. If a matched sibling donor (MSD) is available, the success of hematopoietic stem cell transplantation (HSCT) in curing SCD currently supports exploring this option earlier rather than later in afflicted patients [2, 3]. Severe manifestations that are either potentially fatal or afford poor quality of life (Table 1) merit consideration of transplant from unrelated donors using the best available stem cell source. This intervention is directed at a cure and prevention of ongoing organ damage that leads to permanent morbidity or early mortality that often constitutes the natural history of this disease even with current levels of supportive care [4]. Putting this balance in perspective for a family includes explaining the aim to cure while taking a risk regarding the possibility of immediate mortality or chronic morbidity (graft-versus-host disease) versus early mortality in the third or fourth decades (untransplanted) with significant performance and life quality issues prior to fatality, a difficult decision [5]. How these balance issues are perceived often depends on family dynamics and support, prior exposure to patients with SCD, age of the patient, education, and availability of resources. The cost and acceptability of a decision needs to be based on the above factors and the balance between a potentially curative one-time intervention (HSCT) and recurrent hospitalizations, lifelong morbidity, and a poor quality of life [6]. If HSCT is considered, umbilical cord blood (UCB) is a stem cell source option because of easy accessibility, although there are limitations to using cord products as discussed later, such as cell dose requirements and human leukocyte antigen (HLA) matching, that inhibit success and compromise outcomes [7]. Advances targeted at overcoming the disadvantages of cord blood, however, make this stem cell source an evolving potential for SCD transplants.
Table 1.
Indications for unrelated donor transplants in sickle cell disease

Abbreviations: AVN, avascular necrosis; QOL, quality of life; TCD, transcranial doppler; VOE, veno-occlusive episodes.
Optimizing Cord Blood as a Donor Source
Until gene therapy becomes a reality in SCD patients, the quest to optimize transplant procedures and stem cell sources will need to continue, especially for those with severe disease manifestations. For more than two decades, transplant was considered only if patients with severe SCD had a MSD (normal or with trait) available, which was the case for less than 14% of SCD patients [8, 9]. In the absence of a MSD, donor options include a matched unrelated donor, a mismatched unrelated donor, UCB product, or a haplo-identical family donor. Each of these stem cell sources is associated with advantages and complications innate to the graft source and transplant method used. The advantages of a cord product include easy availability, a source that is enriched for hematopoietic stem and progenitor cells, and the ability to transplant a partially HLA-mismatched product because of a lower incidence of graft-versus-host disease (GVHD); disadvantages include increased risk of graft rejection, a fixed cell dose, delayed engraftment/immune reconstitution, and associated morbidity/mortality. Center for International Blood and Marrow Transplant Research (CIBMTR) data from 2011 suggest that approximately 40% of patients of African origin can find a suitable (HLA matched at five or six of six loci: A, B, DRB1) cord product from accredited cord banks. High-resolution typing is traditionally reserved only for the DRB1 locus; class I typing is typically undertaken at low or intermediate resolution, a convenience/luxury that allows for better availability of product. Of note, in malignant disorders, transplants matched with six of six loci (A, B, and DRB1) had better outcomes than those matched with eight of eight loci [10]. Transplants with one or two antigens mismatched fared significantly better with a total nucleated cell (TNC) dose of >5 × 107/kg, whereas smaller products, especially those mismatched at two or more loci, had higher treatment-related mortality (TRM) or GVHD, as did products with a TNC of <2.5 × 107/kg and/or mismatched at ≥2 loci [11, 12]. They should be used with caution, especially in nonmalignant disorders that are not immediately fatal. However, products mismatched at two loci are likely to be identified at higher frequency and can be considered in a trial setting designed to improve outcomes despite the mismatch, by optimizing cell dose (TNC >5 × 107/kg), considering double cord combinations, or enhancing engraftment by cell expansion techniques discussed below. Measuring colony-forming unit is a useful parameter for predicting cord viability and neutrophil and platelet engraftment, but the lack of standardized methods to do so makes this less universally acceptable [13]. Matching cord products to noninherited maternal antigens results in lower TRM after umbilical cord blood transplant (UCBT), presumably because of T-cell tolerance to maternal haplotypes developed during the fetal period [14]. Manipulation (such as isolation of human placenta-derived stem cells) of cord stem cells and expansion of product ex vivo prior to infusion using growth factors, prostaglandins, nicotinamide, or CD26/DPPIV inhibitors are in early phase clinical trials with unclear but potential benefits of enhancing engraftment [15]. Others such as single or double cord transplants with mesenchymal stromal cell-based, cytokine, copper chelator, or Notch-ligand based expansion have been reported in experimental clinical trial settings with early promise [16, 17]. Successful interventions in these directions will eventually be applicable to SCD transplants with UCB. SCD-specific experimental trials are already under way using some of these technologies to explore beneficial options. A targeted effort to increase cord blood banking focusing on minority populations has been under way with assistance from the CIBMTR and helps to expand cord bank inventory [18]. Individual cord blood banks are making their own efforts using tools such education and information sessions in obstetric practice and minority groups.
UCB Transplantation for SCD: The Past, Present, and Future
Although the SCD population in the United States is more than 72,000, only approximately 500 transplant procedures were reported to the CIBMTR by 2011, over a span of 10–15 years. Of these, 85% were MSD transplants, and 80% were in children less than 16 years of age. Deterrents besides the lack of suitable donors have been the complications of HSCT: infection, GVHD, late effects, and mortality. SCD transplant patients additionally have unique complications such as hypertension, hemorrhagic, or ischemic strokes driven by vasculopathy, a lowered seizure threshold, sinusoidal obstruction syndrome (SOS), and progressive renal and/or pulmonary dysfunction caused by pre-existing organ damage; these complications need heightened awareness, vigilant monitoring, adequate prophylaxis, and timely intervention, making for a high-risk transplant. Myeloablative sibling donor cord blood transplants are less common compared with a larger number performed for thalassemia. However, when performed in children, SCD cord transplants have excellent outcomes, with >90% disease-free survival (DFS) and low mortality [19]. Mixed chimerism is common in both ablative and nonablative settings but still curative, although prolonged immune suppression is often necessary in the very-low-intensity nonablative setting [20]. Donor erythropoiesis has survival advantage in SCD transplants accounting for the success of mixed chimerism in curing the disease [21]. Late graft rejections after mixed donor chimerism have been shown to be rare in thalassemia, and the expectation is that SCD will be similar [22]. The level of chimerism that is necessary to afford a cure is currently under more detailed investigation. A state of persistent mixed chimerism post-transplant, although adequate to curb disease manifestations, can invoke autoimmunity caused by aberrant immune interactions between the donor and host, a complication that requires awareness and monitoring for early intervention. Although a predisposition to autoimmunity has not been described in hemoglobinopathy transplants, this phenomenon is described in primary immune deficiency disorder transplants such as Wiskott-Aldrich syndrome [23].
Unrelated donor cord blood transplants for SCD have fared less well to date, but there are a few lessons learned. Registry data from Europe and the United States were reported in 2011 by Ruggeri et al. [24]. Of 16 patients with SCD reported, the majority were mismatched transplants. Nine of 16 underwent myeloablative transplantation that was primarily busulfan-based. Although overall survival was 94%, the DFS was 50%, with graft rejection the primary cause of failure even with myeloablation. Previous reports with fewer patients fared no better, and GVHD and TRM have been additional causes for concern [25].
The future success of unrelated donor UCBT for SCD depends on modifications based on recent advances and lessons previously learnt. As the natural history of sickle cell disease is tracked with longitudinal studies, it is apparent that disease progression occurs despite supportive care contributing to ongoing strokes, pulmonary hypertension, cardiovascular complications, chronic debilitating pain, all of which are complications that progress at adolescence and worsen during early adulthood with eventual early mortality. Hematologists, especially those caring for this older population of patients, are discussing transplant options with their patients with increasing frequency. Pediatric hematologists, however, are more hesitant to discuss transplantation (especially unrelated donor transplant) in children, who generally fare better than adults with SCD, because of the associated risks of the procedure. In this regard, improving cord blood as a stem cell source is an area that holds promise because it is more accessible. Finding safe and successful conditioning regimens is another. Successful unrelated cord transplants with myeloablation have been described for thalassemia, although all reports have not been equally promising [24, 26]. Myeloablative regimens have organ toxicities to be wary of in SCD patients, especially in patients who are older and who have more advanced disease (neurologic, hepatic SOS, pulmonary toxicity, and renal failure); late effects are a major problem in the young (neurologic, neurocognitive, and performance issues, sterility, second cancers, and growth inhibition in children transplanted at adolescence) [2, 27, 28]. Nonablative regimens, in contrast, are well tolerated but associated with high rates of graft rejection [29–31]. Reduced-intensity regimens that are immunoablative are an alternate intermediate approach now being evaluated at several centers, including ours, for a middle-of-the-road approach where early experience with hemoglobinopathy is promising using immunoablation with alemtuzumab or anti-thymocyte globulin (ATG) [32, 33]. Early results from our center support fertility preservation in female recipients following a conditioning regimen of alemtuzumab, fludarabine, and melphalan with five normal pregnancies in recipients to date. In a national trial of unrelated SCD transplants using this regimen, it was determined that cord blood transplants had a higher risk of graft rejection, although this regimen was tolerated well [34]. This has led to the development of modified reduced-intensity transplant protocols for UCBT at our and other centers to maintain the benefit of this approach but facilitate engraftment of UCB and was tested recently in a trial of reduced-intensity transplantation for thalassemia that included UCB as a stem cell source (the Unrelated Donor Thalassemia [URTH] trial). Early results have been promising for achieving engraftment, but larger numbers, a longer period of evaluation, and follow-up for late effects are still required. Immunoablation with alemtuzumab comes with the risk of invasive infections, and we have taken the approach of early immune ablation pretransplant to facilitate immune reconstitution [35]. This allows early immune reconstitution from the donor by 6 months post-transplant with bone marrow products [36]. However, in the early post-transplant phase, recipients are susceptible to multiple infectious complications, particularly viral reactivation (cytomegalovirus [CMV] or adenovirus) and bacterial/fungal sepsis caused by the profound lymphodepletion of B and T lineage cells by alemtuzumab [37]. Other ablative but “reduced toxicity” transplant regimens such as those using treosulfan have been successful with marrow but are yet to be tested for SCD in the cord blood setting [38]. Supportive care details, such as effective iron chelation prior to and if necessary post-transplant, targeted cell therapy for infections (especially viral infections such as adenovirus and CMV), and diligent infection monitoring and prophylaxis through the period of vulnerability, are helpful in providing better outcomes, especially with an immunoablative regimen. Double cord transplants may benefit the older and larger-sized recipient, but the risk of GVHD needs to be carefully evaluated in this population [39].
Conclusion
Easy availability and a lower degree of HLA matching associated with less GVHD retain UCBT as an important modality in the treatment of SCD. Innovative progress made with optimizing cord products, conditioning, and supportive care, as well as growing inventories in cord banks with rigorous quality control for storage, can only continue to improve UCBT outcomes. The acceptability of adverse outcomes (TRM, GVHD, and graft rejection) is essential and will vary between groups as conceptualized in Figure 1. The benefits and curative aspects of transplant need to be discussed alongside a detailed description of risks such as organ toxicity, GVHD, and mortality based on recipient age, disease state, transplant method, and donor source, so that caregivers and patients have a balanced view of the disease and intervention as they consider this option. Emphasis should also be placed on providing adequate social support to families as they seek and undertake transplant options at experienced referral centers often at a distance from their hometowns. It is important from outcome and safety perspectives to perform SCD transplants and expand applicability strictly in multicenter trial settings to define results and continue to improve upon them.
Figure 1.
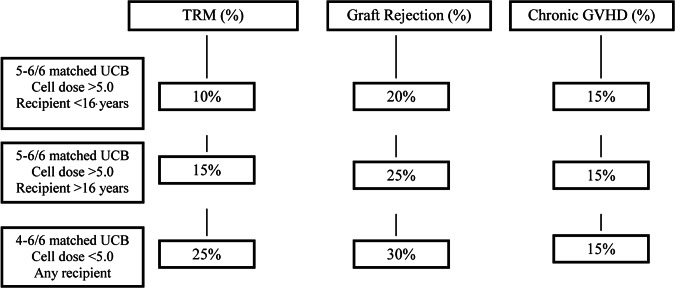
Conceptualization of acceptable transplant risks with unrelated UCB transplant. Abbreviations: GVHD, graft-versus-host disease; TRM, treatment-related mortality; UCB, umbilical cord blood.
Disclosure of Potential Conflicts of Interest
The author indicates no potential conflicts of interest.
Author Contributions
S.S.: conception and design, manuscript writing, final approval of manuscript.
References
- 1.Scothorn D, Price C, Schwartz D. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. J Pediatr. 2002;140:348–354. doi: 10.1067/mpd.2002.122498. [DOI] [PubMed] [Google Scholar]
- 2.Walters M, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16:263–272. doi: 10.1016/j.bbmt.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eggleston B, Patience M, Edwards S, et al. Effect of myeloablative bone marrow transplantation on growth in children with sickle cell anemia: Results of the multicenter study of haematopoietic cell transplantation for sickle cell anemia. Br J Haematol. 2007;136:673–676. doi: 10.1111/j.1365-2141.2006.06486.x. [DOI] [PubMed] [Google Scholar]
- 4.Fitzhugh C, Lauder N, Jonassaint J, et al. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am J Hematol. 2010;85:36–40. doi: 10.1002/ajh.21569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth M, Krystal J, Manwani D, et al. Stem cell transplant for children with sickle cell anemia: Parent and patient interest. Biol Blood Marrow Transplant. 2012;18:1709–1715. doi: 10.1016/j.bbmt.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Shenoy S. Has stem cell transplantation come of age in the treatment of sickle cell disease? Bone Marrow Transplant. 2007;40:813–821. doi: 10.1038/sj.bmt.1705779. [DOI] [PubMed] [Google Scholar]
- 7.Freed J, Talano J, Small T, et al. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘Whom, when and how.’. Bone Marrow Transplant. 2012;47:1489–1498. doi: 10.1038/bmt.2011.245. [DOI] [PubMed] [Google Scholar]
- 8.Mentzer W, Heller S, Pearle P, et al. Availability of related donors for bone marrow transplantation in sickle cell anemia. Am J Pediatr Hematol Oncol. 1994;16:27–29. [PubMed] [Google Scholar]
- 9.Walters M, Patience M, Leisenring W, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7:665–673. doi: 10.1053/bbmt.2001.v7.pm11787529. [DOI] [PubMed] [Google Scholar]
- 10.Eapen M, Rubinstein P, Zhang M, et al. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukemia: A comparison study. Lancet. 2007;369:1947–1954. doi: 10.1016/S0140-6736(07)60915-5. [DOI] [PubMed] [Google Scholar]
- 11.Barker J, Scaradavou A, Stevens C. Combined effect of total nucleated cell dose and HLA match on transplantation outcome in 1061 cord blood recipients with hematologic malignancies. Blood. 2010;115:1843–1849. doi: 10.1182/blood-2009-07-231068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamani N, Spellman S, Hurley CK, et al. State of the art review: HLA matching and outcome of unrelated donor umbilical cord blood transplants. Biol Blood Marrow Transplant. 2008;14:1–6. doi: 10.1016/j.bbmt.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Page K, Zhang L, Mendizabal A, et al. Total colony-forming units are a strong independent predictor of neutrophil and platelet engraftment after unrelated umbilical cord blood transplantation: A single-center analysis of 435 cord blood transplants. Biol Blood Marrow Transplant. 2011;17:1362–1374. doi: 10.1016/j.bbmt.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 14.Rocha V, Spellman S, Zhang M, et al. Effect of HLA-matching recipients to donor noninherited maternal antigens on outcomes after mismatched umbilical cord blood transplantation for hematologic malignancy. Biol Blood Marrow Transplant. 2012;18:1890–1896. doi: 10.1016/j.bbmt.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norkin M, Lazarus H, Wingard J. Umbilical cord blood graft enhancement strategies: Has the time come to move these into the clinic? Bone Marrow Transplant. 2012 doi: 10.1038/bmt.2012.163. (in press) [DOI] [PubMed] [Google Scholar]
- 16.Delaney C, Ratajczak M, Laughlin M. Strategies to enhance umbilical cord blood stem cell engraftment in adult patients. Expert Rev Hematol. 2010;3:273–283. doi: 10.1586/ehm.10.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Lima M, McNiece I, Robinson S, et al. Cord-blood engraftment with ex vivo mesenchymal-cell coculture. N Engl J Med. 2012;367:2305–2315. doi: 10.1056/NEJMoa1207285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S, Kamani N, Confer D. Principles and tools for selection of umbilical cord blood and unrelated adult donor grafts. Biol Blood Marrow Transplant. 2008;14(suppl 1):112–119. doi: 10.1016/j.bbmt.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Locatelli F, Rocha V, Reed W, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003;101:2137–2143. doi: 10.1182/blood-2002-07-2090. [DOI] [PubMed] [Google Scholar]
- 20.Hsieh M, Kang E, Fitzhugh C, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361:2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh M, Wu C, Tisdale J. In mixed hematopoietic chimerism, the donor red cells win. Haematologica. 2011;96:13–15. doi: 10.3324/haematol.2010.035576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lisini D, Zecca M, Giorgiani G, et al. Donor/recipient mixed chimerism does not predict graft failure in children with beta-thalassemia given an allogeneic cord blood transplant from an HLA-identical sibling. Haematologica. 2008;93:1859–1867. doi: 10.3324/haematol.13248. [DOI] [PubMed] [Google Scholar]
- 23.Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: An international collaborative study. Blood. 2011;118:1675–1684. doi: 10.1182/blood-2010-11-319376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruggeri A, Eapen M, Scaravadou A, et al. Umbilical cord blood transplantation for children with thalassemia and sickle cell disease. Biol Blood Marrow Transplant. 2011;17:1375–1382. doi: 10.1016/j.bbmt.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adamkiewicz T, Szabolcs P, Haight A, et al. Unrelated cord blood transplantation in children with sickle cell disease: Review of four-center experience. Pediatr Transplant. 2007;11:641–644. doi: 10.1111/j.1399-3046.2007.00725.x. [DOI] [PubMed] [Google Scholar]
- 26.Jaing T, Hung I, Yang C, et al. Unrelated cord blood transplantation for thalassaemia: A single-institution experience of 35 patients. Bone Marrow Transplant. 2012;47:33–39. doi: 10.1038/bmt.2011.39. [DOI] [PubMed] [Google Scholar]
- 27.Walters M, Sullivan K, Bernaudin F, et al. Neurologic complications after allogeneic marrow transplantation for sickle cell anemia. Blood. 1995;85:879–884. [PubMed] [Google Scholar]
- 28.Khalil A, Zaidman I, Elhasid R, et al. Factors influencing outcome and incidence of late complications in children who underwent allogeneic hematopoietic stem cell transplantation for hemoglobinopathy. Pediatr Hematol Oncol. 2012;29:694–703. doi: 10.3109/08880018.2012.725198. [DOI] [PubMed] [Google Scholar]
- 29.Horan J, Liesveld J, Fenton P, et al. Hematopoietic stem cell transplantation for multiply transfused patients with sickle cell disease and thalassemia after low-dose total body irradiation, fludarabine, and rabbit anti-thymocyte globulin. Bone Marrow Transplant. 2005;35:171–177. doi: 10.1038/sj.bmt.1704745. [DOI] [PubMed] [Google Scholar]
- 30.Iannone R, Casella J, Fuchs E, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9:519–528. doi: 10.1016/s1083-8791(03)00192-7. [DOI] [PubMed] [Google Scholar]
- 31.Jacobsohn D, Duerst R, Tse M, et al. Reduced intensity hematopoietic stem-cell transplantation for the treatment of non-malignant diseases in children. Lancet. 2004;364:156–162. doi: 10.1016/S0140-6736(04)16628-2. [DOI] [PubMed] [Google Scholar]
- 32.Mazur M, Kurtzberg J, Halperin E, et al. Transplantation of a child with sickle cell anemia with an unrelated cord blood unit after reduced intensity conditioning. J Pediatr Hematol Oncol. 2006;28:840–844. doi: 10.1097/MPH.0b013e31802d3e53. [DOI] [PubMed] [Google Scholar]
- 33.Krishnamurti L, Blazar B, Wagner J. Bone marrow transplantation without myeloablation for sickle cell disease. N Engl J Med. 2001;344:68. doi: 10.1056/NEJM200101043440119. [DOI] [PubMed] [Google Scholar]
- 34.Kamani N, Walters M, Carter S, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: Results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) Biol Blood Marrow Transplant. 2012;18:1265–1272. doi: 10.1016/j.bbmt.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shenoy S, Grossman W, DiPersio J, et al. A novel reduced-intensity stem cell transplant regimen for non-malignant disorders. Bone Marrow Transplant. 2005;35:345–352. doi: 10.1038/sj.bmt.1704795. [DOI] [PubMed] [Google Scholar]
- 36.Bednarski J, Le C, Murray L, et al. Immune reconstitution following reduced intensity stem cell transplantation for non-malignant disorders in children: Poster presentation at Tandem Meeting 2013. Biol Blood Marrow Transplant. 2013;19:S200. [Google Scholar]
- 37.Schmidt-Hieber M, Schwarck S, Stroux A, et al. Immune reconstitution and cytomegalovirus infection after allogeneic stem cell transplantation: The important impact of in vivo T cell depletion. Int J Hematol. 2010;91:877–885. doi: 10.1007/s12185-010-0597-6. [DOI] [PubMed] [Google Scholar]
- 38.Bernardo M, Piras E, Vacca A, et al. Allogeneic hematopoietic stem cell transplantation in thalassemia major: Results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood. 2012;120:473–476. doi: 10.1182/blood-2012-04-423822. [DOI] [PubMed] [Google Scholar]
- 39.Sauter C, Abboud M, Jia X, et al. Serious infection risk and immune recovery after double-unit cord blood transplantation without antithymocyte globulin. Biol Blood Marrow Transplant. 2011;17:1460–1471. doi: 10.1016/j.bbmt.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]