

Abstract
CARs are recombinant receptors that provide both antigen-binding and T cell activating functions. A multitude of CARs has been reported over the past decade, targeting an array of cell surface tumor antigens. Their biological functions have dramatically changed following the introduction of tri-partite receptors comprising a costimulatory domain, termed second generation CARs. These have recently demonstrated clinical benefit in patients treated with CD19-targeted autologous T cells. CARs may be combined with costimulatory ligands, chimeric costimulatory receptors or cytokines to further enhance T cell potency, specificity and safety. CARs represent a new class of drugs with exciting potential for cancer immunotherapy.
Introduction
CARs are recombinant receptors for antigen, which, in a single molecule, redirect the specificity and function of T lymphocytes and other immune cells. The general premise for their use in cancer immunotherapy is to rapidly generate tumor-targeted T cells, bypassing the barriers and incremental kinetics of active immunization.(1, 2) Once expressed in T cells, the CAR-modified T cells acquire supra-physiological properties and act as “living drugs” that may exert both immediate and long-term effects. The engineering of CARs into T cells requires that T cells be cultured to allow for transduction and expansion. The transduction may utilize a variety of methods, but stable gene transfer is required to enable sustained CAR expression in clonally expanding and persisting T cells. In principle, any cell surface molecule can be targeted through a CAR, thus over-riding tolerance to self-antigens and the antigen recognition gaps in the physiological T cell repertoire that limit the scope of T cell reactivity. Various T cell subsets, as well as T cell progenitors and other immune cells such as natural killer (NK) cells, can be targeted with a CAR. Redirecting immune reactivity towards a chosen antigen is not however the only purpose of smarter CARs, which are designed to accomplish much more than to target and initiate T cell activation. CARs with different strengths and quality of signaling have the potential to modulate T cell expansion and persistence, as well as the strength of T cell activation within the tumor microenvironment, features that dramatically alter the efficacy and safety of tumor-targeted T cells. In this regards, CARs provide a broader range of functional effects than transduced T cell receptors (TCRs), wherein strength of signaling, which is for the most part determined by the TCR’s affinity for antigen, is the principal determinant of T cell fate. CARs and TCRs have their respective advantages and disadvantages.(1-4) While the flexibility and “dynamic range” of CARs is attractive, current CARs are limited to recognizing cell surface antigens, whereas TCRs recognize both cell surface and intracellular proteins. CARs however do not require antigen processing and presentation by HLA, and are therefore more broadly applicable to HLA-diverse patient populations. We discuss here the targeting and signaling properties of CARs, focusing on their effects on T cell specificity, potency and safety. Other general aspects of adoptive T cell therapy that apply not only to the use of CARs but other T cell therapies as well, including T cell expansion methodologies, T cell subset selection and host conditioning, are beyond the scope of this review. Owing to the extraordinary potential of T cell engineering and the modular nature of their structure, CARs are rapidly evolving and show great promise for their successful utilization in a wide range of immunotherapies.
CAR targeting
CARs are recombinant receptors that typically target native cell surface antigens.(4) Unlike the physiological TCR, which engages HLA-peptide complexes, CARs engage molecules that do not require peptide processing or HLA expression to be recognized. CARs therefore recognize antigen on any HLA background, in contrast to TCRs, which need to be matched to the patient’s haplotype. Furthermore, CARs can target tumor cells that have down-regulated HLA expression or proteasomal antigen processing, two mechanisms that contribute to tumor escape from TCR-mediated immunity.(5) Another feature of the broad applicability of CARs is their ability to bind not only to proteins but also to carbohydrate and glycolipid structures, again expanding the range of potential targets. A survey of antigens targeted to date by CARs is shown in Table 1.
Table 1.
Antigens targeted by CARs
| Target Antigen | Associated Malignancy | Receptor Type (Other specificity) | In vivo studies | Reference |
|---|---|---|---|---|
| α-Folate receptor | Ovarian cancer | ScFv-FcεRIγ | Phase I | (1) |
| epithelial cancers | scFv-41BB-CD3ζ | + | (2) | |
| CAIX | Renal-cell carcinoma | scFv-CD4- FcεRIγ | Phase I | (3-5) |
| Renal cell carcinoma | G250-FcεRIγ | - | (6-8) | |
| CD19 | B cell malignancies | scFv-CD3ζ (EBV) | - | (9) |
| B cell malignancies | scFv-CD3ζ | + | (10, 11) | |
| B cell malignancies | scFv-CD28-CD3ζ | + | (12-16) | |
| Refractory Follicular Lymphoma | scFv-CD3ζ | Phase I | (17, 18) | |
| B cell malignancies | scFv-CD28-CD3ζ | + | (19-22) | |
| ALL | scFv-41BB-CD3ζ | - | (23) | |
| ALL | scFv-41BB-CD3ζ | + | (24) | |
| B cell malignancies | scFv-CD3ζ (Influenza MP-1) | + | (25) | |
| B cell malignancies | scFv-CD3ζ (VZV) | - | (26) | |
| ALL | FMC63-CD28-41BB- CD3ζ | +, - | (27-29) | |
| B cell malignancies | FMC63-41BB-CD3ζ | + | (30) | |
| Follicular lymphoma | FMC63-CD28-CD3ζ | NCT00924326 | (31) | |
| B cell malignancies | FMC63-CD28-CD3ζ | NCT00924326 | (32) | |
| CLL & ALL | SJ25C1-CD28-CD3ζ | (NCT00466531 NCT01044069) | (33) | |
| CLL | FMC63-41BB-CD3ζ | NCT01029366 | (34, 35) | |
| Lymphoma | scFv-CD3z + scFv-CD28-CD3ζ | Phase I | (36) | |
| CD20 | Lymphomas | scFv-CD28-CD3ζ | - | (37) |
| B cell malignancies | scFv-CD4-CD3ζ | - | (38) | |
| B-cell lymphomas | scFv-CD3ζ | - | (39, 40) | |
| Mantle cell lymphoma, indolent B cell lymphomas | scFv-CD28-41BB-CD3ζ | NCT00621452 | (41, 42) | |
| CD22 | B cell malignancies | scFV-CD4-CD3ζ | - | (38) |
| CD23 | CLL | scFv-CD28-CD3ζ | + | (43) |
| CD24 | Pancreatic adenocarcinoma | scFv- CD28-FcεRIγ | + | (44) |
| CD30 | Lymphomas | scFv-FcεRIγ | - | (45) |
| Hodgkin lymphoma | scFv-CD3ζ (EBV) | + | (46) | |
| scFv-CD28-CD3ζ (EBV) | + | (47) | ||
| CD33 | AML | scFv-CD28-CD3ζ | - | (48) |
| cFv-41BB-CD3ζ | ||||
| scFv-CD28-CD3ζ (EBV) | + | (49) | ||
| CD38 | Non Hodgkin lymphoma | scFv-41BB-CD3ζ | + | (50) |
| CD44v7/8 | Cervical carcinoma | scFv-CD8-CD3ζ | + | (51) |
| CEA | Colorectal cancer | scFv-CD3ζ | + | (52-56), |
| scFv-FcεRIγ | + | (55, 57) | ||
| scFv-CD3ε | - | (58) | ||
| scFv-CD28-CD3ζ | - | (59) | ||
| scFv-CD28-CD3ζ | + | (60, 61) | ||
| EGFRvIII | Glioblastoma | scFv-CD28-41BB- CD3ζ | NCT01454596 | (62) |
| EGP-2 | Multiple malignancies | scFv-CD3ζ | - | (63) |
| scFv-FcεRIγ | - | (63),(64) | ||
| EGP-40 | Colorectal cancer | scFv-FcεRIγ | - | (65) |
| EphA2 | Glioblastoma | scFv-CD28-CD3ζ | + | (66) |
| erb-B2 | Breast and others | scFv-CD28-CD3ζ | + | (67, 68) |
| scFv-CD28-CD3ζ (Influenza) | + | (69) | ||
| scFv-CD28mut.-CD3ζ | + | (70) | ||
| Prostate cancer | scFv-FcεRIγ | + | (71) | |
| Colon cancer | (72) | |||
| Various tumors | scFv-CD28-41BB- CD3ζ | + | (73, 74) | |
| erb-B 2,3,4 | Breast and others | Heregulin-CD3ζ | - | (75),(76) |
| scFv-CD3ζ | + | (77) | ||
| FBP | Ovarian cancer | scFv-FcεRIγ | + | (78-80) |
| Ovarian cancer | scFv-FcεRIγ (alloantigen) | + | (81) | |
| Fetal acethylcholine e receptor | Rhabdomyosarcoma | scFv-CD3ζ | - | (82) |
| GD2 | Neuroblastoma, Melanoma | scFv-CD3ζ | - | (9, 10) |
| scFv-CD3ζ | NCT00085930 | (83, 84) | ||
| scFv-CD28-OX40-CD3ζ | -, + | (74, 85, 86) | ||
| scFv-CD3ζ (VZV) | - | (26) | ||
| Ewing sarcoma | scFv-CD28-CD3ζ | + | (87) | |
| GD3 | Melanoma | scFv-CD3ζ, ScFv-CD3ε | - | (88) |
| scFv-CD28-CD3ζ | + | (89) | ||
| Her-2 | Medulloblastoma | scFv-CD3ζ | + | (90) |
| scFv-CD28-CD3ζ | + | (91) | ||
| Pancreatic adenocarcinoma | scFv-CD28-41BB- CD3ζ | + | (44) | |
| Glioblastoma | Phase I | (92) | ||
| Osteosarcoma | scFv-CD28-CD3ζ | + | (93) | |
| Ovarian | scFv-CD28-CD3ζ | + | (94) | |
| HMW-MAA | Melanoma | scFv-CD3ζ, ScFv-CD28-CD3ζ | - | (95) |
| IL-11Rα | Osteosarcoma | scFv-CD28-CD3ζ | + | (96) |
| IL-13R-α2 | Glioma | IL-13-CD28-4-1BB- CD3ζ | + | (97) |
| Glioblastoma | IL-13- CD3ζ | + | (98, 99) | |
| Medulloblastoma | IL-13- CD3ζ | + | (100) | |
| KDR | Tumor neovasculature | scFv-FcεRIγ | - | (101) |
| κ-light chain | B-cell malignancies (B-NHL, CLL) | scFv-CD3ζ | + | (102) |
| scFv-CD28-CD3ζ | + | (102) | ||
| Lewis Y | Various carcinomas | scFv-FcεRIγ | - | (103) |
| Epithelial derived tumors | scFv-CD28-CD3ζ | + | (104-106) | |
| L1-cell adhesion molecule | Neuroblastoma | scFv- CD3ζ | Phase I | (107, 108) |
| MAGE-A1 | Melanoma | scFV-CD4-FcεRIγ | - | (109) |
| scFV-CD28-FcεRIγ | ||||
| Mesothelin | Mesothelioma | scFv-41BB-CD3ζ | + | (73, 110, 111) |
| Murine CMV infected cells | Murine CMV | Ly49H- CD3ζ | + | (112) |
| MUC1 | Breast, Ovary | scFV-CD28-OX40- CD3ζ | + | (113, 114) |
| MUC16 | Ovary | scFV-CD28-CD3ζ | (115) | |
| NKG2D Ligands | Myeloma, ovarian & other tumors | NKG2D-CD3ζ | + | (116-121) |
| NY-ESO-1 (157-165) | Multiple myeloma | scFv-CD28-CD3ζ | + | (122) |
| Oncofetal antigen (h5T4) | Various tumors | scFV-CD3ζ (vaccination) | + | (123) |
| PSCA | Prostate carcinoma | 7F5-β2-CD3ζ | - | (124) |
| scFv-CD3ζ | (125) | |||
| PSMA | Prostate cancer/ tumor vasculature | scFv-CD3ζ | + | (126, 127) |
| Prostate/tumor vasculature | scFv-CD28-CD3ζ | - | (128) | |
| scFv-CD3ζ | + | (129) | ||
| ROR1 | B-CLL and mantle cell lymphoma | scFv-CD28-CD3ζ | + | (130) |
| Targeting via mAb IgE | Various tumors | FcεRI-CD28-CD3ζ | + | (131) |
| TAG-72 | Adenocarcinomas | scFv-CD3ζ | + | (132),(133) |
| VEGF-R2 | Tumor neovasculature | scFv-CD3ζ | - | (134) |
| Biotinylated molecules | Various tumors-ovarian | BBIR-z/CD28z | + | (135) |
The moieties used to bind to antigen fall in three general categories, either scFv’s derived from antibodies, Fab’s selected from libraries, or nature ligands that engage their cognate receptor (see Fig. 1, first generation CARs). Successful examples in each of these categories–too many to cite–have been reported (Table 1). scFv’s derived from murine immunoglobulins are commonly used, as they are easily derived from well-characterized monoclonal antibodies. They however may prove to be more immunogenic than Fab’s derived from human libraries or invariant human ligands.
Figure 1. Three generation of CARs.
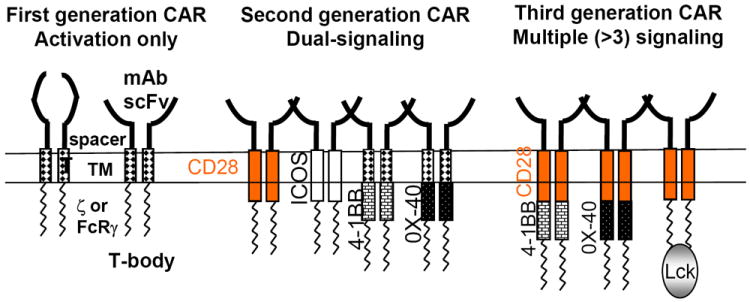
Left: First generation CARs, including activating receptors such as CD8/CD3z fusion receptors and T-bodies; middle: Second generation CARs providing dual-signaling to direct combined activating and costimulatory signals; right: Third generation CARs comprising more complex structures with 3 or more signaling domains.
The rules for selecting optimal epitopes for CAR targeting are still little known. The position of the epitope and its distance to the cell surface are expected to affect the binding to the antigen and the optimal formation of T cell-target conjugates and synapses,(6) but there is still little knowledge of the overall rules governing optimal epitope selection. Empirical observations indicate that the structure of the “spacer region” between an scFv and the transmembrane region (Fig. 1) can affect CAR specificity, but no definitive principles have yet emerged.(7) CAR length and protrusion from the T cell membrane are likely to affect synapse formation. The optimal affinity of CARs is also little defined. Few studies have attempted to address this question, which is of major importance in the case of TCRs(8, 9) and likely to impact CAR function as well. Informative studies comparing multiple CARs recognizing the same epitope with different affinities are still lacking. Finally, the effect of antigen density is not yet well defined. CARs typically target highly expressed antigens, but little is known about minimum thresholds. It is uncertain whether CARs are as exquisitely sensitive as TCRs.(9) If not, lesser sensitivity could represent a limitation in their activity against tumors expressing low antigen levels but may also turn into an advantage where avoidance of low-level antigen expression on normal cells is desirable. Thus, the antigen-binding moiety of the CAR is not just a targeting device but is integral to CAR function, which is largely but not solely defined by the signaling components incorporated into the CAR’s cytoplasmic domain.
CAR signaling
The first fusion receptors shown to have T cell-activating potential on their own were chimeric molecules between CD3-ζ or Fc receptor γ and CD8, CD4, CD25 or CD16 (Fig. 1, first generation CARs), which were shown to initiate phosphatidylinositol and tyrosine kinase pathways together with calcium influx in human T cell leukemias.(10-13) The addition of a hapten-specific scFv derived from amurine antibody to the extracellular portion of such fusions, termed a T-body (Fig. 1, first generation CARs), effectively redirected cytolysis by murine T cell hybridomas.(14) While CD3-ζ chain aggregation is sufficient to enable lytic activity in CTL lines, it is important to bear in mind that the strength of signal required for cytotoxicity is lower than that needed for other T cell functions. This likely underscores the limited therapeutic responses reported with activating receptors, the anti-tumoral effects of which are often confined to local administration models (15, 16) or short-term systemic models.(17) In transgenic mice, T cells expressing CARs that only comprise an activation domain in their cytoplasmic domain are prone to undergoing anergy.(18)
Once we could efficiently transduce human primary T cells, we found that CD3-ζ CARs failed to elicit a robust cytokine response, including interleukin-2, and support T cell expansion upon repeated exposure to antigen.(19) It would take the design of a tri-partite fusion receptor, possessing both activating and costimulatory properties (Fig. 1, second generation CARs), to obtain absolute T cell expansion of human peripheral blood T cells upon repeated exposure to antigen.(20) Significantly, these essential functions cannot be investigated in leukemic or immortalized T cell lines,(21) but only in primary T cells, which CAR investigators have now solidly embraced as the gold standard for evaluating CAR function in vitro or in vivo. Eventually dubbed second-generation CARs, receptors encompassing the CD3-ζ chain and the cytoplasmic domain of a costimulatory receptor such as CD28, 4-1BB, DAP10, OX40 or ICOS, were eventually reported (Table 1). The superior activity of dual-signaling receptors over the activating-only receptors was observed in several models utilizing mouse or human T cells.(22-24) The key attribute of dual-signaling receptors is to confer greater strength of signaling and persistence to the T cells, resulting in their overall greater potency. The enhanced persistence imparted by dual-signaling CARs has been confirmed in patients treated with a mixture of T cells transduced with either a CD28/CD3ζ or CD3ζ-only CAR.(25) Second generation CARs come in varied configurations, but exhaustive comparisons are still lacking. Some CD28 and 4-1BB–based second generation CARs were compared in animal models, but either one proved to be superior to the other in different contexts. In one study, Carpenito et al found that two CD28 and 4-1BB-based CD19-specific CARs had the same therapeutic activity, but noted that the T cells expressing the 19-BB CAR accumulated to greater levels over time, possibly in antigen-independent fashion.(26) This difference was not observed in another model.(27) More comparative studies are needed, noting that such studies must take into account the variability between CARs within any one given category. For example, different CD28/CD3ζ CARs differ in their ability to elicit interleukin (IL)-2 secretion.(20, 28) Furthermore, the location of the targeted epitope, its density, the affinity of the CAR and other topological effects of CAR structure affect CAR signaling, as discussed above. Comparisons will thus need to include multiple representatives of the evaluated categories to reach generalizable conclusions.
A third generation of CARs, encompassing two costimulatory domains combined with an activation domain in their cytoplasmic domain (Fig. 1, third generation CARs), has been described, which appears to confer yet greater potency to tumor targeted T cells in some mouse models.(26, 27, 29, 30) These more complex structures warrant further study as well. A first clinical study utilizing a CD20-specific CD28/4-1BB/CD3ζ did not shown dramatic responses,(31) but this early result should not in anyway detract from the potential value of these “triple-decker” CARs. Overall, more investigation is needed to attain a better understanding of optimal CAR signaling to promote sustained T cell function and survival, preventing premature death, rapid exhaustion or undue proliferation.
Potentiation and complementation of CAR function: costimulatory ligands, chimeric costimulatory receptors and cytokines
Costimulatory support can be engineered into T cells otherwise than through a CAR. (Fig. 2) The coexpression of chimeric costimulatory receptors (CCRs), costimulatory receptor ligands and cytokines, have all been utilized to modulate the function and/or survival of CAR-transduced T cells.
Figure 2. Strategies to provide costimulatory support to CAR-modified T cells.
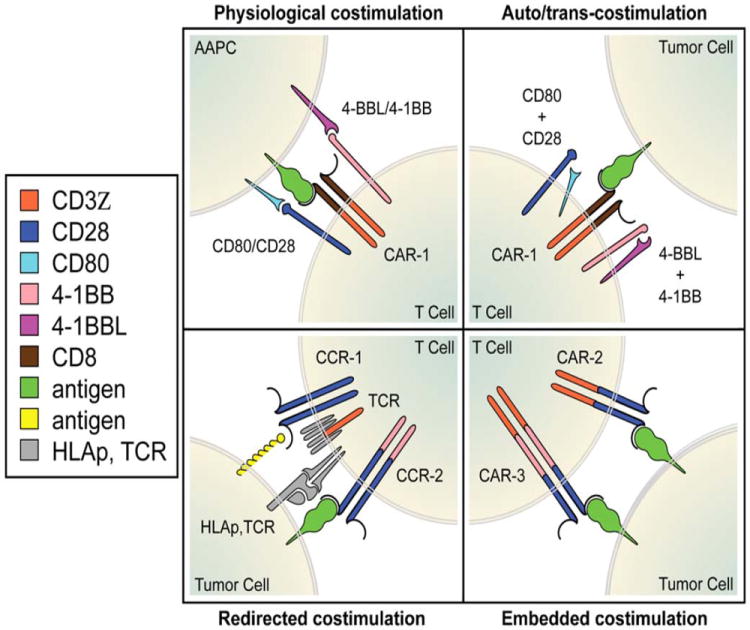
From upper left: UL, physiological costimulatory ligand display by professional or artificial antigen presenting cells; UR, auto- and trans-costimulation by T cells expressing costimulatory ligands; LR, embedded costimulation provided by second or third generation CARs; LL, redirected costimulation mediated by an antigen-specific chimeric costimulatory receptor (CCR).
Costimulatory ligands
The constitutive expression of costimulatory ligands on the T cell surface (Fig 2) provides a powerful means to potentiate CAR-targeted T cells. Several ligands for Ig super-family and TNF receptor family costimulatory receptor, including CD80, CD86, 4-1BBL, OX40L and CD70, have been shown to enhance T cell proliferation and cytokine secretion upon antigen engagement.(32) The combination of two ligands, in particular CD80 and 4-1BBL, results in sustained in vivo T cell expansion and persistence, associated with the rejection of massive, established tumor burdens.(32) Both auto- and trans-costimulation have been shown to contribute to enhanced T cell activity in this context, which may be useful to enhance adoptive cell therapies utilizing CAR- or TCR-transduced T cells. The occurrence of costimulatory ligands found on tumor cells is also likely to influence the activity of CAR-modified T cells, whether they are activating (e.g., CD80, CD40L, 4-1BBL) or inhibitory (e.g., PD-L1).
Chimeric costimulatory receptors
CCRs mimic costimulatory signals but, unlike CARs, do not provide a T cell activation signal. Their purpose is to provide costimulation, e.g. a CD28-like signal,(33) in the absence of the natural costimulatory ligand on the antigen-presenting cell (Fig.2). They thus provide a means for the tumor to direct counterfeit costimulation specifically within the tumor microenvironment. CCRs targeting the glycolipid GD2, MUC16, PSMA and the α-folate receptor have been described, utilized in conjunction with a TCR or a CAR to augment T cell reactivity against dual-antigen expressing T cells, reinforcing T cell activation in the absence of natural costimulatory ligands and in antigen-dependent fashion.(20, 33-35) Under particular conditions, CCRs may also be utilized to improve selective tumor targeting, as further discussed below.
Cytokines
Another approach to enhance the potency of CAR-targeted T cells is to further genetically modify the T cells to secrete pro-inflammatory or pro-proliferative cytokines. Its purpose is not only to provide autocrine support to enhance the function, proliferation and/or persistence of CAR-expressing T cells, but also to favorably alter the tumor microenvironment and recruit endogenous innate and cognate immune effectors. The expression of T cell-encoded cytokines additionally aims to limit the systemic toxicity of many cytokines. Preclinical reports investigating γc cytokines or IL-12 show great promise for this approach.
T cell-encoded IL-15 increases the viability and proliferation of human peptide-specific T cells despite withdrawal of exogenous IL-2. (36) Improved in vitro and in vivo expansion of human Epstein Barr Virus specific cytotoxic T cells (EBV-CTLs) following retroviral gene transfer of the IL-2 or IL-15 cDNA has also been reported.(37) The report of an isolated IL-15 modified CD8+ T cell clone exhibiting logarithmic proliferation for over 1 year in the absence of exogenous cytokine support cautions against this approach, (38) although this concern may be mitigated by using a suicide gene to potentially remove T cells via drug-induced apoptosis(37). Cytokine modified T cells used as antigen presenting cells to expand tumor targeted T cells, expressing either IL-7 and IL-12 (39) or IL-21, (40) successfully expanded tumor-targeted T cells, with a more favorable central memory phenotype in the latter case. Comparisons between cytokines expressed at different levels in different assays or tumor models are complex to interpret. Nonetheless, we compared CD19 CAR-targeted human 19z1+ T cells that constitutively expressed either IL-2, IL-7, IL-15 or IL-21 under standardized conditions and found that all four γc cytokines enhanced tumor rejection in a xenotransplant model of human CD19+ tumor, more so, in this context, with IL-7 and IL-21 than IL-2 and IL-15 (41).
In an immune competent syngeneic tumor model, CD19-targeted, CAR-modified T cells expressing IL-12 showed greater efficacy than CAR-modified T cells alone.(42) Significantly, IL-12 modified T cells eradicated CD19+ tumors in the absence of any prior conditioning and, additionally, exhibited resistance to regulatory T cell (Treg) inhibition. In a murine melanoma model, transgenic Pmel-1 CD8+ T cells, as well as Pmel-1 TCR-transduced murine T cells that were modified to express IL-12, eradicated established tumors with significantly greater potency than T cells expressing the Pmel-1 TCR alone(43). Similarly improved outcomes were obtained in tumor bearing mice treated with IL-12 secreting T cells targeted to tumor by an anti-VEGF receptor-2 CAR.(44) In both latter models, the effect of IL-12 appears to act at least in part by altering myeloid cells in the tumor microenvironment.(44, 45)
The titration of cytokine secretion by T cells is important because of the potential toxicity of elevated systemic levels. One may address this concern by appropriately calibrating promoter strength or through conditional cytokine release following T cell activation utilizing nuclear factor of activated T cells (NFAT)-inducible promoters.(46) Using this approach to control cytokine secretion, two trials treating metastatic NY-ESO-1+ tumors with autologous TCR-targeted T cells or tumor-infiltrating T cells secreting IL-12, are under way at the NCI (NCT01457131, NCT01236573).
CARs in the clinic
The CD19 paradigm
The most investigated target to date is CD19, an attractive target for CAR-based therapy as it is present in most B cell leukemias and lymphomas but not in any normal tissue other than the B cell lineage.(47, 48) CD19+ malignancies were the first cancers to be eliminated by CAR-engineered human T cells administered intravenously to systemic tumor-bearing mice.(49) Successful B cell tumor eradication was eventually obtained with different CD19 CARs(15, 22-24), paving the way for several on-going clinical trials. The targeting of CD19 has thus become a paradigm for evaluating CAR technology.(50) We estimate that at least 50 patients with leukemia or lymphoma have been treated at the time this review is written, 28 of which were reported from 5 centers in the past year.(25, 51-56) The reported clinical outcomes were recently reviewed elsewhere (57, 58) and are briefly summarized here.
The largest series and most dramatic early results were reported from the National Cancer Institute (NCI), Memorial Sloan-Kettering Cancer (MSKCC) and the Abramson Family Cancer Research Institute at the University of Pennsylvania (UPenn). These clinical trials followed the same overall procedures, including patient T cell apheresis, retroviral or lentiviral CAR transduction, T cell expansion and host conditioning prior to T cell infusion. They however differ in several regards, including not only CAR design (same CD28/CD3z dual-signaling domain utilized at the NCI and MSKCC, 4-1BB/CD3z utilized at UPenn), but also T cell manufacturing, conditioning chemotherapy, tumor burden, tumor chemo-sensitivity, and T cell dosage, which are reviewed in detail in ref. (57) Kochendorfer and the NCI group reported on 8 patients (4 with CLL, 3 with follicular lymphoma, and one with marginal zone lymphoma) conditioned with fludarabine and cyclophosphamide, and further given IL-2 after T cell infusion. Amongst the 4 CLL patients, one achieved a complete response (CR) and another stable disease (SD). Four of the 8 treated patients exhibited B cell aplasias.(53) Brentjens and colleagues reported on 8 patients with CLL and 1 patient with B cell acute lymphoblastic leukemia (B-ALL), whose diseases were resistant to the milder cyclophosphamide conditioning regimen used in 5 of the 8 CLL patients (the first 3 were given T cells without any prior conditioning; no significant response was obtained). In the CLL cohort, 2 patients had stable disease and one patient demonstrated a substantial lymph node reduction. None of the CLL patients developed B cell aplasia, in contrast to the one patient with relapsed B-ALL. (47) June and colleagues (54, 55) treated 3 patients with bulky CLL that were conditioned with bendamustine, a highly active agent in these in these patients. Two of them achieved dramatic, long-lasting CR’s. The reasons for the different outcomes in these 15 CLL patients treated at 3 different centers, which include significant differences in CAR design, conditioning intensity and the selection of chemosensitive patients amongst many variables,(57) still remain to be elucidated. Altogether, better responses were observed following more active conditioning, resulting in a 25% CR rate in 12 CLL patients treated with T cells following chemotherapy conditioning. Much will undoubtedly be learned about the role of the CAR and other parameters by comparing biological and clinical outcomes using similarly manufactured T cells in similarly selected patients.
Solid tumors
One next frontier for CAR-based therapies is to take on solid tumors. Early attempts with first generation CARs did not yield very encouraging data,(59) although one recent study targeting the GD2 ganglioside in children with neuroblastoma showed 2 CRs in 13 patients.(60) Solid tumors present a different set of challenges compared to B cell malignancies: overall lesser sensitivity to T cell mediated cytotoxicity, a microenvironment that presents with an array of immunosuppressive mechanisms differing between tumor types, and a paucity of target antigens with an expression profile as favorable as CD19. Despite an impressive number of investigated targets (Table 1), few target candidates are tumor-specific, or restricted to the tumor and a “dispensable” normal cell type or a tissue that is sheltered from an immune attack. In this perspective, identifying valid targets to achieve efficacious tumor rejection while ensuring patient safety is an essential goal that requires further investigation. Nonetheless, several trials utilizing first and second generation CARs are under way and listed on the US clinicaltrials.gov web site.
CAR safety
The two main safety concerns associated with the use of CARs are the targeted destruction of normal tissues and cytokine storms associated with large-scale immune responses. The toxicity of the different conditioning regimens used in conjunction with adoptive T cell therapies is also a significant issue to consider but is beyond the scope of this review.
On-target, off-tumor responses
The immune-mediated rejection of normal tissues that express the targeted antigen is referred to as an “on-target, off-tumor” response. This occurrence is best illustrated in the B cell aplasias induced by CD19-targeted CARs.(52-54) Whereas B cell aplasia can be effectively managed by administering intravenous immunoglobulin, such collateral damage may not be tolerable in many other instances. This may for example be the case for her2,(61) which is expressed at low level in several normal tissues, including heart and pulmonary vasculature. Other examples, for which no toxicities have been reported to date, include PSMA, which is highly expressed in castrate-resistant, metastatic prostate cancer,(62) but is detected in astrocytes type II, the renal proximal tubule and the jejunum brush border; ROR1, which is expressed in a subset of leukemias and lymphomas, but is also detected in adipocytes.(63) T cells are very effective at destroying normal tissues that express the targeted antigen, as exemplified by the ocular and vestibular effects of MART-1-specific T cells(64) and the cholestatic effect of T cells targeted to carbonic anhydrase IX.(65) It is presently unknown whether the very low level expression of antigens such as PSMA and ROR1 on normal tissues will expose these to immune destruction. This problem would be easily resolved if there were truly tumor-specific cell surface molecules to target, but such molecules are so far very rare. The identification of restricted CAR targets is therefore a high priority.
Cytokine storms
The second major concern is that of “cytokine storms” associated with intense anti-tumor responses mediated by large numbers of activated T cells.(53-55) These typically cause high fever and hypotension, potentially resulting in organ failure. Their management may require steroids, vasopressors and/or supportive therapy delivered in the intensive care unit. Grupp and colleagues have observed that IL-6 blockade utilizing Tocilizumab may be effective in steroid-refractory circumstances, without compromising T cell efficacy.(66) Unlike many conventional drug-induced side effects, this toxicity cannot be controlled by simply reducing drug dosage, as proliferating T cells will increase in numbers and eventually reach critical levels where a synchronous cytokine response exceeds tolerability. Split T cell dosing or short-lived T cells may partially reduce this effect, but more fundamental solutions are needed to reduce and ideally prevent the occurrence of overwhelming T cell activity..
Emerging solutions to improve CAR safety
Recognizing that CAR-modified T cells are in general well tolerated, their broader use requires having solid strategies to treat or, better, prevent on-target, off-tumor effects and cytokine storms. On therapeutic option is to utilize suicide genes to have a means to eliminate an excessive response. Herpes simplex virus thymidine kinase(67) and inducible caspase-9(68) are clinically tested systems that could be used to halt deleterious responses.(69) The downsides to this approach are that it is reactive, not preventative, and that active T cells will be eliminated, possibly curtailing the therapy. A better understanding of cytokine storms may offer novel prospects for reducing toxicity without compromising therapy and limiting the use of corticosteroids.(66) Significantly, we find, in ALL patients treated with a CD19-specific CD28/CD3z CAR, that stronger cytokine responses occur in patients with large tumor burdens but not in those with minimal residual disease at the time of T cell infusion, a finding that suggests that reducing tumor burden by alternative means prior to T cell infusion will reduce the risk of T cell-induced cytokine-mediated toxicity following a subsequent T cell infusion (unpublished observations). Ultimately, the design of T cells that are effective, highly tumor-specific, and regulated in their maximal accumulation and activation (so as to prclude toxic cytokine elevation), will represent a valuable advance for the use of CARs. One approach to improve tumor selectivity, based on combinatorial antigen recognition, is reviewed below.
New directions in the CAR industry
New technologies or concepts other than the design of better CARs and their combination with costimulatory ligands, chimeric costimulatory receptors or cytokines, are emerging to broaden or improve the use of CARs. These include improved CAR delivery systems, the design of CARs that recognize intracellular antigens, and combinatorial antigen recognition to increase T cell specificity and potency.
CAR delivery
The mechanics of T cell transduction are beyond the scope of this review but are briefly addressed here. CARs began to be investigated in meaningful ways when methods for the transduction of human primary T cells became available(70-72). For the past 15 years, virtually all CAR studies have relied on retroviral vectors, including gamma-retroviral and lentiviral vectors.(73) Most current clinical trials utilize retroviral vectors derived from murine leukemia virus or human immune deficiency virus-1. Although retroviral vectors can induce insertional oncogenesis in hematopoietic progenitors(74, 75), T cells appear to be far less susceptible to retroviral vector-induced transformation(76-79). Transposases, which also provide random vector integration(80), are starting to be evaluated in the context of CAR therapy.(81) The relative advantages/disadvantages of these different integrating systems have not yet been elucidated, but will hinge on CAR expression levels, silencing over time, safety features, ease of manufacturing and usage, and cost. Although T cell transformation secondary to insertional mutagenesis has not been reported to date, site-directed vector integration into genomic safe harbors(82) may eventually enable to achieve long-term CAR expression without any risk of insertional oncogenesis. Alternative approaches that do not rely on transgene integration, which utilize RNA electroporation,(83, 84) or cell surface conjugation,(85) result in transient CAR expression, precluding effective T cell persistence beyond a week or two. The usefulness of transiently CAR-expressing T cells, which would presumably require multiple infusions to provide meaningful tumor responses but may reduce destruction of normal tissues or prevent T cell accumulation to levels exposing to the risk of cytokine storms, remains to be established.
Another key aspect of CAR delivery is the addressee and identifying what T cells, expanded under what conditions, are better suited for optimal tumor eradication. As stated above, this topic is beyond the scope of this review, recognizing that different T cell subsets (CD4+ or CD8+ αβ T cells, γδ T cells, naïve, central memory, effector memory, virus-specific T cells and the recently described stem-like memory T cells) (86, 87) warrant further investigation to delineate whether different CAR designs are best suited for different T cell types. CARs are also functional in Tregs(88) and in the progeny of transplanted T cell progenitors(89).
CAR-like TCRs and TCR-like CARs
The transfer of TCRs into T cells poses two particular challenges that CARs elude: the risk of mispairing between endogenous and transduced TCR chains(90), and competition for rate-limiting CD3 complex(91), which is required for TCR signaling. Several approaches have been proposed to prevent TCR chain mispairing, including partial murinization of the constant regions, the addition of disulfide bonds and altering the knob-in-hole directional interaction between constant regions. Another approach is to add signaling domains to the intracellular portions of the transduced TCR (92), similarly to first generation CARs, which Willemsen and Debets showed could at once avert TCR mispairing and reduce association with CD3.(92, 93)
Conversely, HLA-peptide complexes can be targeted by antibody structures that mimic TCR recognition. CARs may be advantageous in this regards as they do not interfere nor compete with the native TCR and CD3, and can be further endowed with costimulatory capabilities. Human Fab fragments specific to peptide/MHC molecules have been derived from phage display libraries.(94). While many preferentially bind to MHC, (95) some high-affinity Fabs with greater binding affinity for the peptide have been generated and shown by crystallographic analysis to have a binding footprint to MHC/peptide complexes similar to that of TCRs(96). The therapeutic potential and toxicity of these TCR-like CARs remain to be established.
Combinatorial antigen recognition
T cells may also be rendered more tumor-selective through combinatorial antigen recognition. We recently described a strategy that integrates combinatorial antigen recognition, split signaling and, critically, balanced strength of T cell activation and costimulation, to generate T cells that eliminate target cells that express a combination of antigens while sparing cells that express each antigen individually.(97) In this approach, T cell activation requires TCR or CAR-mediated recognition of one antigen, while costimulation is independently mediated by a CCR(33) specific for a second antigen. To achieve tumor selectivity, we diminished the efficiency of T cell activation to a level where it was ineffective without rescue provided by simultaneous CCR recognition of the second antigen.(98) Novel approaches to enforce tumor specificity in the face of truly unique target antigens are an important direction for future immunotherapies.
Perspectives
While there remains a vast number of important biological questions to address – optimizing CAR signaling, defining optimal targets, working out optimal combinatorial strategies, identifying the best and most practical processes for T cell subset selection and T cell manufacturing, reducing T cell-mediated toxicity and the toxicity of host conditioning – the first clinical successes of CAR therapy are being registered. The prospect of meeting the challenging premise of adoptive T cell therapy – to achieve specific tumor destruction with one or few T cell infusions and limited collateral damage to normal tissues– may be within reach. The targeting of B cell malignancies through CD19 has emerged as the paradigm for the CAR field. At the present, it also stands as an exception. The identification of safe targets in a broad range of tumor types, eventually in combinatorial fashion, and harnessing CAR technology for the treatment of solid tumors, are future challenges for all adoptive T cell therapies including those utilizing CARs. As this review aims to convey, there are many exciting strategies in the pipeline, and as many reasons to be optimistic about the prospects for CAR therapies. Providing access to targeted T cell therapies to all medical centers and their patients will pose additional biological, logistical and economic challenges, which are beyond the scope of this review. The fact that models for broad access to targeted T cell therapies are increasingly discussed is testimony to the therapeutic potential and rising credibility of CARs.
Supplementary Material
Statement of Significance.
Chimeric antigen receptors (CARs) are a new class of drugs with great potential for cancer immunotherapy. Upon their expression in T lymphocytes, CARs direct potent, targeted immune responses that have recently shown encouraging clinical outcomes in a subset of patients with B cell malignancies. This review focuses on the design of CARs, including the requirements for optimal antigen recognition and different modalities to provide costimulatory support to targeted T cells, which includethe use of second and third generation CARs, costimulatory ligands, chimeric costimulatory receptors and cytokines.
Acknowledgments
Our work is supported by NIH, MSKCC’s Experimental Therapeutics Center, the Terry Fox Foundation, the Alliance for Cancer Gene Therapy, the Mallah Foundation, the Majors Foundation and the Sanders Fund.
Footnotes
Conflict of Interest Statement:
I, Michel Sadelain, confirm there is no conflict of interest.
References
- 1.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 2.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer cell. 2003;3:431–7. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–23. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou G, Levitsky H. Towards curative cancer immunotherapy: overcoming posttherapy tumor escape. Clinical & developmental immunology. 2012;2012:124187. doi: 10.1155/2012/124187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dustin ML, Depoil D. New insights into the T cell synapse from single molecule techniques. Nature Reviews Immunology. 2011;11:672–84. doi: 10.1038/nri3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bridgeman JS, Hawkins RE, Hombach AA, Abken H, Gilham DE. Building better chimeric antigen receptors for adoptive T cell therapy. Curr Gene Ther. 2010;10:77–90. doi: 10.2174/156652310791111001. [DOI] [PubMed] [Google Scholar]
- 8.Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. 2009;126:165–76. doi: 10.1111/j.1365-2567.2008.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards LJ, Evavold BD. T cell recognition of weak ligands: roles of signaling, receptor number, and affinity. Immunol Res. 2011;50:39–48. doi: 10.1007/s12026-011-8204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- 11.Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64:1037–46. doi: 10.1016/0092-8674(91)90327-u. [DOI] [PubMed] [Google Scholar]
- 12.Letourneur F, Klausner RD. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc Natl Acad Sci U S A. 1991;88:8905–9. doi: 10.1073/pnas.88.20.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romeo C, Amiot M, Seed B. Sequence requirements for induction of cytolysis by the T cell antigen/Fc receptor zeta chain. Cell. 1992;68:889–97. doi: 10.1016/0092-8674(92)90032-8. [DOI] [PubMed] [Google Scholar]
- 14.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–4. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–44. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 16.Hwu P, Yang JC, Cowherd R, Treisman J, Shafer GE, Eshhar Z, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995;55:3369–73. [PubMed] [Google Scholar]
- 17.Chmielewski M, Rappl G, Hombach AA, Abken H. T cells redirected by a CD3zeta chimeric antigen receptor can establish self-antigen-specific tumour protection in the long term. Gene Ther. 2012 doi: 10.1038/gt.2012.21. [DOI] [PubMed] [Google Scholar]
- 18.Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181:1653–9. doi: 10.1084/jem.181.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1:123–7. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 21.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161:2791–7. [PubMed] [Google Scholar]
- 22.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–35. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 23.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 25.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–20. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–41. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010;21:75–86. doi: 10.1089/hum.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–25. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 31.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–50. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–9. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 33.Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. 1998;188:619–26. doi: 10.1084/jem.188.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. Journal of clinical immunology. 2012 doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 35.Duong CP, Westwood JA, Berry LJ, Darcy PK, Kershaw MH. Enhancing the specificity of T-cell cultures for adoptive immunotherapy of cancer. Immunotherapy. 2011;3:33–48. doi: 10.2217/imt.10.81. [DOI] [PubMed] [Google Scholar]
- 36.Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol. 2005;175:7226–34. doi: 10.4049/jimmunol.175.11.7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood. 2007;109:5168–77. doi: 10.1182/blood-2006-06-029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster AE, Leen AM, Lee T, Okamura T, Lu A, Vera J, et al. Autologous designer antigen-presenting cells by gene modification of T lymphocyte blasts with IL-7 and IL-12. J Immunother. 2007;30:506–16. doi: 10.1097/CJI.0b013e318046f3b1. [DOI] [PubMed] [Google Scholar]
- 40.Kaka AS, Shaffer DR, Hartmaier R, Leen AM, Lu A, Bear A, et al. Genetic modification of T cells with IL-21 enhances antigen presentation and generation of central memory tumor-specific cytotoxic T-lymphocytes. J Immunother. 2009;32:726–36. doi: 10.1097/CJI.0b013e3181ad4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115:3508–19. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725–34. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–83. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121:4746–57. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ponomarev V, Doubrovin M, Lyddane C, Beresten T, Balatoni J, Bornman W, et al. Imaging TCR-dependent NFAT-mediated T-cell activation with positron emission tomography in vivo. Neoplasia. 2001;3:480–8. doi: 10.1038/sj.neo.7900204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li YS, Wasserman R, Hayakawa K, Hardy RR. Identification of the earliest B lineage stage in mouse bone marrow. Immunity. 1996;5:527–35. doi: 10.1016/s1074-7613(00)80268-x. [DOI] [PubMed] [Google Scholar]
- 48.Li YS, Hayakawa K, Hardy RR. The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. J Exp Med. 1993;178:951–60. doi: 10.1084/jem.178.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279–86. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 50.Kohn DB, Dotti G, Brentjens R, Savoldo B, Jensen M, Cooper LJ, et al. CARs on track in the clinic. Mol Ther. 2011;19:432–8. doi: 10.1038/mt.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–20. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davila ML, Brentjens R, Wang X, Riviere I, Sadelain M. How do CARs work? Early insights from recent clinical studies targeting CD19. Oncoimmunology. 2012;1:1–7. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooper LJ, Jena B, Bollard CM. Good T cells for bad B cells. Blood. 2012;119:2700–2. doi: 10.1182/blood-2011-12-398719. [DOI] [PubMed] [Google Scholar]
- 59.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81–5. [PubMed] [Google Scholar]
- 63.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116:4532–41. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seaman BJ, Guardiani EA, Brewer CC, Zalewski CK, King KA, Rudy S, et al. Audiovestibular dysfunction associated with adoptive cell immunotherapy for melanoma. Otolaryngol Head Neck Surg. 2012;147:744–9. doi: 10.1177/0194599812448356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 66.Grupp SA, Porter DL, Teachey D, Barrett DM, Chew A, Suppa E, et al. CD19-Redirected Chimeric Antigen Receptor T (CART19) Cells Induce a Cytokine Release Syndrome (CRS) and Induction of Treatable Macrophage Activation Syndrome (MAS) That Can Be Managed by the IL-6 Antagonist Tocilizumab (toc). 54th ASH Annual Meeting and Exposition; Atlanta, GA. 2012. Abstract 2604. [Google Scholar]
- 67.Lupo-Stanghellini MT, Provasi E, Bondanza A, Ciceri F, Bordignon C, Bonini C. Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum Gene Ther. 2010;21:241–50. doi: 10.1089/hum.2010.014. [DOI] [PubMed] [Google Scholar]
- 68.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sadelain M. Eliminating cells gone astray. N Engl J Med. 2011;365:1735–7. doi: 10.1056/NEJMe1109971. [DOI] [PubMed] [Google Scholar]
- 70.Mavilio F, Ferrari G, Rossini S, Nobili N, Bonini C, Casorati G, et al. Peripheral blood lymphocytes as target cells of retroviral vector-mediated gene transfer. Blood. 1994;83:1988–97. [PubMed] [Google Scholar]
- 71.Bunnell BA, Muul LM, Donahue RE, Blaese RM, Morgan RA. High-efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proc Natl Acad Sci U S A. 1995;92:7739–43. doi: 10.1073/pnas.92.17.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gallardo HF, Tan C, Ory D, Sadelain M. Recombinant retroviruses pseudotyped with the vesicular stomatitis virus G glycoprotein mediate both stable gene transfer and pseudotransduction in human peripheral blood lymphocytes. Blood. 1997;90:952–7. [PubMed] [Google Scholar]
- 73.Suerth JD, Schambach A, Baum C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr Opin Immunol. 2012;24:598–608. doi: 10.1016/j.coi.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 74.Kustikova OS, Schiedlmeier B, Brugman MH, Stahlhut M, Bartels S, Li Z, et al. Cell-intrinsic and vector-related properties cooperate to determine the incidence and consequences of insertional mutagenesis. Mol Ther. 2009;17:1537–47. doi: 10.1038/mt.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Riviere I, Dunbar CE, Sadelain M. Hematopoietic stem cell engineering at a crossroads. Blood. 2012;119:1107–16. doi: 10.1182/blood-2011-09-349993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, et al. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–86. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- 77.Recchia A, Bonini C, Magnani Z, Urbinati F, Sartori D, Muraro S, et al. Retroviral vector integration deregulates gene expression but has no consequence on the biology and function of transplanted T cells. Proc Natl Acad Sci U S A. 2006;103:1457–62. doi: 10.1073/pnas.0507496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–35. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang X, Wilber A, McIvor RS, Zhou X. DNA transposons for modification of human primary T lymphocytes. Methods Mol Biol. 2009;506:115–26. doi: 10.1007/978-1-59745-409-4_9. [DOI] [PubMed] [Google Scholar]
- 81.Jin Z, Maiti S, Huls H, Singh H, Olivares S, Mates L, et al. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011;18:849–56. doi: 10.1038/gt.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. 2012;12:51–8. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 83.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–9. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Birkholz K, Hombach A, Krug C, Reuter S, Kershaw M, Kampgen E, et al. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009;16:596–604. doi: 10.1038/gt.2008.189. [DOI] [PubMed] [Google Scholar]
- 85.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72:1844–52. doi: 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–84. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 88.Lee JC, Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M, et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 2011;71:2871–81. doi: 10.1158/0008-5472.CAN-10-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zakrzewski JL, Suh D, Markley JC, Smith OM, King C, Goldberg GL, et al. Tumor immunotherapy across MHC barriers using allogeneic T-cell precursors. Nat Biotechnol. 2008;26:453–61. doi: 10.1038/nbt1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16:565–70. doi: 10.1038/nm.2128. 1p following 70. [DOI] [PubMed] [Google Scholar]
- 91.Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009;69:9003–11. doi: 10.1158/0008-5472.CAN-09-1450. [DOI] [PubMed] [Google Scholar]
- 92.Sebestyen Z, Schooten E, Sals T, Zaldivar I, San Jose E, Alarcon B, et al. Human TCR that incorporate CD3zeta induce highly preferred pairing between TCRalpha and beta chains following gene transfer. J Immunol. 2008;180:7736–46. doi: 10.4049/jimmunol.180.11.7736. [DOI] [PubMed] [Google Scholar]
- 93.Roszik J, Sebestyen Z, Govers C, Guri Y, Szoor A, Palyi-Krekk Z, et al. T-cell synapse formation depends on antigen recognition but not CD3 interaction: studies with TCR:zeta, a candidate transgene for TCR gene therapy. Eur J Immunol. 2011;41:1288–97. doi: 10.1002/eji.200940233. [DOI] [PubMed] [Google Scholar]
- 94.Denkberg G, Reiter Y. Recombinant antibodies with T-cell receptor-like specificity: novel tools to study MHC class I presentation. Autoimmun Rev. 2006;5:252–7. doi: 10.1016/j.autrev.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 95.Hulsmeyer M, Chames P, Hillig RC, Stanfield RL, Held G, Coulie PG, et al. A major histocompatibility complex-peptide-restricted antibody and t cell receptor molecules recognize their target by distinct binding modes: crystal structure of human leukocyte antigen (HLA)-A1-MAGE-A1 in complex with FAB-HYB3. The Journal of biological chemistry. 2005;280:2972–80. doi: 10.1074/jbc.M411323200. [DOI] [PubMed] [Google Scholar]
- 96.Stewart-Jones G, Wadle A, Hombach A, Shenderov E, Held G, Fischer E, et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci U S A. 2009;106:5784–8. doi: 10.1073/pnas.0901425106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2012;31:71–5. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hanada K, Restifo NP. Double or nothing on cancer immunotherapy. Nat Biotechnol. 2013;31:33–4. doi: 10.1038/nbt.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.