

Abstract
We describe and characterize an improved implementation of ETD on a modified hybrid linear ion trap-Orbitrap instrument. Instead of performing ETD in the mass-analyzing quadrupole linear ion trap (A-QLT), the instrument collision cell was modified to enable ETD. We partitioned the collision cell into a multi-section RF ion storage and transfer device to enable injection and simultaneous separate storage of precursor and reagent ions. Application of a secondary (axial) confinement voltage to the cell end lens electrodes enables charge-sign independent trapping for ion-ion reactions. The approximately two-fold higher quadrupole field frequency of this cell relative to that of the A-QLT, enables higher reagent ion densities and correspondingly faster ETD reactions, and, with the collision cell’s longer axial dimensions, larger populations of precursor ions may be reacted. The higher ion capacity of the collision cell permits the accumulation and reaction of multiple full loads of precursor ions from the A-QLT followed by FT Orbitrap m/z analysis of the ETD product ions. This extends the intra-scan dynamic range by increasing the maximum number of product ions in a single MS/MS event. For analyses of large peptide/small protein precursor cations, this reduces or eliminates the need for spectral averaging to achieve acceptable ETD product ion signal-to-noise levels. Using larger ion populations, we demonstrate improvements in protein sequence coverage and aggregate protein identifications in LC-MS/MS analysis of intact protein species as compared to the standard ETD implementation.
Introduction
Electron capture and electron transfer dissociation (ECD and ETD, respectively), are unique in their proclivity to cleave the inter-residue N-Cα bonds of peptides with relative indifference to amino acid composition.(1–3) In addition, both reactions become faster and more efficient as precursor charge increases. These unique features have enabled researchers to study large, highly charged protein molecules that were previously difficult or impossible to characterize using dissociation methods involving vibrational activation.(4–7)
Electron transfer dissociation has been demonstrated on a variety of single and multiple mass analyzer instruments including quadrupole ion trap (1, 2), triple quadrupole(8, 9), quadrupole-time-of-flight (Q-TOF)(10–13), linear ion trap-Orbitrap (14, 15), and quadrupole-FT-ICR instruments (16). With this variety of instrument types, there has been a corresponding diversity in the ion confinement devices wherein the ETD reaction has been performed. ETD reactions have been performed in Paul Traps (3D RF quadrupole ion traps) (17–19), linear RF multipole ion traps (linear quadrupole and hexapole traps) (8, 9, 16), and RF ion pipes (RF stacked ring ion guides) (10). The more successful implementations of ETD, including all of the commercial implementations, involve trapping of reagent ions.
The types of traps used for ETD have primarily been determined by what pre-existing instrument ion path component could be conveniently and effectively modified and repurposed to perform ETD without compromising its usual function. This means ETD is mostly performed within ion traps whose design was optimized for mass analysis, or within ion guides (usually originally designed and optimized for use as collision cells) which are converted into linear ion traps by altering the applied voltages. The first implementation of ETD involving FT Orbitrap analysis of product ions utilized a modified commercial hybrid quadrupole linear ion trap–Orbitrap instrument, LTQ Orbitrap XL.(14) In that and subsequent commercial implementations of ETD on hybrid QLT-Orbitrap instruments, the ETD reaction was conducted in the quadrupole linear ion trap m/z analyzer (A-QLT) in a substantially identical manner to how it was performed with the corresponding commercial standalone quadrupole linear ion trap instruments. The overall approach has been little altered from that used for the original demonstration of ETD.(2) Features of an ideal implementation of ETD would include efficient accumulation of both precursor cations and reagent anions, minimization of ETD reaction times, complete control of the initiation and subsequent quenching of ETD reactions, ability to expose precursor ions to IR activation before and during the ETD reaction, high trapping capacity for both precursor and reagent ions, retention of widest possible m/z range of product ions and efficient transfer of product ions to the m/z analyzer.
The introduction of a quadrupole collision cell into the designs of recent generations of commercial hybrid QLT-Orbitrap instruments (20) offers the opportunity to perform ETD in a device better suited for ion-ion reactions than a quadrupole ion trap of design specifically optimized for m/z analysis. Here, we describe a modified hybrid dual cell quadrupole linear ion trap-Orbitrap mass spectrometer, LTQ Orbitrap Velos ETD, (Thermo Fisher Scientific, Bremen Germany). As presented previously at various scientific conferences,(21, 22) we modified the standard RF quadrupole collision cell to enable ETD and activated-ion ETD (AI-ETD) (23, 24) as well as collision cell-type CID (HCD). This multi-dissociation cell (MDC) was created by partitioning the collision cell quadrupole ion guide into four segments with independently controllable DC biases. This partitioning of the cell enables injection and simultaneous separate storage of precursor cations and reagent anions. Provision was also made for the application of secondary RF (axial) confinement voltage to the cell end plate lens electrodes to enable charge-sign independent trapping (CSIT) during the ETD reaction. The approximately two fold higher operating quadrupole field frequency of this cell, relative to that of the A-QLT, enables higher reagent ion densities and correspondingly higher ETD reaction rates. The higher trapping field frequency, in combination with the collision cell’s longer axial dimensions, permits reaction of larger populations of precursor ions. These modifications have not compromised the function of the MDC as a conventional collision cell. Conversion from operation of as a collision cell to an ETD reaction cell is accomplished solely by altering the voltages applied to the device’s electrode.
In addition to facilitating faster ion-ion reactions, the MDC enables advanced scan functions; for example, multiple loads of precursor ions can be injected into the MDC, allowing for very large ion populations (~5×106 charges) to be accumulated prior to ETD activation. Similar scan functions have recently been described, during which multiple loads of precursor cations were injected into an FT-ICR prior to ECD activation.(25–27) These scan functions are particularly well suited for top-down analysis, as analysis of even modest-sized whole proteins typically necessitates spectral averaging, requiring long acquisition times. Using the MDC, it is possible to instead use multiple cycles of ion injection, isolation, and transfer to the MDC, to accumulate a large precursor ion population. Following the multiple cycles of precursor accumulation, single ETD activation and m/z analysis events are performed. We demonstrate this procedure decreases the overall time required to obtain high quality ETD spectra of large peptides or proteins. We directly compare analytical figures of merit for ETD performed in the MDC against ETD performed in the A-QLT pertaining to product ion generation, signal-to-noise, ion statistics, MS/MS duty cycle, and top-down LC-MS/MS data quality and quantity. Our results illustrate substantial improvements in protein sequence coverage and aggregate protein identifications facilitated by interrogating large precursor populations using multi-dissociation cell ETD.
Experimental Procedures
Mass Spectrometry Instrumentation
All experiments were performed on an ETD-enabled, hybrid, dual cell-quadrupole ion trap-Orbitrap mass spectrometer (Velos-Orbitrap, Thermo Fisher Scientific, San Jose, CA). The primary modification was the installation of a modified multi-dissociation cell, MDC, in place of the pre-existing collision (HCD) cell (Figure 1). The MDC retained the basic layout of the HCD cell it replaced, with its quadrupole ion guide having the same length and r0 (2.75 mm). The rod electrodes of the MDC quadrupole were divided into four sections with lengths, starting from end of the closest to the C-Trap, of 19 mm, 48.45 mm, 47.65 mm and 16.85 mm respectively. New quadrupole electronics were installed which could deliver sufficiently high RF voltages at 2,365 kHz to provide for a low m/z cutoff of ~90 Th (Mathieu q= 0.4 for m/z 202) during ETD reactions. Each section of the MDC quadrupole was driven by a separate filer of the of the new RF system’s tuned circuit transformer coil permitting the sections to have independently controlled DC biases whilst having common applied RF voltages. An additional RF electronics system was used to provide RF up to ~ 500 V0-P at ~1222 kHz to the MDC end lens plate electrodes to provide the axial confinement for charge sign independent trapping (CSIT) of ions during ETD reactions. To support the control of the MDC device’s additional voltages and to enable newly possible scan functions, we extensively modified the instrument control code.
Figure 1.

Multi-dissociation cell installed onto a hybrid QLT-Orbitrap mass spectrometer. In place of the pre-existing HCD cell (A), we installed a custom ion-ion reaction vessel (multi-dissociation cell, B–D). The multi-dissociation cell (MDC) comprises four separate sections, allowing for axial manipulation of ion populations. The MDC occupies the same space within the mass spectrometer as a normal collision cell (E).
As there was no provision on the instrument for a direct pressure measurement of the MDC cell pressure, MDC cell pressure was set and monitored according its contribution to the Orbitrap chamber pressure as measured by a Penning ionization gauge (delta pressure). Typical MDC Nitrogen pressure/flow settings for ETD experiments corresponded to a delta pressure of ~0.3×10−10 Torr for kinetics studies and ~0.1×10−10 Torr for protein analyses.
For MS/MS events using FT analysis, the A-QLT performed most of its usual functions, namely accumulation, trapping and isolation of precursor cations. Subsequent to isolation, cations were transmitted to the MDC. This sequence could be repeated several times, such that multiple populations of selected precursor cations would be delivered to the MDC. This multiple fill scheme increased the overall number of precursor ions subjected to ETD and thus product ions analyzed in each Orbitrap FT m/z analysis event. Following accumulation, cation precursors were sequestered in the front sections of the MDC, while the DC voltages of the back sections were set to positive values suitable for accumulation of ETD reagent anions. Anions generated in the reagent ion source were transmitted to and accumulated in the rear sections of the MDC. ETD was initiated by applying axial confining RF voltages to the end lenses of the MDC, and by setting all the DC bias voltage of the MDC to 0 V. The ETD reaction was quenched by setting the center two sections to negative DC offsets. Using the identical procedure for analysis of CID product ions produced in the collision cell, the ETD product ions were then transferred to the C-Trap and injected to the Orbitrap for FT m/z analysis.
In all experiments the radical anion of Fluoranthene (m/z 202) was the primary ETD reagent ion. There was no provision for m/z selection of the reagent ions for ETD in the MDC so the Fluoranthene radical anion comprised only ~95 % of the reagent ions delivered to the MDC for ion-ion reactions. The other 5% of the reagent ions were the Fluoranthene Nitrogen ion molecule reaction product anion, m/z 216, and various lower level background anions. The lack of purity of the reagent ion population would led to somewhat more prominent intact charge reduced product m/z peaks produced by proton transfer reactions with the reagent impurity anions. Supplementary Figure 1 provides evidence of this occurring as reaction of the +15 Histone H3.3 precursor in the MDC (10 Multiple Fills, 60 ms reaction time) produces a higher abundance of +13 and +14 charge-reduced precursors relative to the unreacted precursor than the same precursor is subjected to A-QLT ETD (10 microscans, 6 ms reaction time).
Sample preparation
Recombinant, isotopically labeled (13C2-leucine) human histone H3.3 was acquired through collaboration with New England BioLabs Inc. (Ipswich, MA). Histones were desalted using 50 mg C8 Sep-Pak cartridge (Waters Corp., Milford, MA) and reconstituted in 50% ACN with 0.2% formic acid to a concentration of 5 pmol/μL for direct infusion experiments. Wild-type Saccharomyces cerevisiae was cultured, lysed, and protein was extracted as previously described.(28) Protein estimation on the supernatant was performed by BCA and diluted to 5 ug/uL with lysis buffer and stored at −80°C. Protein lysate was reduced with dithiothreitol (DTT) at 37 °C for 30 min and alkyla ted with iodoacetamide for 45 min in the dark. Protein aliquots (30 μL diluted to 300 μL with lysis buffer) were passed through a 30 kDa nominal molecular weight centrifugal cut-off filter (Microcon YM-30, Millipore Corp., Billerica, MA) to deplete high-molecular weight proteins. The eluate (~250 μL) was buffer-exchanged against 3 volumes of 50 mM phosphate buffer (pH 7.2) in 5% acetonitrile using a 5 kDa nominal molecular weight centrifugal cut-off filter (Spin-X® UF, Corning Life Science, Corning, NY). The retentate (~ 50 μL), enriched in proteins between approximately 5–30 kDa, was diluted to an estimated 1 μg/μL in exchange buffer.
MS/MS parameters for LC analyses
Orbitrap MS1 analyses were performed with the 60k resolution setting and 3 microscans (transient averaging) with an AGC target value of 1×106 charges. Orbitrap MS/MS analyses with ETD conducted in the ion trap were performed at the 60k resolution setting and 6 microscans (transient averaging) with an AGC target value of 5×105 charges. The ETD reagent AGC target value was 5×105 charges. Orbitrap MS/MS analyses with ETD conducted in the MDC were performed at the 60k resolution setting and 1 microscans (no transient averaging) and 6 fills were used with each fill representing ~ 5×105 charges (effective AGC target value of 3×106 charges). The ETD reagent AGC target value was 5×105 charges and the injection time calculated to reach this value was used for both A-QLT and MDC ETD; however, the actual reagent population in the MDC was estimated to be ~ 1.0×106 – 1.5×106 charges. The increase was related to improved transmission efficiency to the MDC relative to the A-QLT due to considerably fewer ion optical elements between the reagent ion source and the MDC than between the reagent ion source and the A-QLT.
Charge-state dependent reaction time scaling was custom coded into the instrument firmware with reaction times determined from prior LC scouting runs and optimized reaction times from protein infusion experiments. For example, for A-QLT ETD, a +5, +10, and +15 precursor populations were reacted for 17.5 ms, 5.5 ms, and 4.0 ms, respectively. The same charge states were reacted for 50 ms, 28 ms, and 16 ms, respectively, using MDC ETD. Unassigned charge states were allowed to be sampled for MS/MS, but +1, +2, and +3 charge states were excluded. Unassigned precursor charge states were assumed to be highly charged and were reacted for 3.5 ms (QLT ETD) or 6 ms (MDC ETD). MDC ETD reaction times are ~ 3 – 4X longer than A-QLT ETD reaction times because a larger number of ions (6–10X) are reacted during MDC ETD. However, this additional reaction time is negligible when comparing multiple fills to microscans as each additional microscan consists of an ETD reaction followed by the collection of a transient (768 ms at 60k resolving power).
The 2 most intense precursors were selected by data-dependent analysis with dynamic exclusion enabled (exclusion time: 30 s, repeat count: 1, exclusion width: +/− 1.5 Th). The isolation width for both ETD experiments was 5 Th to capture the entire precursor isotopic cluster and to limit isolation space charge effect. The first mass was fixed at m/z 225. The maximum MS and MS/MS ion injection times were set to 300 ms and 750 ms, respectively. The MS/MS max injection time was relatively long, as compared to peptide analysis, but only ~ 8% of scans reached this value. Raw data files are available for download at http://www.chem.wisc.edu/~coon/Downloads/MDC%20Data/.
Liquid chromatography
Micro-capillary columns with an integrated ESI emitter were manufactured as previously described. (29) Analytical columns were prepared from fused silica tubing (360 μm o.d. x 75 μm i.d.) slurry-packed to 15 cm in length with Magic C18AQ, 5 μm, 300 Å particles (Michrom Bioresources Inc., Auburn, CA). Precolumns, with cast chemical frits, were made from 360 μm o.d. x 75 μm i.d. fused silica and slurry-packed with 5 cm of the same material used for analytical columns.(30) Gradient elution was achieved using a nanoACQUQLTY Ultra Performance LC® system (Waters Corporation, Milford, MA) with analytical flow rates of 300 nL min−1. Mobile phase A consisted of 0.2% formic acid and mobile phase B contained 99.8% ACN/0.2% formic acid. Sample concentration/desalting onto the precolumn was carried out at 1.5 μL/min for 10 min with 5% mobile phase B. The gradient began at 10% B and increased to 25% B at 30 min. The gradient then linearly increased from 25%B to 50%B at 80 min.
Automated database searching
Automated database searching was performed using ProSightPC 2.0 SP1 (Thermo Fisher).(31, 32) Spectra were converted to neutral monoisotopic masses using the Xtract algorithm only considering product ions with S/N greater than 2.5 with a maximum charge of 20. Top-down spectra were searched against the “top_down_simple” Saccharomyces cerevisiae database downloaded from the ProSightPC database website (ftp://prosightpc.northwestern.edu/). The database contained 15,303 basic protein sequences with 29,050 protein forms. Searches were performed by first using the Absolute Mass search mode with a 3.0 Da precursor and 15 ppm fragment ion tolerances using monoisotopic masses. The minimum number of matching fragments to be considered for identification was set to 10. Spectra that failed to produce a protein-spectrum match or matches with E-value scores less than 10−4 were searched in Biomarker mode with 10 ppm precursor and fragment ion tolerances with a minimum of 7 fragment ions for a protein-spectrum match. Include Modified Forms was enabled along with the Δm option in Biomarker mode.
Results and Discussion
Characterization of ETD in the Multi-dissociation Cell
The differing physical and operational parameters of the A-QLT and MDC which have the potential to cause difference in ETD functionality would be the use of different damping/collision gases and pressures (A-QLT: Helium at ~3×10−3 Torr, MDC: Nitrogen at > 1 ×10−3 Torr) (33), the axial dimensions of the two devices (A-QLT: ~62 mm, MDC: 133 mm), and quadrupole trapping field frequencies (A-QLT:1144 kHz, MDC: 2365 kHz).
The choice of nitrogen as the collision/damping gas rather than helium was driven by the desire to preserve the functionality of the MDC for collision cell type CID (HCD). Further the function of the C-Trap relies on being pressurized by nitrogen flowing from the MDC cell (or HCD cell). The damping/collision gas type and pressure potentially could influence the equilibrium reagent cloud radius and density, and therefore the reaction kinetics. Gas type and pressure could also influence the rate of collisional cooling of the intact charge reduced precursor ions after electron transfer and therefore influence the partitioning in the generation of ETD and ETnoD products. We made no effort characterize the extent of such effects for various MDC nitrogen pressures. The fixed helium pressure in the high pressure cell (HPC) of the A-QLT (~3×10−3 Torr) was determined by the instrument’s designers though co-optimization of ion injection and m/z isolation efficiency rather than any attribute specific to ETD. Similarly the setting of the MDC nitrogen pressures for our work was determined by co-optimization of transfer efficiency from the A-QLT and the efficiency of transfer to the C-Trap and Orbitrap.
The greater length of the MDC cell (~ 133 mm) relative to the length of the HPC of dual cell A-QLT (~62 mm) has both advantageous and disadvantageous aspects. For comparable trapping field conditions (ion Mathieu q-value/LMCO, field frequency, and damping gas type and pressure, etc.) increased axial dimension means that proportionally more reagent ions would be required to achieve a given reagent ion density, so injection times would be correspondingly be longer. However, longer axial dimension increases the path length for collisional stabilization of injected ions and can lead to higher injection efficiency. In this specific instance, the ion optics path between the MDC cell and the reagent source contained far fewer elements than the path between the reagent source and the A-QLT (Figure 1E). The efficiency of transfer of reagent ions was expected to be 2–4 fold higher to the MDC than to the HPC of the A-QLT. In our work the injection times/Reagent Target settings (as determined by and set for the A-QLT) required to reach maximal ETD reaction rates were comparable or slightly lower for the MDC than for A-QLT (see below).
The different number and length of the segments between the two devices has an influence on the total number of precursor ions available for ETD. For ETD in the A-QLT the precursor ions are sequestered (trapped) in the front section of the three section HPC quadropole while the reagent ions were injected into the center section of the HPC. The front section of the 3 section HPC quadrupole was ~ 12 mm long with an r0 of ~4.75 mm. The relatively short length the front section (~12 mm) in combination with the device’s r0 (~4.75 mm) created a very restricted precursor trapping region (perhaps ~ 3 – 5 mm length) which limited the storage (sequestration) space charge capacity to 3 ×105 – 1 ×106 charges. During ETD reagent ion injection and accumulation in the rear section of the MDC, the precursor ions were sequestered in the front two sections of the MDC (combined length ~68 mm). The storage space charge capacity of these combined sections of the MDC quadrupole probably exceeded 6 ×106 charges.
The different quadrupole trapping field frequencies would account for the large differences in the ETD functionality between the A-QLT and the MDC. When operated with identical low m/z cutoffs, the MDC, with its higher quadrupole field frequency, should provide stronger radial confinement of ions and, thus, enable higher reagent ion cloud density for the same number of trapped reagent ions per unit length. For ETD reaction, in either device, the ETD reagent ion population was always in sufficient excess of precursor ion population ions such that kinetics of the ETD reaction were pseudo first order. Under these conditions population of precursor Np(t) ions would be expected to decay with reaction time, t, as
| (Equation 1) |
where [R] is the average number density of the portion of the reagent ion cloud that overlaps with the precursor ion cloud, and k is ETD ion-ion reaction rate constant. Based on the treatment of ion-ion kinetics of McLuckey and Stephenson (34, 35), we expect that the rate constant, k, could be expressed as
| (Equation 2) |
where Zp, mp, Zr and mr are the charge states and masses of the precursor and reagent ion species respectively and the quantity c(|v|) is a function of the distribution the magnitude of differential velocities, |v|, of precursor and reagent ions in the overlapping ion clouds. In both the A-QLT and MDC trapped ions are subject to large numbers of collisions with a collision/damping gas and are expected to assume near thermal velocity distributions (36), so we assumed that c(|v|) was about the same for reactions in both devices. This is perhaps a dubious assumption as the factor c(|v|) varies as |v|−3 for overlapping ion clouds where |v| is uniform (crossed mono-energic beams). Thermal velocities vary with mass (m−½) so the rate constant, k, depends on mp and mr both explicitly and implicitly though c(|v|). Since the mass of the reagent anion (202 Da for the Fluoranthene anion) was usually be considerable smaller than the precursor peptide or protein cation masses (such as the 3+ Angiotenisn I cations (m/zp ≈ 433 Th, mp ≈1299 Da), k is relatively weakly dependent upon the precursor ion mass.
As all known ETD reagent anions are singly charged, ETD reaction rates will be primarily influenced by the precursor charge state (quadratic dependence), reagent ion mass and the average number density of the portion of the reagent ion cloud that overlaps with the precursor ion cloud. For relatively low numbers of stored ions, the ion cloud diameter is determined by the kinetic energy of the trapped ions, the m/z of the ions and the trapping field intensity (for RF-only 2D quadrupole fields, this is compactly defined by ion’s Mathieu q value) and the quadrupole trapping field frequency, f. For low numbers of trapped ions, implying the absence of significant effects due to mutual repulsion of ions (i.e., space charge effects), the average ion number density in the cloud scales proportionally with the number of trapped ions. Using the adiabatic theory of ion motion (pseudo-potential approximation) the maximum radius of ion motion, rmax, for a given ion kinetic energy, KEavg, (averaged over an RF cycle) can be expressed as
| Equation 3 |
where qion and mion are the Mathieu q and mass of the ion respectively. This expression indicates that for a “low density” reagent ion cloud of fixed ion number with a near thermal distribution of kinetic energies and maintained at a fixed q in the field (i.e., operating with fixed LMCO), the cloud radius will vary as 1/f so the average number density of the ions in the cloud will vary as f2. As the number of trapped ions are further increased, ion-ion repulsion will cause the ion cloud to expand and the average number density in the ion cloud will grow less than linearly with trapped ion number. Eventually, further increases in trapped ion numbers will be accommodated completely by growth in the ion cloud radius without significant increase in ion number density in bulk of the ion cloud.(36) The estimated saturation number density, [Nion]max, can be given as
| Equation 4 |
where ε0 is the permittivity of free space, e is the elemental charge, and Zion is the ion charge state. Therefore, ETD reaction rates, k[R], in both the A-QLT and MDC were expected to increase linearly with reagent ion number (as controlled by the Reagent Target setting) for relatively low reagent ion numbers and then saturate at higher reagent ion numbers. We systematically measured ETD reaction rates of the 3+ cation of Angiotensin I in both devices over wide range of Reagent Target settings. The low mass cutoff for all experiments was maintained at ~m/z 89 so the reagent ion Mathieu q was always ~ 0.4 during the reactions. The reaction rates were determined by a semi-logarithmic least squares fit of the recorded abundance of the surviving precursor m/z peak as a function of reaction time. Both the A-QLT data and MDC kinetics data were in good qualitative match to the predictions based on adiabatic theory – linear increase with Reagent Target eventually saturating at a maximal reaction rate. The saturation reaction rate for the A-QLT was ~50/sec. For the MDC the maximal reaction rate was ~ 130 /sec (Supplementary Figure 2). According to adiabatic theory, the maximal ion densities should vary quadratically with trapping field frequency. We saw a ~2.6 fold increase in reaction rates, rather than the predicted 4.3 fold increase expected based on the MDC and A-QLT frequencies. A Mathieu q-value of 0.4 is at the far upper limit for applicability of the adiabatic model, so high populations of reagent ions may been subject to a fair amount of RF heating in the MDC resulting in increased reagent velocities and a corresponding reduction in the magnitude of the c(|v|) term. The extent of this effect would likely be amplified by the use of the higher molecular weight collision/damping gas (nitrogen rather than helium). This could account for the lower than predicted increase in saturation reaction rate.
When analyzing whole protein MDC ETD data it was evident that a larger amount of charge reduced precursor exists as compared to A-QLT ETD. We suspect this is from the presence of the nitrogen adduct of Fluoranthene (m/z 216) which preferentially performs PTR. This adduct is usually removed during the reagent anion selection step when performing A-QLT ETD, but because the MDC is situated after the C-trap there is no means for anion selection.
Characterization of Multiple Precursor Fills Scan Function
The isolation space charge limit in the A-QLT is ~ 5 × 105 charges when isolating with a 3 Th window.(37) While both the activation and storage space charge capacities are many-fold larger (38), the total charge of the precursor ion population is restricted to this value when ions are isolated and reacted in the in the same device. For Orbitrap FT analyses, 5×105 precursor charges are more than sufficient to produce peptide MS/MS spectra with high signal-to-noise; however, it is rarely adequate to produce quality MS/MS spectra of intact proteins without substantial signal (transient) averaging. A principle reason for this is the much larger number of potential backbone cleavages and the larger number of charge states for each the possible product ion structure. Further, product ions are of higher average molecular weight which means that the signal for each product ion structure may be distributed across several isotopic m/z peaks as well as between different charge states. For ETD, which is relatively non-selective in backbone cleavage points, this means that that the number of different dissociation product ions (m/z peaks) grows faster than linearly with precursor molecular weight. The number of expected product ion m/z peaks of from a 15000 Da precursor ion, is well more than 10 fold greater than what expected to be produced from a 1500 Da precursor ion. The MDC has an estimated storage space charge limit of 2 × 107 charges, accommodating a precursor population many-fold greater than what can be isolated and then reacted in the A-QLT. To build up a large precursor ion population in the MDC for MS/MS, multiple loads of protein precursors are iteratively isolated in the A-QLT and transmitted to the MDC; a process we refer to as multiple fills (Supplementary Figure 3). The ions are then activated in one ETD reaction with subsequent analysis in the Orbitrap.
To investigate the utility of multiple fills, recombinant human histone H3.3 was infused and the +15 charge state (m/z 1015.2) was subjected to ETD conducted in the A-QLT (A-QLT ETD) or MDC (MDC ETD) (Figure 2). As a starting point, H3.3 was analyzed using A-QLT ETD with a 5×105 MS/MS automatic gain control (AGC) target value with product ion analysis in the Orbitrap (60k resolving power, 6 ms activation) using 10 microscans (transient averaging). We then performed ETD in the MDC using 10 precursor fills (1 fill ≈ 5×105 charges) with product ion analysis in the Orbitrap using 1 microscan, or 10 fills with 4 Orbitrap microscans, corresponding to an equivalent per specturm number of precursor charges (5×106) or similar acquisition time (~9 s), respectively. The activation duration for MDC ETD (60 ms) was scaled to generate approximately the same relative amount of intact charge-reduced precursor to unreacted precursor as was observed in the reference A-QLT ETD spectra (Figure 2B). Note that MDC ETD activation could be reduced to 20–30 ms without appreciable decreases in product ion S/N (as computed by the data system). However, we determined that the ratio of charge-reduced to unreacted precursor was a fair strategy to normalize for reaction completeness. The scan cycle depicting precursor injection, activation, and product ion analysis for each ETD MS/MS experiment is given in Figure 2A. The major contributor to the MS/MS cycle is the time required to record the ion image current signal transient for product ion m/z analysis in the Orbitrap (60k res. = 768 ms).(39) High MS and MS/MS resolving power is required to facilitate precursor mass determination and product ion assignment from the abundant and highly charged species generated from ETD. To increase product ion S/N for A-QLT ETD, multiple transients could be averaged, but at the expense of a corresponding increase in time required to obtain the product ion spectrum. By comparison, only a small time penalty is incurred by using multiple fills in increase product ion S/N for MDC ETD as the time required to accumulate, transmit, and store a load of precursor ions is relatively short (~ 50 ms for a fill of 5×105 charges) for these infusion experiments.
Figure 2.

Comparison of A-QLT and MDC ETD for Histone H3.3. (A) The major scan events contributing to the MS/MS duty cycle for conducting ETD in the MDC using multiple fills and ion trap ETD for histone infusion. The major scan events are divided into precursor injection (5×105 AGC target), precursor activation by ETD, and Orbitrap product ion analysis (60k resolution, 768 ms transient). (B) MS/MS spectra of histone H3.3 (+15, m/z 1015.2, 60k resolving power) analyzed using MDC ETD with 10 fills and 4 microscans (top), MDC ETD with 10 fills and 1 microscan (middle), and A-QLT ETD using 10 microscans. Reaction completeness for each ETD reaction was determined from the relative ratios of charge-reduced precursors to the unreacted precursor. A comparison of the top and bottom spectra represent similar acquisition times (7.9 s and 9.2 s), while a comparison of the middle and bottom spectra represent the same total number of precursors analyzed during the course of acquisition (~ 5×106 charges). (C) Annotated portion of the histone H3.3 spectra. A product ion was considered a match if the S/N was > 3 and with a mass error less than 10 ppm. (D) Distribution of product ion S/N from (A).
Product ions from the 600–900 m/z range in the histone spectra were examined in detail to directly compare ETD MS/MS approaches detailed above (Figure 2B). A-QLT ETD with 10 microscans produced 47 product ions (S/N ≥ 3) in this region with an average S/N of 12. MDC ETD with 10 fills and 1 microscan generated 49 product ions (S/N ≥ 3) with an average S/N of 14, while MDC ETD with 10 fills and 4 microscans produced 53 product ions (S/N ≥ 3) with an average S/N of 27. For MDC ETD data quality was improved in terms of both product ion generation and S/N relative to A-QLT ETD. Superior S/N was achieved for MDC ETD with 10 fills and 4 microscans where more than half of the product ions had S/N > 16 (Figure 2C). MDC ETD with 10 fills and 1 microscan also produced a higher quality MS/MS spectrum than A-QLT ETD with 10 microscans in ~25% of the scan cycle time.
Comparison of Multiple Precursor Fills and Transient Averaging
To characterize both the effect of the number of fills for MDC ETD and the effect of microscans for A-QLT ETD on product ion S/N, 10 representative product ions from the histone spectra were measured as the number of fills (with 1 microscan) or microscans were increased from 2 up to 10 (Figure 3A). The average S/N of A-QLT ETD product ions increased from 8.4 to 20.2. This observation is in agreement with what is predicted from signal averaging (S/N = 18.8) where S/N increases with the square root of the number of average measurements. The average S/N of MDC ETD product ions increased from 18.3 to 41.6. If the only source of noise was electrical in nature (thermal noise) then S/N should proportionally increase with each additional fill.(40–42) The expected average S/N should be 91.5 after 10 fills. However, chemical noise from electrospray ionization is a major contributor to noise as measured by the system and increases with each fill decreasing the expected S/N by more than half.(43) Yet, the increase in product ion S/N per each additional fill for MDC ETD was more than double than that from each additional microscan for A-QLT ETD. After 10 microscans and more than 9 s of acquisition, the average product ion S/N was 20.2. Comparable S/N (18.3) was realized in less than 1.4 s using MDC ETD with 2 fills. An additional fill boosted the S/N to 24.7 in less than 1.5 s of total acquisition.
Figure 3.
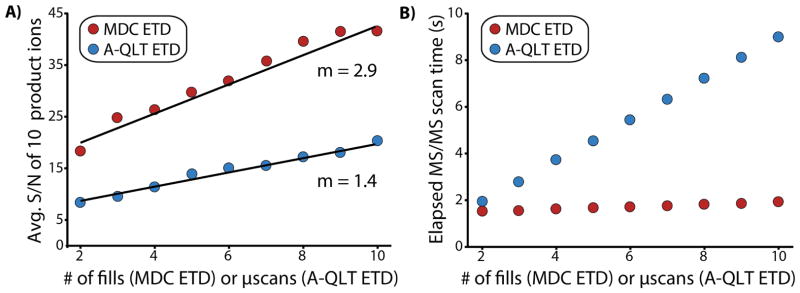
Comparison of transient averaging and multiple fills for selected product ions of Histone H3.3. The average S/N of 10 representative product ions from Fig. 4 as a function of the number of microscans (A-QLT ETD) or fill (MDC ETD). B) The average elapsed MS/MS scan time to perform A-QLT ETD using microscans or MDC ETD using multiple fills. These values are from histone infusion experiments where the average time required to accumulate 5×105 precursor charges for MS/MS was ~ 4.5 ms. The Orbitrap resolving power was set to 60k (768 ms transient).
There is limit to the number of fills that can be utilized for MDC ETD. The magnitude of the increase in product ion S/N diminished after 9 fills in this experiment. In general, the S/N increase generally levels off at ~ 10 fills for all MDC ETD experiments regardless of precursor attributes (charge state, m/z, length). We believe that this effect is due to the extraction space charge limit of the C-Trap because the we expect storage space charge capacities of both the MDC and the C-Trap to exceed the ~1×107 charges that would correspond to 10 fills.
Collecting microscans significantly lengthens the MS/MS duty cycle due to the time required to make multiple measurements of the time-domain signal transient. By comparison, the amount time required to accumulate additional fills is relatively small (Figure 3B). For the infusion experiments, each consecutive microscan added an additional 880 ms to the time required to produce a MS/MS spectrum (effective scan time), whereas each consecutive fill added 50 ms. The highest quality data, however, was achieved through the use of both multiple fills and transient averaging. Minimization of effective scan time is not a primary concern for protein infusion experiments. Effective scan times of more than 8 s are unacceptably long for LC analyses of complex mixtures. This is where the MDC ETD/multiple fills strategy has the greatest benefit.
Comparison of A-QLT and MDC ETD for Top Down LC-MS/MS
Proteins extracted from yeast lysate were filtered to remove proteins greater than 30 kDa, the current upper mass limit for routine analysis on this instrument type. To provide a direct comparison of the analysis of similar numbers of precursor charges the sample was analyzed in duplicate over an 80-minute gradient using A-QLT ETD with 6 microscans or MDC ETD with 6 fills and 1 microscan. Performing the ETD reaction in the MDC resulted in a 132% increase in the number of protein-spectrum matches (PrSMs) identified, a 36% increase in the number proteoforms identified, and a 36% increase in the number of unique proteins (accession numbers) that could be confidently identified using ProSightPC (Figure 4A). A distribution of all intact protein molecular weights can be found for both A-QLT and MDC ETD in Supplementary Figure 4. Different charge states of the same proteoforms were identified multiple times leading to the large difference in the number of PrSMs and proteoforms that were identified. The average elapsed MS/MS scan times were 2.5 s and 7.6 s for MDC ETD and A-QLT ETD, respectively. Clearly, performing MDC ETD for intact protein analysis is advantageous for LC analyses due to for its shorter effective scan time.
Figure 4.

Results of LC-MS/MS analyses of yeast lysate enriched in proteins < 30 kDa interrogated with either MDC ETD or A-QLT ETD (reported values are the average of duplicates) (A). MDC ETD facilitated the identification of more protein-spectrum matches (PrSMs), proteoforms, and unique protein accession numbers compared to A-QLT ETD. The 60S ribosomal protein L30 was identified with both MDC ETD (B) and A-QLT ETD (C). MDC ETD produced 71% more fragment ions leading to a better E-value and better sequence coverage compared to A-QLT ETD using ProSightPC 2.0 SP1 for raw file processing and database searching.
In addition to shorter effective scan time, MDC ETD systematically produced spectra of higher signal-to-noise ratio. Spectral database correlation using ProSightPC relies upon the conversion of product ions to neutral monoisotopic masses through charge-state determination/deconvolution and deisotoping.(31, 44, 45) Critical to the correct assignment of monoisotopic masses is the ability to generate product ions with isotope distributions that match theoretical isotope distributions.(46–51) Determination of monoisotopic masses is confounded by low S/N and poor ion statistics. Low-level ions buried in the noise may never rise to a detectable level despite extensive transient averaging; however, these low-level ions may be boosted to a detectable level if the initial precursor ion population is sufficiently large as demonstrated in the histone infusion experiments. A portion of the ribosome spectrum generated by MDC ETD and A-QLT ETD is shown in Figure 5A. The isotope distribution from two product ions, c243+ and c465+, were compared to the theoretical isotope distribution of each ion based upon the elemental composition of each (Figure 5B). In both cases, product ions from MDC ETD have a greater number of isotopic peaks appearing above the instrument’s applied S/N threshold and their relative heights more closely matched those of the theoretical distributions. The analysis of a larger product ion population, rather than performing transient averaging, led to better ion statistics and increased the intrascan dynamic range. Improved ion statistics and intrascan dynamic range also improve the automated detection of isotopic clusters and aid in the ability to perform peak subtraction of overlapping product ion isotopic clusters to make use of all of analytical information available in the MS/MS spectrum.(49)
Figure 5.

Comparison of yeast 60S ribosomal protein fragmentation. A) Annotated portion of the yeast 60S ribosomal protein L30 MS/MS spectrum generated by MDC ETD (6fills, 1 microscan, top) and A-QLT ETD (6 microscans, bottom). B) The analysis of the isotope distributions of the two product ions demonstrate that the improved ion statistics engendered by A-QLT ETD lead to a larger intrascan dynamic range with isotope distributions more closely matching theoretical distributions modeled in Xcalibur.
For example the 60S ribosomal protein L30 (11.3 kDa, accession # A6ZUW4) was identified with both MDC ETD and A-QLT ETD at 1.41 and 1.44 ppm mass error, respectively. Of the 206 possible protein backbone cleavages resulting in the production of c- and z-type product ions, 65 product ions were identified from the MDC ETD MS/MS spectrum compared to 38 for A-QLT ETD (Figure 4B–C). The expectation-value (E-value) score for the MDC ETD spectrum is substantially improved leading to a more confident match.
Conclusions
By performing the ETD reactions in a multi-dissociation cell (MDC) – combination ETD and HCD cell - rather than in the analyzing quadruple linear ion trap (A-QLT) we have enabled ETD of 6–10 fold larger initial populations of precursor ions. Further the ~2 fold higher operating frequency of the MDC enables 2.4 fold higher reaction rates for reactions performed at comparable low mass cutoffs (LMCO). We have demonstrated an advanced scan function utilizing multiple cycles of precursor injection and isolation in the A-QLT followed by transfer to and accumulation in the MDC, There this aggregated ion population is subjected to ETD and then extracted to the Orbitrap for high resolution m/z analysis. Using this multi-fill approach for LC-ETD-MS/MS analysis of intact proteins yields more protein identifications with higher confidence due to the increased number and higher quality of MS/MS spectra. Further studies will continue a comparison of microscans (A-QLT ETD) and multiple fills (MDC ETD) on newer generation Orbitrap instruments that provide higher resolution for equivalent transient collection times.(39, 52)
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support from Thermo Fisher Scientific, NSF CAREER grant 0747990, and NIH grant R01 GM080148. C.M.R was funded by an NSF Graduate Research Fellowship and NIH Traineeship (T32GM008505).
References
- 1.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. (Translated from English) Proc Natl Acad Sci U S A. 2004;101(26):9528–9533. doi: 10.1073/pnas.0402700101. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF. Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. (Translated from English) International Journal of Mass Spectrometry. 2004;236(1–3):33–42. (in English) [Google Scholar]
- 3.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. (Translated from English) Journal of the American Chemical Society. 1998;120(13):3265–3266. (in English) [Google Scholar]
- 4.Kelleher NL. Top-down proteomics. (Translated from English) Anal Chem. 2004;76(11):196A–203A. (in English) [PubMed] [Google Scholar]
- 5.Chi A, Bai DL, Geer LY, Shabanowitz J, Hunt DF. Analysis of intact proteins on a chromatographic time scale by electron transfer dissociation tandem mass spectrometry. (Translated from English) Int J Mass Spectrom. 2007;259(1–3):197–203. doi: 10.1016/j.ijms.2006.09.030. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsybin YO, et al. Structural Analysis of Intact Monoclonal Antibodies by Electron Transfer Dissociation Mass Spectrometry. (Translated from English) Analytical Chemistry. 2011;83(23):8919–8927. doi: 10.1021/ac201293m. (in English) [DOI] [PubMed] [Google Scholar]
- 7.Fornelli L, et al. Analysis of Intact Monoclonal Antibody IgG1 by Electron Transfer Dissociation Orbitrap FTMS. (Translated from English) Molecular & cellular proteomics: MCP. 2012;11(12):1758–1767. doi: 10.1074/mcp.M112.019620. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Hager JW, Xia Y, Londry FA, McLuckey SA. Positive Ion Transmission Mode Ion/Ion Reactions in a Hybrid Linear Ion Trap. Analytical Chemistry. 2004;76(17):5006–5015. doi: 10.1021/ac049359m. [DOI] [PubMed] [Google Scholar]
- 9.Liang X, Hager JW, McLuckey SA. Transmission Mode Ion/Ion Electron-Transfer Dissociation in a Linear Ion Trap. Analytical Chemistry. 2007;79(9):3363–3370. doi: 10.1021/ac062295q. [DOI] [PubMed] [Google Scholar]
- 10.Rand K, Pringle S, Morris M, Engen J, Brown J. ETD in a Traveling Wave Ion Guide at Tuned Z-Spray Ion Source Conditions Allows for Site-Specific Hydrogen/Deuterium Exchange Measurements. (Translated from English) Journal of the American Society for Mass Spectrometry. 2011;22(10):1784–1793. doi: 10.1007/s13361-011-0196-7. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia Y, et al. Implementation of Ion/Ion Reactions in a Quadrupole/Time-of-Flight Tandem Mass Spectrometer. Analytical Chemistry. 2006;78(12):4146–4154. doi: 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia Y, Thomson BA, McLuckey SA. Bidirectional Ion Transfer between Quadrupole Arrays: MSn Ion/Ion Reaction Experiments on a Quadrupole/Time-of-Flight Tandem Mass Spectrometer. Analytical Chemistry. 2007;79(21):8199–8206. doi: 10.1021/ac071448m. [DOI] [PubMed] [Google Scholar]
- 13.Huang T-y, McLuckey SA. Top-down protein characterization facilitated by ion/ion reactions on a quadrupole/time of flight platform. PROTEOMICS. 2010;10(20):3577–3588. doi: 10.1002/pmic.201000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAlister GC, et al. A Proteomics Grade Electron Transfer Dissociation-Enabled Hybrid Linear Ion Trap-Orbitrap Mass Spectrometer. Journal of Proteome Research. 2008;7(8):3127–3136. doi: 10.1021/pr800264t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAlister GC, Phanstiel D, Good DM, Berggren WT, Coon JJ. Implementation of Electron-Transfer Dissociation on a Hybrid Linear Ion Trap-Orbitrap Mass Spectrometer. Analytical Chemistry. 2007;79(10):3525–3534. doi: 10.1021/ac070020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan DA, et al. Electron transfer dissociation in the hexapole collision cell of a hybrid quadrupole-hexapole Fourier transform ion cyclotron resonance mass spectrometer. Rapid Communications in Mass Spectrometry. 2008;22(3):271–278. doi: 10.1002/rcm.3356. [DOI] [PubMed] [Google Scholar]
- 17.Hartmer R, Lubeck M. New Approach for Characterization of Post Translational Modified Peptides Using Ion Trap MS with Combined ETD/CID Fragmentation. LC-GC Europe. 2005;18:11–13. [Google Scholar]
- 18.Sobott F, et al. Comparison of CID Versus ETD Based MS/MS Fragmentation for the Analysis of Protein Ubiquitination. (Translated from English) Journal of the American Society for Mass Spectrometry. 2009;20(9):1652–1659. doi: 10.1016/j.jasms.2009.04.023. (in English) [DOI] [PubMed] [Google Scholar]
- 19.Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA. Electron Transfer Ion/Ion Reactions in a Three-Dimensional Quadrupole Ion Trap: Reactions of Doubly and Triply Protonated Peptides with SO2•. Analytical Chemistry. 2005;77(6):1831–1839. doi: 10.1021/ac0483872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olsen JV, et al. A Dual Pressure Linear Ion Trap Orbitrap Instrument with Very High Sequencing Speed. Molecular & Cellular Proteomics. 2009;8(12):2759–2769. doi: 10.1074/mcp.M900375-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell JD, et al. Single scan, top-down intact protien analysis of a Velos Orbitrap modified with a dedicated high-capacity ion/ion reaction cell. Procceedings of the 59th ASMS Conference on Mass Spectrometry and Allied Topics; Denver, CO. 2011. [Google Scholar]
- 22.Yang C, et al. Profiling histone H3 isoforms in Human embryonic stem cells using a Velos Orbitrap modified with a segmented reaction cell. Procceedings of the 59th ASMS Conference on Mass Spectrometry and Allied Topics; Denver, CO. 2011. [Google Scholar]
- 23.Ledvina AR, et al. Activated-Ion Electron Transfer Dissociation Improves the Ability of Electron Transfer Dissociation to Identify Peptides in a Complex Mixture. (Translated from English) Anal Chem. 2010;82(24):10068–10074. doi: 10.1021/ac1020358. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ledvina AR, et al. Infrared Photoactivation Reduces Peptide Folding and Hydrogen-Atom Migration following ETD Tandem Mass Spectrometry. (Translated from English) Angew Chem- Int Edit. 2009;48(45):8526–8528. doi: 10.1002/anie.200903557. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaub TM, et al. High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 tesla. (Translated from English) Anal Chem. 2008;80(11):3985–3990. doi: 10.1021/ac800386h. (in English) [DOI] [PubMed] [Google Scholar]
- 26.Tsybin YO, Witt M, Baykut G, Hakansson P. Electron capture dissociation Fourier transform ion cyclotron resonance mass spectrometry in the electron energy range 0–50 eV. (Translated from English) Rapid Commun Mass Spectrom. 2004;18(14):1607–1613. doi: 10.1002/rcm.1525. (in English) [DOI] [PubMed] [Google Scholar]
- 27.Hakansson K, et al. Combined electron capture and infrared multiphoton dissociation for multistage MS/MS in a Fourier transform ion cyclotron resonance mass spectrometer. (Translated from English) Anal Chem. 2003;75(13):3256–3262. doi: 10.1021/ac030015q. (in English) [DOI] [PubMed] [Google Scholar]
- 28.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Meth. 2008;5(11):959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McAlister GC, et al. Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal Chem. 2012;84(6):2875–2882. doi: 10.1021/ac203430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ficarro SB, et al. Improved electrospray ionization efficiency compensates for diminished chromatographic resolution and enables proteomics analysis of tyrosine signaling in embryonic stem cells. Anal Chem. 2009;81(9):3440–3447. doi: 10.1021/ac802720e. [DOI] [PubMed] [Google Scholar]
- 31.Zamdborg L, et al. ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res. 2007;35(suppl_2):W701–706. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LeDuc RD, Kelleher NL. Curr Protoc Bioinform. Vol. 19. John Wiley & Sons, Inc; 2007. Using ProSight PTM and related tools for targeted protein identification and characterization with high mass accuracy tandem MS data; pp. 13.16.11–13.16.28. [DOI] [PubMed] [Google Scholar]
- 33.McAlister GC, Phanstiel DH, Brumbaugh J, Westphall MS, Coon JJ. Higher-energy Collision-activated Dissociation Without a Dedicated Collision Cell. Molecular & Cellular Proteomics. 2011;10(5) doi: 10.1074/mcp.O111.009456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Compton PD, Strukl JV, Bai DL, Shabanowitz J, Hunt DF. Optimization of Electron Transfer Dissociation via Informed Selection of Reagents and Operating Parameters. (Translated from English) Anal Chem. 2012;84(3):1781–1785. doi: 10.1021/ac202807h. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLuckey SA, Stephenson JL, Asano KG. Ion/Ion Proton-Transfer Kinetics: Implications for Analysis of Ions Derived from Electrospray of Protein Mixtures. Analytical Chemistry. 1998;70(6):1198–1202. doi: 10.1021/ac9710137. [DOI] [PubMed] [Google Scholar]
- 36.Tolmachev AV, Udseth HR, Smith RD. Modeling the ion density distribution in collisional cooling RF multipole ion guides. International Journal of Mass Spectrometry. 2003;222(1):155–174. [Google Scholar]
- 37.McAlister GC, Phanstiel DP, Coon JJ. A dual reaction cell, ETD-enabled orbitrap mass spectrometer for top-down proteomics. 56th Proceedings of the American Society for Mass Spectrometry and Allied Topics Conference.2008. [Google Scholar]
- 38.Schwartz J, Senko M, Syka J. A two-dimensional quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2002;13(6):659–669. doi: 10.1016/S1044-0305(02)00384-7. [DOI] [PubMed] [Google Scholar]
- 39.Michalski A, et al. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics. 2012;11(3) doi: 10.1074/mcp.O111.013698. pages numbers not available. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makarov A, et al. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal Chem. 2006;78(7):2113–2120. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- 41.Perry RH, Cooks RG, Noll RJ. Orbitrap mass spectrometry: Instrumentation, ion motion and applications. Mass Spectrom Rev. 2008;27(6):661–699. doi: 10.1002/mas.20186. [DOI] [PubMed] [Google Scholar]
- 42.Alexander M. Practical Aspects of Trapped Ion Mass Spectrometry. IV. CRC Press; 2010. Theory and practice of the Orbitrap mass analyzer; pp. 251–272. [Google Scholar]
- 43.Cech NB, Enke CG. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom Rev. 2001;20(6):362–387. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- 44.Taylor GK, et al. Web and database software for identification of intact proteins using “top down” mass spectrometry. Anal Chem. 2003;75(16):4081–4086. doi: 10.1021/ac0341721. [DOI] [PubMed] [Google Scholar]
- 45.LeDuc RD, et al. ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004;32(suppl_2):W340–345. doi: 10.1093/nar/gkh447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yergey JA. A general approach to calculating isotopic distributions for mass spectrometry. Int J Mass Spec Ion Physics. 1983;52(2–3):337–349. doi: 10.1002/jms.4498. [DOI] [PubMed] [Google Scholar]
- 47.Rockwood AL, Van Orden SL, Smith RD. Rapid calculation of isotope distributions. Anal Chem. 1995;67(15):2699–2704. doi: 10.1021/ac951158i. [DOI] [PubMed] [Google Scholar]
- 48.Senko MW, Beu SC, McLafferty FW. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J Am Soc Mass Spectrom. 1995;6(4):229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 49.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Am Soc Mass Spectrom. 2000;11(4):320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 50.Renard B, Kirchner M, Steen H, Steen J, Hamprecht F. NITPICK: peak identification for mass spectrometry data. BMC Bioinformatics. 2008;9(1):355. doi: 10.1186/1471-2105-9-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaitly N, et al. Decon2LS: An open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics. 2009;10:87. doi: 10.1186/1471-2105-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tran JC, et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480(7376):254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.