

Abstract
Background
Immune complex deposition in the subepithelial zone of glomerular capillaries can lead to membranous glomerulopathy.
Objective
To present the case of a 23-year-old man with X-linked agammaglobulinemia (XLA) who developed idiopathic membranous glomerulopathy while receiving intravenous immunoglobulin (IVIG).
Methods
We performed an immunological workup, genetic testing, and a renal biopsy.
Results
XLA was confirmed with less than 0.02% CD19+ cells in the blood after sequence analysis revealed a nonfunctional BTK gene. He presented with microhematuria, which persisted for 3 years and spanned treatment with 5 different preparations of intravenous gammaglobulin. Immunohistochemistry revealed membranous glomerulopathy.
Conclusion
Although endogenous serum immunoglobulin (Ig) production is severely impaired in XLA, rare B lymphocytes that have managed to mature can produce functional IgG antibodies. The pathogenic immune complexes could reflect IVIG reacting with polymorphic autoantigens, an endogenous IgG-producing clone reacting with a common idiotype present in the IVIG, or both.
Keywords: X-linked agammaglobulinemia (XLA), Bruton agammaglobulinemia, Membranous glomerulopathy, Microhematuria, Intravenous gammaglobulin
Introduction
Loss-of-function mutations in the Bruton tyrosine kinase (BTK) gene underlies the prototypic humoral immunodeficiency X-linked agammaglobulinemia (XLA) [1,2]. BTK plays a critical role in enabling maturation of pre B lymphocytes to immature B lymphocytes. In its absence, B-lymphocyte development, and thus immunoglobulin (Ig) production, is impaired [3,4]. We present an unusual case of XLA in a man with membranous glomerulopathy (MG), an immune complex disease [5] that persisted in spite of sequential treatment with 5 different gammaglobulin preparations.
Case Description
The patient, who was of European descent, suffered severe oropharyngeal Herpes simplex at age 11 months. By age 5 he had suffered repeated sinusitis, bronchitis, pneumonia, septic arthritis, and Haemophilus influenzae type b pyothorax. B lymphocyte counts and serum Ig levels were severely depressed, and replacement gammaglobulin therapy was initiated.
The patient first presented at the Clinical Immunology Service of the University of Alabama at Birmingham, Birmingham, Alabama, USA at age 23. While previously receiving Gammar-P IV (King of Prussia, PA, USA), he was recently switched to 0.34 gm/kg of Polygam SD (Baxter, Deerfield, IL, USA). He complained of recurrent sinusitis and chronic conjunctivitis. Serum Ig levels were as follows: IgM, 8 mg/dL (reference range, 50–225); IgG, 806 mg/dL (reference range, 775–1850); IgA <8 mg/dL (reference range, 75–450); and IgE <2 IU/mL (reference range, 3–423). Complement levels were as follows: C3, 88 mg/dL (reference range, 70–150); and C4, 18.2 mg/dL (reference range, 10–50). Antinuclear antibody titers, rheumatoid factor titers, and the erythrocyte sedimentation rate were normal. Flow cytometric analysis of blood confirmed a virtual absence of IgM+, CD19+, CD20+, and CD21+ cells (<0.02% of the lymphocyte fraction). Natural killer cell and T-lymphocyte counts were normal, with a CD4/CD8 ratio of 1.7.
Sequence analysis of BTK revealed a 10.8-kb tandem duplication of exons 6–18, which created a frameshift with a premature stop codon. Duplication appeared to result from unequal homologous recombination within a 49-bp interval of sequence identity between an Alu Sg site at the end of intron 5 (bp 57,977–58,025; Accession number U78027) and an Alu Sx site within intron 18 (bp 68,800–68,848).
Microscopic hematuria (MH) was identified during screening for participation in a phase III intravenous immunoglobulin (IVIG) study of Gamunex (Talecris, Research Triangle Park, NC, USA). Other than a remote history of acute hematuria after blunt trauma during childhood, the patient denied any prior history. Family and personal history were unremarkable for nephrolithiasis, cystitis, nephritis, hearing disorders, easy bruising, or hemarthrosis. He denied present dysuria, hesitancy, or urethral discharge. He was normotensive and afebrile. The conjunctiva of both eyes were inflamed. Serum creatinine was 1.1 mg/dL (reference range, 0.7–1.3 mg/dL). Urinalysis revealed a specific gravity of 1.019, pH 5.0, and trace blood with 3–10 red blood cells and 0–5 white blood cells per high power field.
The patient was referred to the nephrology department. Mild hypercalciuria was noted and MH was confirmed. Creatinine clearance was 98 cc/min and the glomerular filtration rate (GFR) calculated using the modification of diet in renal disease (MDRD) formula was normal at 97 cc/min/1.73 m2. A 24-hour urine protein determination revealed excretion of 149 mg of protein (normal <150 mg/24 hours). Given the absence of gross renal disease and absence of symptoms, additional renal studies were not performed. The patient was entered into the first study receiving Gamunex (0.34 g/kg/4 weeks).
Upon completion of the study, he was placed on Sandoglobulin (Novartis, East Hanover, NJ, USA) (0.34 g/kg q 4 weeks). Two years later he was screened for a second phase III IVIG protocol, testing a different formulation of IVIG from a different manufacturer. Screening again revealed MH.
Repeat evaluation by the nephrology department revealed a creatinine clearance rate of 83 cc/min and an MDRD GFR at 80.5 cc/min/1.73 m2. Urine protein excretion was in the normal range (92 mg/24 hours; reference range, <150 mg/24 hours). A renal biopsy revealed a number of sparse deposits in various stages of resolution consistent with repeated episodes of antigen-antibody complex formation. Immunofluorescence staining with IgGλ and IgGκ demonstrated equivalent patterns. The findings were felt to be consistent with stage II–III membranous glomerulopathy (Figures 1–3). As the patient remained asymptomatic, no treatment was prescribed.
Figure 1.

Hematoxylin-eosin stain of a glomerulus showing increased mesangial matrix with mild segmental mesangial hypercellularity (arrow). The capillary basement membranes show only mild focal thickening (original magnification, X200 [A] and X400 [B]).
Figure 3.
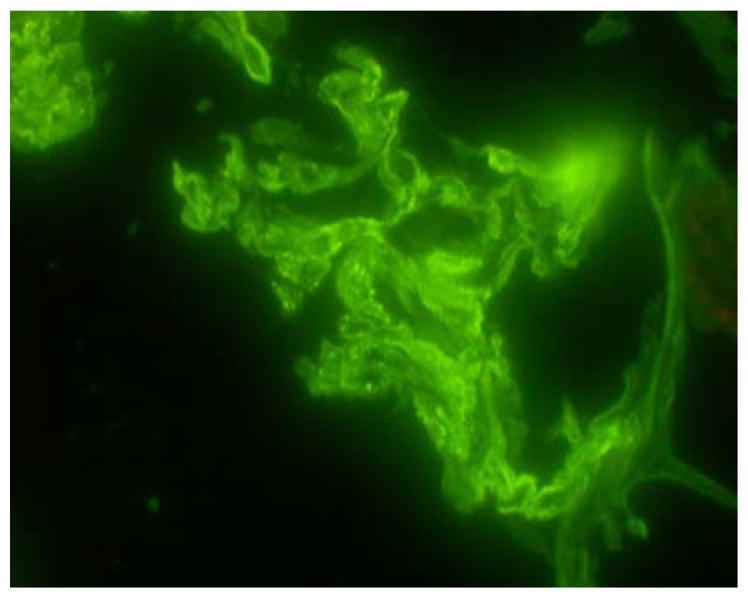
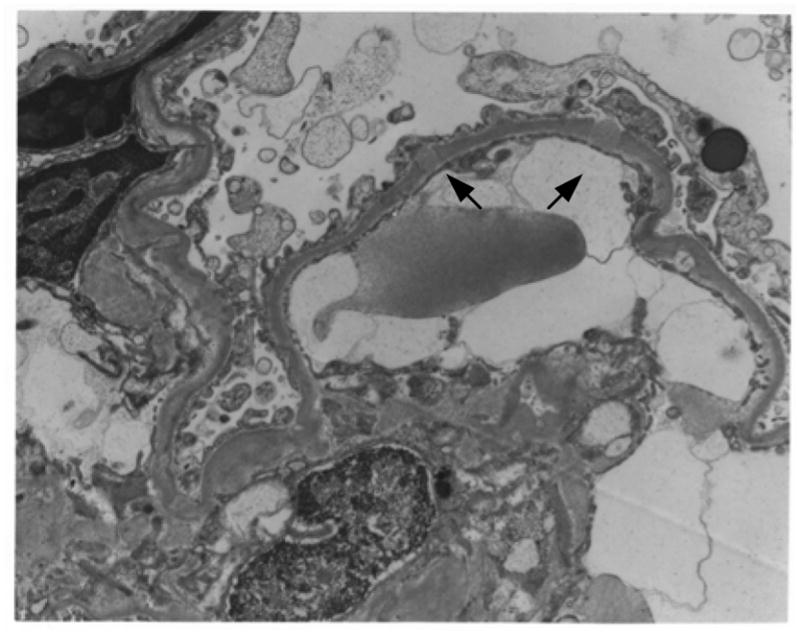
A, Immunofluorescence staining with FITC anti-Igk. Detail of a glomerulus with granular deposits along the capillary loops (original magnification, X400). B, Electron microscopy of an affected glomerulus revealed occasional subepithelial electron dense deposits (arrows) (original magnification X12 600).
The patient entered the second study with IVIG at a dose of 0.4 g/kg/4 weeks. Two years later, the patient remained asymptomatic with normal renal function and minimal proteinuria on Carimune (San Diego, CA, USA) at 0.4 g/kg/4 weeks.
Discussion
Due to transplacental IgG transport, BTK-deficient neonates exhibit normal serum IgG levels. Infections typically begin by 6 months of age, when normal catabolism depletes maternal Ig. Recurrent sinopulmonary infections, meningitis, osteomyelitis, and septic arthritis most often involve encapsulated pyogenic bacteria. Viral infections resolve normally, but absence of neutralizing antibody can lead to recurrence [1]. Conjunctivitis is observed in up to 1 in 5 adult patients [6].
More than 500 different BTK mutations have been reported in patients with XLA. The classic XLA phenotype is characterized by a peripheral B-lymphocyte compartment that represents less than 1% of circulating peripheral lymphocytes and near complete absence of endogenous Ig. Consistent correlations between the type of BTK mutation and the severity of the clinical presentation have not been identified [2].
The goal of therapy is to provide replacement Ig at levels sufficient to provide passive protection against infections [1]. Adverse reactions to IVIG can be immediate or delayed. Immediate reactions include headache, flushing, back pain, nausea, vomiting, and, rarely, circulatory collapse [7]. Nephrotoxicity is an example of a delayed response, with peak onset 5 days after infusion [8]. Altered renal function has been attributed to the increased osmotic load provided by preparations of IVIG that use sucrose or similar agents to inhibit IgG aggregation in the IVIG preparation.
MG, a cause of nephrotic syndrome in adults [4], is characterized by thickening of the basement membrane due to subepithelial deposition of immune complexes. Clinically, over 80% of patients present with proteinuria, and MH is apparent in half of the patients. Men are more commonly affected, with peak incidence between 30 and 50 years [9]. MG is often idiopathic, but has been associated with systemic diseases including systemic lupus erythematosus, infections, and malignancy [4]. It is thought to be result of chronic antigen-antibody complex formation with complement activation and formation of the membrane attack complex. Recently, M-type phospholipase A2 receptor has been identified as a major target antigen in primary human MG [10].
Treatment options include observation and conservative therapy, corticosteroids in combination with cytotoxic agents, and IVIG [11,12]. The renal dysfunction in our XLA patient persisted despite treatment with 5 different preparations of IVIG, each with its own stabilization agent and separate donor pools. Therefore, it was unlikely that the glomerulopathy reflected an idiopathic reaction to a specific IVIG preparation or contamination of the IVIG by IgG from a donor with active MG.
While XLA patients have greatly reduced numbers of circulating B lymphocytes, those cells that are present are capable of producing antigen-specific antibody, including allergen-specific IgE and antigen-specific IgM and IgG [13]. Patients with primary B or T lymphocyte immunodeficiency may be predisposed to the development of pathogenic B or T lymphocyte clones with high affinity/avidity for self-antigen [14]. We observed an equal mixture of IgGκ and IgGλ in subepithelial deposits, but were unable to assign its source. We speculate that the patient’s pathogenic immune complexes may reflect either IVIG reacting with a rare polymorphic variant of an endogenous protein that may or may not be expressed in his kidneys, the patient’s own endogenous IgG reacting with a common idiotype present in the IVIG, or both.
To our knowledge, we describe the first reported case of MG in a patient with XLA who was receiving IVIG. However, ours is not the first XLA patient to present with renal immune complex deposition. Membranoproliferative glomerulonephritis (MPG) was previously reported in another XLA patient receiving IVIG [15]. MG and MPG differ in that MPG is associated with both subepithelial and mesangial immune complex deposits, whereas only subepithelial deposits are observed in MG. The antigens responsible for idiopathic MPG are unknown, but a viral origin is suspected [16].
Our case differs from the case of MPG reported above in several key respects. The MPG patient had higher endogenous levels of IgM, IgG, and IgA than ours and presented at the age of 3 years, whereas our patient presented as an adult. Both MG and MPG are immune complex–mediated disorders with evidence of complement activation. However, the MPG patient presented with a low C3 level, which is typical of MPG, whereas our patient had normal C3 and C4 levels, which is typical of MG [17].
Nevertheless, these patients share key features. Both produced pathogenic immune complexes and both were treated with multiple forms and lots of IVIG, which had no effect on the course of their disease. Both patients appear to have developed renal dysfunction as a result of the endogenous production of rare pathogenic IgG clones. Surviving B lymphocytes in XLA lack BTK and, thus, experience abnormal signal transduction. Suppression of XLA B lymphocytes producing pathogenic Ig using IVIG may be more difficult than in B lymphocytes with a normal BTK function.
We conclude that the B lymphocyte deficiency engendered by BTK dysfunction does not eliminate the possibility of antibody-induced renal disease in grossly agammaglobulinemic patients.
Figure 2.
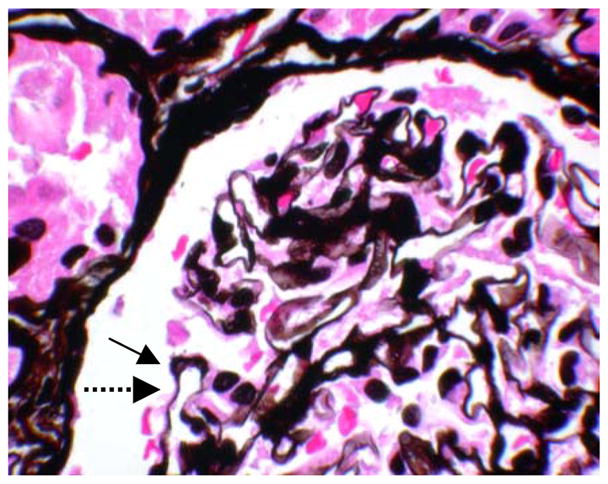
Jones methenamine silver stain of glomerulus. Note the holes in the glomerular basement membranes, which are cut en face (solid arrow). Compare with normal appearance of glomerular basement membrane (dashed arrow) (original magnification X400).
Acknowledgments
This work was supported in part by an NIH training grant AI07051 and by NIH research grants AI079741 and AI090902.
Footnotes
The authors declare that they have no competing financial interests.
References
- 1.Schroeder HW. Primary antibody deficiencies. In: Rich R, Fleisher TA, Shearer WT, Schroeder HW Jr, Frew AJ, Weyand CM, editors. Clinical Immunology Principles and Practice. 3. Philadelphia: Elsevier; 2008. pp. 518–20. [Google Scholar]
- 2.Broides A, Yang W, Conley ME. Genotype/phenotype correlations in X-linked agammaglobulinemia. Clinical Immunology. 2006;118:195–200. doi: 10.1016/j.clim.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Lindvall JM, Blomberg KEM, Väliaho J, Vargas L, Heinonen J, Berglöf A, Mohamed A, Nore B, Vihinen M, Smith CIE. Bruton’s tyrosine kinase: cell biology, sequence, conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunological Reviews. 2005;203:200–15. doi: 10.1111/j.0105-2896.2005.00225.x. [DOI] [PubMed] [Google Scholar]
- 4.Conley ME, Broides A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T, Shurtleff S. Genetic analysis of patients with defects in early B-cell development. Immunological Reviews. 2005;203:216–234. doi: 10.1111/j.0105-2896.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 5.Nachman PH, Jennette JC, Falk RJ. Primary glomerular disease. In: Brenner BM, editor. Brenner and Rector’s the kidney. 8. Philadelphia: Saunders; 2007. pp. 1007–14. [Google Scholar]
- 6.Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks W, Conley ME, Cunningham-Rundles C, Ochs HD. X-linked agammaglobulinemia: Report on a United States registry of 201 patients. Medicine. 2006;85:193–202. doi: 10.1097/01.md.0000229482.27398.ad. [DOI] [PubMed] [Google Scholar]
- 7.Ballow M. Immuoglobulin therapy: replacement and immunomodulation. In: Rich R, Fleisher TA, Shearer WT, Schroeder HW Jr, Frew AJ, Weyand CM, editors. Clinical Immunology Principles and Practice. 3. Philadelphia: Elsevier; 2008. pp. 1265–80. [Google Scholar]
- 8.Levy J, Pusey C. Nephrotoxicity of intravenous immunoglobulin. Q J Med. 2000;93:751–55. doi: 10.1093/qjmed/93.11.751. [DOI] [PubMed] [Google Scholar]
- 9.Wasserstein A. Membranous glomerulonephritis. J Am Soc Nephrol. 1997;8:664–74. doi: 10.1681/ASN.V84664. [DOI] [PubMed] [Google Scholar]
- 10.Beck LH, Jr, Bonegio RGB, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ. M –Type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du Buf-Verejiken P, Branten A, Wetzels J. Idiopathic membranous nephropathy: outline and rational of a treatment strategy. Am J Kidney Dis. 2005;46:1012–29. doi: 10.1053/j.ajkd.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 12.Palla R, Cirami C, Panichi V, Bianchi AM, Parrini M, Grazi G. Intravenous immunoglobulin therapy of membranous nephropathy: efficacy and safety. Clin Nephrol. 1991;35:98–104. [PubMed] [Google Scholar]
- 13.Nonoyama S, Tsukada S, Yamadori T, Miyawaki T, Jin YZ, Watanabe C, Morio TYJ, Ochs H. Functional analysis of peripheral blood B cells in patients with X-linked agammaglobulinemia. J immunol. 1998;161:3925–29. [PubMed] [Google Scholar]
- 14.Goyal R, Bulua AC, Nikolov NP, Schwartzberg PL, Siegel RM. Rheumatologic and Autoimmune Manifestations of Primary Immunodeficiency Disorders. Curr Opin Rheumatol. 2009;21(1):78–84. doi: 10.1097/BOR.0b013e32831cb939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshino A, Honda M, Kanegane H, Obata K, Matsukura H, Sakazume S, Katada Y, Miyawaki T, Ueda Y, Nagai T. Membranoproliferative glomerulonephritis in a patient with X-linked agammaglobulinemia. Pediatr Nephrol. 2006;21:36–8. doi: 10.1007/s00467-005-2029-z. [DOI] [PubMed] [Google Scholar]
- 16.Alpers C. The kidney. In: Kumar V, Abbas A, Fausto N, Louis St, Saunders WB, editors. Robbins and Cotran’s Pathologic Basis of Disease. 7. Philadelphia: W.B. Saunders Company; 2005. pp. 984–87. [Google Scholar]
- 17.Wasserstein A. Membranous glomerulonephritis. J Am soc nephrol. 1997;8:664–674. doi: 10.1681/ASN.V84664. [DOI] [PubMed] [Google Scholar]