

Abstract
Hypercholesterolemia frequently occurs in patients treated with efavirenz who cannot be treated adequately with statins because of drug interactions. These patients may benefit from cholesterol-lowering therapy with ezetimibe. This study determined the influence of single-dose and multiple-dose efavirenz (400 mg/day for 9 days) on the pharmacokinetics and sterol-lowering of ezetimibe (10 mg) in 12 healthy subjects. In addition, the influence of efavirenz on genome-wide intestinal expression and in vitro function of ABCB1, ABCC2, UGT1A1, and OATP1B1 was studied. Efavirenz (multiple dose) had no influence on the pharmacokinetics and lipid-lowering functions of ezetimibe. Intestinal expression of enzymes and transporters (e.g., ABCB1, ABCC2, and UGT1A1) was not affected by chronic efavirenz. Efavirenz (single dose) slightly increased ezetimibe absorption and markedly decreased exposure to ezetimibe-glucuronide (single dose and multiple dose), which may be explained by inhibition of UGT1A1 and ABCB1 (in vitro data). Ezetimibe had no effect on the disposition of efavirenz. Consequently, ezetimibe may be a safe and efficient therapeutic option in patients with HIV infection.
Highly active antiretroviral therapy (HAART) has markedly improved the survival of patients with HIV infection over the past decade.1,2
Unfortunately, HAART regimens are associated with several side effects, among which the development of a lipodystrophy syndrome with hypercholesterolemia remains a major clinical issue. One highly effective and frequently prescribed first-line component of HAART regimens is efavirenz,3,4 a non-nucleoside reverse-transcriptase inhibitor that was reported to increase serum cholesterol levels in nearly 50% of HIV-infected patients.5,6 Therefore, comedication with low-density lipoprotein (LDL)-cholesterol–lowering statins has been recommended to prevent increased cardiovascular morbidity and mortality.5,7–10 However, the lipid-lowering effect of several statins was shown to be significantly reduced after comedication with efavirenz.11,12 The reason for this interaction is most likely activation of the nuclear constitutive androstane receptor (CAR), for which efavirenz is a potent activator in vitro and in vivo.13–17 With CAR-type enzyme induction, efavirenz may regulate several drug metabolizing enzymes and transporter proteins that are involved in the disposition of statins (e.g., CYP3A4, ABCB1, and ABCC2) and in its own pharmacokinetics (auto-induction of CYP3A4 and CYP2B6).6,18,19
To avoid the undesired pharmacokinetic interaction with the antilipemic prophylaxis, efavirenz could be alternatively combined with ezetimibe, which efficiently inhibits the intestinal uptake of cholesterol. Ezetimibe may be a safe and efficient therapeutic option in HIV-infected patients, as indicated by several small clinical trials.20–25 Ezetimibe is not a substrate of cytochrome P450 (CYP) enzymes, which are regulated by CAR, but it is extensively metabolized by intestinal UGT1A1 and is a substrate of the drug transporters ABCB1, ABCC2, and OATP1B1.26–28
However, efavirenz might also affect disposition and lipid-lowering effects of ezetimibe because expression of UGT1A1, ABCB1, and ABCC2 is known to be regulated via CAR and because efavirenz can modulate ABC transporter functions.29–31 Whether ezetimibe may affect the pharmacokinetics and antiviral effects of efavirenz has not been investigated.
Therefore, it was the aim of this study to investigate the influence of chronic administration of efavirenz on the pharmacokinetics and lipid-lowering effects of ezetimibe. To acquire deeper insights into this potential drug–drug interaction, genome-wide intestinal expression was monitored and in vitro experiments on the function of UGT1A1, ABCB1, ABCC2, and OATP1B1 were performed.
We provide evidence that ezetimibe may be a safe and efficient alternative to statins in the treatment of hyperlipidemia associated with efavirenz HAART regimens.
RESULTS
Clinical study
Influence of efavirenz on the pharmacokinetics and pharmacodynamics of ezetimibe
Our clinical study investigated the influence of single-dose and multiple-dose administration of efavirenz on the pharmacokinetics and lipid-lowering effects of ezetimibe in 12 healthy subjects (Figure 1). Single-dose efavirenz slightly influenced the disposition of ezetimibe, as indicated by a significantly higher maximum plasma concentration (Cmax) (1.2-fold, P = 0.049) and area under the plasma concentration–time curve (AUC)0–6 h (1.4-fold, P = 0.005). Conversely, exposure to ezetimibe-glucuronide was somewhat, but significantly, decreased. Consequently, ezetimibe combined with single-dose efavirenz was found to not be bioequivalent to administration of ezetimibe alone (Table 1). Pretreatment with multiple doses of efavirenz substantially diminished serum Cmax and AUC of ezetimibe-glucuronide by about 43% (P = 0.012) and 30% (P = 0.005), respectively. Consistent with this, the urinary excretion of the glucuronide was reduced significantly (35%, P = 0.003) by efavirenz at unchanged renal clearance. However, all other pharmacokinetic parameters of ezetimibe and ezetimibe-glucuronide remained unchanged (Table 1, Figure 2).
Figure 1.
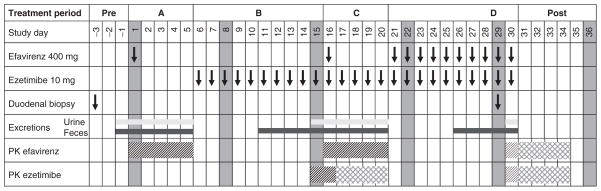
Gantt chart of the study design. PK, pharmacokinetic.
Table 1.
Pharmacokinetic data of ezetimibe and ezetimibe-glucuronide with and without comedication of 400 mg efavirenz in 12 healthy subjects
| Units | Ezetimibe | Ezetimibe + efavirenz single dose | Ratio (90% CI) | Ezetimibe + efavirenz multiple dose | Ratio (90% CI) | |
|---|---|---|---|---|---|---|
| Ezetimibe | ||||||
| AUC0–24 h | ng × h/ml | 51.9 ± 21.7 | 58.6 ± 23.8 | 1.146 (1.034, 1.270) | 51.1 ± 23.1 | 0.975 (0.850, 1.118) |
| AUC0–6 h | ng × h/ml | 13.6 ± 5.5 | 18.5 ± 8.1* | 1.346 (1.201, 1.508) | 14.6 ± 6.5 | 1.052 (0.880, 1.256) |
| Cmax | ng/ml | 4.82 ± 2.74 | 5.54 ± 2.65 | 1.194 (1.031, 1.382) | 4.46 ± 2.07 | 0.951 (0.730, 1.240) |
| CLR | ml/min | 1.38 ± 0.65 | 1.39 ± 1.35 | — | 1.41 ± 0.89 | — |
| Ezetimibe-glucuronide | ||||||
| AUC0–24 h | ng × h/ml | 466 ± 223 | 411 ± 195* | 0.885 (0.809, 0.968) | 325 ± 152* | 0.700 (0.608, 0.806) |
| AUC0–6 h | ng × h/ml | 205 ± 90 | 182 ± 87 | 0.872 (0.774, 0.982) | 141 ± 58* | 0.691 (0.592, 0.806) |
| Cmax | ng/ml | 78.9 ± 43.1 | 60.9 ± 29.5 | 0.776 (0.625, 0.963) | 45.3 ± 14.6* | 0.606 (0.469, 0.783) |
| CLR | ml/min | 24.7 ± 8.77 | 23.3 ± 7.61 | — | 21.7 ± 5.91 | — |
| Ezetimibe total | ||||||
| Aeurine | μg | 622 ± 195 | 565 ± 357 | — | 407 ± 210* | — |
| Aefeces | mg | 6.57 ± 2.17 | 8.03 ± 1.21 | — | 6.87 ± 2.99 | — |
Arithmetic means ± SD are given.
AUC, area under the concentration–time curve; CI, confidence interval; Cmax, maximum plasma concentration; CLR, renal clearance.
P < 0.025 for comparison with ezetimibe alone, Wilcoxon test with Bonferroni correction. Ratios of the pharmacokinetic parameters and 90% CIs are presented for equivalence analysis.
Figure 2.

Mean serum concentration–time profiles of ezetimibe (above) and ezetimibe-glucuronide (below) under steady-state conditions after repeated administration of 10 mg ezetimibe alone (open circles) and in combination with single-dose (closed circles) and multiple-dose 400 mg efavirenz (squares) in 12 healthy subjects (arithmetic means ± SD are given).
Chronic administration of ezetimibe caused a significant reduction of serum concentrations of the plant sterols campesterol and sitosterol (by ~40%) and of total serum cholesterol by ~10–15% (Figure 3). The lathosterol/cholesterol ratio, a marker for endogenous cholesterol synthesis, was significantly increased after treatment with ezetimibe. Acute (study day 16) and chronic (study days 21–30) comedication with efavirenz did not influence the serum levels of plant sterols, cholesterol, or lathosterol, or the respective sterol/cholesterol ratios.
Figure 3.

Influence of ezetimibe (EZE) and efavirenz (EFA) on the relative serum concentrations of cholesterol, campesterol, and sitosterol and the lathosterol/cholesterol ratio in 12 healthy subjects (arithmetic means ± SD are given). *Significant (p < 0.05) differences compared to baseline values on study day 1.
Influence of ezetimibe on the pharmacokinetics of efavirenz
Single-dose pharmacokinetics of efavirenz was not influenced by chronic comedication with ezetimibe (Figure 4). None of the following parameters for efavirenz was different in the presence of ezetimibe: serum AUC0–∞ (27.0 ± 12.2 μg × h/ml vs. 29.7 ± 15.4 μg × h/ml, P = 0.814), Cmax (0.66 ± 0.40 μg/ml vs. 0.62 ± 0.24 μg/ml, P = 0.695), time to Cmax (Tmax) (2.6 ± 1.5 h vs. 2.4 ± 1.9 h, P = 0.824), terminal half-life (t½) (106 ± 49.3 h vs. 121 ± 97.0 h, P = 0.973), cumulative renal excretion (83.4 ± 26.9 μg vs. 88.4 ± 18.8 μg, P = 0.530), and renal clearance (0.06 ± 0.03 ml/min vs. 0.06 ± 0.03 ml/min, P = 0.814). Moreover, AUC0–∞ and Cmax of efavirenz were within the accepted range of bioequivalence (0.8–1.25) after comedication with ezetimibe.
Figure 4.

Mean plasma concentration–time profiles of efavirenz after single-dose administration of 400 mg efavirenz alone (open circles) and in combination with 10 mg ezetimibe (closed circles) in 12 healthy subjects (arithmetic means ± SD are given).
Influence of efavirenz on intestinal mRNA expression
Pretreatment with 400 mg efavirenz for 9 days caused 1.5-fold changes in the expression of approximately 4,000 genes in all investigated samples from eight healthy subjects (data not shown). However, a nonuniform regulation pattern could be observed (Figure 5a). Furthermore, the quantitative expression analysis of several CAR-regulated genes involved in intestinal metabolism and transport, including CYP3A4, CYP2B6, UGT1A1, ABCB1, and ABCC2, revealed no change in intestinal gene expression after administration of efavirenz (Figure 5b).
Figure 5.

Influence of repeated administration of efavirenz (400 mg, 9 days) on duodenal mRNA expression in 8 healthy volunteers. (a) Expression pattern of selected ADME genes before and after treatment with efavirenz. The heat map shows log2-transformed, median-centered normalized expression signals. Green indicates low expression and red indicates higher expression. (b) Quantitative analysis of selected genes of metabolism and transport.
Influence of efavirenz on hepatic CYP3A4
Repeated administration of efavirenz increased the hepatic activity of CYP3A4, as concluded from significantly elevated ratios of serum concentrations of 4β-OH-cholesterol/cholesterol (104.9 ± 24.7% on study day 21 vs. 131.8 ± 24.2% on study day 25 vs. 182.9 ± 40.6% on study day 30; compared with the initial ratio).
In vitro studies
Efavirenz competitively inhibited the efflux of calcein AM in ABCB1-transfected Madin Darby canine kidney II cells. ABCB1 activity was inhibited to the same extent by 316 μmol/l efavirenz as by the potent ABCB1 inhibitor PSC833 (10 μmol/l): relative half maximal inhibitory concentration (IC50) value = 123 μmol/l (Figure 6). The accumulation of estradiol-17β-glucuronide into ABCC2-containing inside-out vesicles was also modulated by efavirenz (IC50 = 60.3 μmol/l). Moreover, the in vitro glucuronidation of ezetimibe by UGT1A1 was strikingly inhibited by efavirenz: IC50 value = 9.6 μmol/l (Figure 6). In contrast to this, efavirenz had no influence on the OATP1B1-mediated uptake of bromosulfophthalein into OATP1B1-transfected HEK293 cells (Figure 6).
Figure 6.

Influence of efavirenz on in vitro glucuronidation of ezetimibe by human UGT1A1, ABCB1 function using the calcein AM assay in ABCB1-transfected MDCKII cells, uptake of estradiol-17-β-glucuronide (E17βG) into ABCC2 inside-out vesicles, and the OATP1B1-mediated uptake of bromosulfophthalein (BSP) into OATP1B1-transfected HEK cells. IC50, half maximal inhibitory concentration; MDCKII, Madin Darby canine kidney II.
DISCUSSION
In this study, we demonstrated that multiple doses of the anti-HIV drug efavirenz had no major influence on the pharmacokinetics and lipid-lowering effects of the cholesterol absorption inhibitor ezetimibe in healthy volunteers.
Consistent with this, the intestinal expression of several CAR-regulated metabolizing enzymes and transporters, including ABCB1, ABCC2, CYP3A4, CYP2B6, and UGT1A1, were not altered by chronic premedication of efavirenz for 9 days. In contrast to other prototypic activators of nuclear receptors such as the PXR ligand rifampin, no systematic pattern of intestinal gene induction was observed.27,32 This finding was somewhat surprising because efavirenz is known to be a potent activator of CAR in vitro and in vivo, thereby resulting in some clinically relevant drug interactions with several anti-HIV drugs, statins, and antibiotics.6,11,13,17,33 Conversely, efavirenz was capable of inducing hepatic CYP3A4 activity, as indicated by a significantly increased 4-βOH-cholesterol/cholesterol ratio, which was also demonstrated in previous studies.34
Ezetimibe undergoes extensive UGT1A1-mediated intestinal glucuronidation and is a substrate of the transport proteins ABCB1, ABCC2, and OATP1B1.26–28 The observed lack of interaction between ezetimibe and multiple-dose efavirenz supports our previous finding that ezetimibe undergoes predominately intestinal metabolism and transport.27,35
Surprisingly, acute and chronic comedication with efavirenz significantly decreased the exposure to ezetimibe-glucuronide by ~12–30%. Associated in vitro studies using UGT1A1 supersomes demonstrated for the first time that efavirenz can inhibit the UGT1A1-mediated glucuronidation of ezetimibe. This finding was unexpected, as efavirenz itself is predominantly metabolized by UGT2B7.36 Because the estimated luminal concentrations of efavirenz after oral administration of 400 mg (dose/250 mL, ~5 mmol/l) was some 500-fold greater than the respective IC50 value for the inhibition of ezetimibe-glucuronide formation (~10 μmol/l), this interaction appears to be plausible in vivo.
Moreover, single-dose efavirenz slightly increased the intestinal absorption of the ABCB1 substrate ezetimibe, as indicated by significantly higher AUC0–6 h (1.4-fold) and Cmax, most likely due to inhibition of intestinal ABCB1. Similar interactions have been reported for the combinations of ezetimibe with other ABCB1-modulating drugs, such as sirolimus and tacrolimus.37,38
Indeed, we observed in our in vitro studies that ABCB1 function was significantly inhibited by efavirenz: IC50 value = ~123 μmol/l. This is in line with previous studies that have also reported a significant inhibition of ABC transporters.29,31 On the basis of the current drug-interaction guidelines of the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), significant interactions had to be expected from these in vitro data because the observed IC50 value was considerably lower than the estimated luminal concentrations of efavirenz (123 μmol/l vs. 5 mmol/l).
However, the inhibition of intestinal ABCB1 seems not to be of clinical relevance because the pharmacodynamic effects of ezetimibe were not influenced by efavirenz and this interaction was not observed after chronic comedication with both drugs.
The same is true for ABCC2, for which in vitro function was also significantly inhibited by efavirenz (IC50 = 60.3 μmol/l), but no corresponding in vivo effect could be observed. Although ezetimibe-glucuronide is a high-affinity substrate of this transporter, serum levels of the glucuronide were not elevated as expected, but in fact significantly decreased. This phenomenon may have been caused by the simultaneous inhibition of UGT1A1, which may have masked the excretory function of ABCC2.
Finally, our in vitro studies could exclude any influence of efavirenz on the hepatic uptake of ezetimibe-glucuronide by OATP1B1.
In conclusion, our in vitro studies suggest that all observed changes in the pharmacokinetics of ezetimibe can be plausibly explained by direct modulation of intestinal UGT1A1 and ABCB1.
The reason for the obscure discrepancy between the intestinal and hepatic gene regulation as caused by chronic oral administration of efavirenz remains unknown and needs further investigation. The lack of intestinal gene regulation by efavirenz was already reported by Mouly et al. in 2002.16 In that study, treatment of healthy volunteers with multiple doses of efavirenz (200/400 mg/day for 10 days) increased the activity of hepatic CYP3A4 as indicated by the erythromycin breath test, but it caused no regulation of intestinal ABCB1 and CYP3A4 proteins.
A possible explanation for this phenomenon might be the pharmacokinetic properties of efavirenz. Because efavirenz is rapidly absorbed from the upper intestine (Tmax 2–3 h) and does not undergo significant biliary secretion,6,18 there seems to be only a short duration of exposure of the enterocytes to efavirenz, even after daily oral administration, which may have counteracted intestinal gene regulation.
From the therapeutic point of view, the combination of efavirenz and ezetimibe may be a promising therapeutic option in HIV-infected patients with hypercholesterolemia. In contrast to described drug interactions with statins,11,12 we observed no clinically relevant influence of efavirenz on the disposition and lipid-lowering effect of ezetimibe. With reference to this, monotherapy with ezetimibe was shown to be an equivalent therapeutic option in clinical practice.20–25
Moreover, no influence of ezetimibe on the pharmacokinetics of efavirenz was observed. This was not surprising because efavirenz is not a substrate of transporter proteins but undergoes extensive oxidative metabolism by CYP2B6 and CYP3A4 and conjugation via UGT2B7, which are not affected by ezetimibe in vitro.26
In line with our conclusion, the combination of efavirenz and ezetimibe was shown to be safe and efficient in HIV-infected patients in several smaller clinical trials.20–25 However, the clinical outcome of patients receiving LDL-cholesterol–lowering ezetimibe in monotherapy remains controversial (e.g., SEARCH, SEAS, and ENHANCE studies).39–41 Further clinical studies are needed to demonstrate convincingly the cardiovascular benefit of ezetimibe treatment in patients with hypercholesterolemia. The ongoing IMPROVE-IT study (anticipated completion date 2013) may be a potential candidate to fill this gap in knowledge.
METHODS
Clinical study
Subjects
Twelve healthy Caucasian subjects (all male; age 20–36 years; body mass index 19.9–27.2 kg/m2) were enrolled after providing written informed consent and confirmation of good health by medical histories, physical examinations, and routine clinical–chemical and hematological screenings. All subjects were nonsmokers and took no medication.
Study protocol
The study was performed as a controlled, open, single-dose and steady-state study with the following four study periods (Figure 1): period A (days 1–5), single-dose administration of efavirenz (400 mg, Sustiva capsules; Bristol-Myers Squibb, Munich, Germany) and pharmacokinetic analysis; period B (days 6–15), daily treatment with 10 mg ezetimibe (Ezetrol tablets, MSD Sharp & Dohme, Haar, Germany) prior to pharmacokinetic analysis of ezetimibe at steady state on study day 15; period C (days 16–20), daily treatment with 10 mg ezetimibe with concomitant single-dose administration of 400 mg efavirenz on the 16th treatment day and pharmacokinetic analysis of both drugs (ezetimibe at steady state and efavirenz); and period D (days 21–30), daily treatment with 10 mg ezetimibe and 400 mg efavirenz, and pharmacokinetic analysis of both drugs at steady state on the 30th treatment day.
In each study period, the subjects were admitted to our clinical study unit the evening before drug administration on the next morning. After overnight fasting for at least 10 h, either 10 mg ezetimibe or 400 mg efavirenz alone or the combination of both drugs was administered with 240 ml of tap water. Standard meals were served 5, 8, and 11 h after drug administration and at 8:00 AM, 1:00 PM, and 8:00 PM of the next day. To exclude influences of different intestinal amounts of water on oral drug absorption, drinking of tap water was standardized. In detail, after initial drug administration with 200 ml of tap water, additional drinking of 100 ml and 200 ml was stipulated after 1, 2, 3, 4, 7, and 8 and after 5, 9, 11, 13, and 15 h, respectively. Forearm venous blood (7.5 ml) was sampled before and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, and 120 h after medication in study periods A, C, and D. In study period B, blood was sampled from 0–24 h using the given intervals. Urine and feces were collected according to the study protocol (Figure 1). Serum samples and aliquots of urine and fecal homogenates were stored at −20 °C (ezetimibe) and −80 °C (efavirenz) until quantitative analysis.
Strenuous physical exercise and alcoholic beverages were not allowed between 48 h before drug administration and the last sampling of feces. Intake of grapefruit-, orange-, and poppy seed–containing products was prohibited from 7 days prior to the first drug administration until the last sampling of the study.
Duodenal samples for analysis of intestinal gene expression were obtained via gastroduodenoscopy in the University Hospital Greifswald Clinic of Internal Medicine. Biopsies were taken at least 3 days before the first study day and on study day 29, that is, after chronic administration of efavirenz for 9 days. Duodenal samples were stored in 1.0 ml RNAlater solution (Qiagen, Hilden, Germany) at −80 °C until RNA isolation.
The study was approved by the local ethics committee of the Medical Faculty of the University of Greifswald and by the German Federal Institute for Drugs and Medical Devices (BfArM, No. 4035064); the study was registered in the European (EudraCT No. 2009-011050-17) and US (ClinicalTrials.gov NCT00810303) clinical trial databases. Planning and performance of the study followed the German Medical Act and was in accordance with the International Conference on Harmonisation guidance for good clinical practice.
Intestinal mRNA expression
Duodenal biopsies were obtained prior to drug treatment and following multiple doses of efavirenz (400 mg/day for 9 days). Tissue samples were immediately snap-frozen after collection and stored at −80 °C. Total mRNA was isolated using the RNeasy Mini kit following the manufacturer’s instructions (Qiagen, Hilden, Germany), followed by determination of integrity and amount using the Lab-on-a-Chip technology and the Agilent Bioanalyzer 2100 (Agilent, Waldbronn, Germany). Subsequently, genome-wide intestinal gene expression was measured in eight individuals; pretreatment and posttreatment expression were compared using the Human Gene ST 1.0 microarray (Affymetrix, Santa Clara, CA) according to the manufacturer’s protocol. In brief, total RNA (200 ng) was reverse- transcribed into cDNA and further processed to terminal-labeled DNA fragments. Staining and scanning were performed using the Fluidics Station 450 and Scanner 3000 7G (Affymetrix). Raw CEL files were generated within the GeneChip operating software (Affymetrix). Microarray data analysis was then performed using the Rosetta Resolver system for gene expression data analysis (Rosetta Biosoftware, Cambridge, MA). Raw signals of the probes were summarized using robust multichip analysis, thereby generating probe set–specific signal intensities. Chips were normalized by using the quantile normalization. Fold change was calculated by direct comparison of gene expression before and after efavirenz treatment for each subject.
Quantitative assays for efavirenz, ezetimibe, and serum sterols
Plasma and urine concentrations of efavirenz were measured using a previously published liquid chromatography–tandem mass spectrometry (LC-MS/MS) method with slight modification.42 In brief, a plasma or urine sample (0.2 ml) was mixed with the internal standard (30 μl of 500 ng/ml nevirapine) and extracted with 6 ml of ethyl acetate under alkaline pH (500 μl of 1 mol/l glycine/1 mol/l sodium hydroxide buffer, pH 11.3). The sample was centrifuged at 2890g, and the organic layer was evaporated to dryness. Residue was reconstituted in 50 μl mobile phase, and 25 μl was analyzed using API 2000 MS/MS (Applied Biosystems, Foster City, CA) equipped with a TurboIon spray and coupled with a Shimadzu HPLC (high-performance liquid chromatography) system (Columbia, MD). Efavirenz and nevirapine were quantified using multiple-reaction monitoring at m/z transitions of 316 to 244 and 267 to 226, respectively, in positive ion mode. This assay had a lower limit of quantification of 1 ng/ml, was linear over the range of 1–2,500 ng/ml efavirenz, and had within-day and between-day coefficients of <15% for plasma and <8% for urine.
Ezetimibe and ezetimibe-glucuronide in serum, urine, and feces were quantified by LC-MS/MS as previously described.43 The limits of quantification for ezetimibe in serum, urine, and feces were 0.05, 1.0, and 10 ng/ml, respectively. Within- and between-day precision for this assay in all matrices was within the range of 2.0 to 12.3%, whereas within- and between-day accuracy expressed as relative error was −8.6 to 4.9%.
Serum sterol concentrations were measured in serum samples on study days 1, 2, 6, 10, 15, 17, 21, 25, and 30. Cholesterol was determined by gas chromatography with flame ionization detection; the cholesterol precursor lathosterol and the plant sterols campesterol and sitosterol were analyzed by GC-MS as recently described.44 The limit of quantification was 10 μg/ml for cholesterol and 0.05 μg/ml for lathosterol, campesterol, and sitosterol. The within-day and between-day coefficients of variation for all sterols were <10%. 4β-OH-cholesterol was measured with a highly sensitive and specific isotope dilution methodology (gas chromatography–mass spectrometry) as recently described by Lütjohann et al.45 The lower limit of quantification was 3.0 ng/ml, and the linear range was 0.3–300 ng/ml. Between-day and within-day precision values were 2.1 and 2.7%, respectively, and the between-day and within-day accuracies were 2.9 and 3.3%, respectively.
Pharmacokinetic and statistical evaluation
Cmax values were taken from the concentration–time curves. The AUC was calculated by the trapezoidal rule. t½ was calculated by linear regression of the terminal slope of the concentration–time profiles. Unless indicated otherwise, all data were expressed as mean ± SD. Pharmacokinetic and statistical evaluation was done with SPSS 19 (IBM, Armonk, NY) and SAS 9.1 (SAS Institute, Cary, NC).
Sample differences were evaluated with the nonparametric Wilcoxon test or Mann-Whitney test as appropriate. Significance was assumed if P values were <0.05. To avoid alpha error accumulation by multiple testing, the Bonferroni method was used. Moreover, pharmacokinetic data were analyzed using the bioequivalence approach. Bioequivalence was assumed if the 90% confidence intervals for the test/reference ratios of the AUC and Cmax were within the range of 0.8–1.25.
Twelve volunteers were included in the study in order to detect a significant difference of at least 20% with an 80% power (α-error 5%), considering the substantial intrasubject and intersubject coefficient of variation of former pharmacokinetic studies (nQuery Adviser 5.0, StatSol, Cork, Ireland).27,37
In vitro studies
Inhibitory effect of efavirenz on ABCB1
Competition studies in ABCB1 overexpressing cells were performed using the calcein AM accumulation assay in parental and ABCB1-transfected MDCKII cells as recently described.37 All incubations with efavirenz (1–1,000 μmol/l) were performed in triplicate and compared with the effect of the well-known ABCB1-inhibitor PSC833 (10 μmol/l).
Inhibitory effect of efavirenz on ABCC2
The function of ABCC2 was studied with ABCC2-containing inside-out vesicles generated from Sf9 cells (BD Biosciences, Heidelberg, Germany) as previously published using [3H]estradiol-17β-glucuronide as a reference compound.27 Competitive inhibition was studied in the presence of 1–1,000 μmol/l efavirenz after incubation for 10 min (each N = 3).
Inhibitory effects of efavirenz on OATP1B1
The influence of efavirenz on OATP1B1-mediated cellular uptake was studied as previously described.37 In brief, competitive inhibition of efavirenz (1–1,000 μmol/l) on the cellular uptake of 50 nmol/l [3H]-bromosulfophthalein (BSP, 14 Ci/mmol; Hartmann Analytic, Braunschweig, Germany) in HEK293 cells stably transfected with OATP1B1 was determined after incubation at 37 °C for 5 min (n = 3). After cell lysis, intra-cellular accumulation of radiolabeled BSP was measured and protein concentration was determined. The transporter-mediated net uptake of BSP was obtained by subtracting the BSP uptake of vector-transfected cells from that in transporter-expressing cells. The percentage of uptake inhibition was calculated from experiments in the absence of efavirenz.
Inhibition of ezetimibe glucuronidation by efavirenz
The glucuronidation of ezetimibe was studied using human UGT supersomes for UGT1A1, UGT1A3, UGT2B7, and UGT2B15 (BD Biosciences, Heidelberg, Germany). In each case, 100 μmol/l ezetimibe was incubated with 0.9 mg/ml UGT microsomes, 4.6 mmol/l saccharolactone, 23 μg/ml alamethicin, and 7.4 mmol/l magnesium chloride, alone and in the presence of efavirenz (1–500 μmol/l). After 5 min preincubation at 37 °C, the reaction was started by adding 1.85 mmol/l uridine-diphospho-glucuronic acid (UDPGA), followed by incubation at 37 °C for 120 min. The reaction was stopped using 200 μl ice-cold methanol. Supernatants were obtained after centrifugation (10,000g, 10 min, 4 °C) and stored at −20 °C until drug analysis by LC-MS/MS as described above. All reactions were performed in triplicate.
Acknowledgments
This study was supported by the German Federal Ministry for Education and Research (grant 03IP612, InnoProfile); by the National Institute of General Medical Sciences, the National Institutes of Health (grant RO1GM078501 (Z.D.)); and by an institutional grant by MSD Sharp & Dohme to support the clinical study. The authors express their gratitude to Gitta Schumacher, Sabine Bade, and Danilo Wegner for their excellent technical assistance.
Footnotes
CONFLICT OF INTEREST
The authors declared no conflict of interest.
References
- 1.Thompson MA, et al. Antiretroviral treatment of adult HIV infection: 2010 recommendations of the International AIDS Society-USA panel. JAMA. 2010;304:321–333. doi: 10.1001/jama.2010.1004. [DOI] [PubMed] [Google Scholar]
- 2.Volberding P, et al. Antiretroviral therapy in acute and recent HIV infection: a prospective multicenter stratified trial of intentionally interrupted treatment. AIDS. 2009;23:1987–1995. doi: 10.1097/QAD.0b013e32832eb285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maggiolo F. Efavirenz: a decade of clinical experience in the treatment of HIV. J Antimicrob Chemother. 2009;64:910–928. doi: 10.1093/jac/dkp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rakhmanina NY, van den Anker JN. Efavirenz in the therapy of HIV infection. Expert Opin Drug Metab Toxicol. 2010;6:95–103. doi: 10.1517/17425250903483207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calza L, Manfredi R, Chiodo F. Hyperlipidaemia in patients with HIV-1 infection receiving highly active antiretroviral therapy: epidemiology, pathogenesis, clinical course and management. Int J Antimicrob Agents. 2003;22:89–99. doi: 10.1016/s0924-8579(03)00115-8. [DOI] [PubMed] [Google Scholar]
- 6.Sustiva (efavirenz) product information. New York: Bristol-Myers Squibb; 2009. [Google Scholar]
- 7.Bain AM, White EA, Rutherford WS, Rahman AP, Busti AJ. A multimodal, evidence-based approach to achieve lipid targets in the treatment of antiretroviral-associated dyslipidemia: case report and review of the literature. Pharmacotherapy. 2008;28:932–938. doi: 10.1592/phco.28.7.932. [DOI] [PubMed] [Google Scholar]
- 8.Fichtenbaum CJ. Does antiretroviral therapy increase or decrease the risk of cardiovascular disease? Curr HIV/AIDS Rep. 2010;7:92–98. doi: 10.1007/s11904-010-0045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grinspoon S, Carr A. Cardiovascular risk and body-fat abnormalities in HIV-infected adults. N Engl J Med. 2005;352:48–62. doi: 10.1056/NEJMra041811. [DOI] [PubMed] [Google Scholar]
- 10.Sekhar RV, Balasubramanyam A. Treatment of dyslipidemia in HIV-infected patients. Expert Opin Pharmacother. 2010;11:1845–1854. doi: 10.1517/14656566.2010.487484. [DOI] [PubMed] [Google Scholar]
- 11.Gerber JG, et al. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr. 2005;39:307–312. doi: 10.1097/01.qai.0000167156.44980.33. [DOI] [PubMed] [Google Scholar]
- 12.Rahman AP, et al. Safety and efficacy of simvastatin for the treatment of dyslipidemia in human immunodeficiency virus-infected patients receiving efavirenz-based highly active antiretroviral therapy. Pharmacotherapy. 2008;28:913–919. doi: 10.1592/phco.28.7.913. [DOI] [PubMed] [Google Scholar]
- 13.Faucette SR, et al. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320:72–80. doi: 10.1124/jpet.106.112136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hariparsad N, Nallani SC, Sane RS, Buckley DJ, Buckley AR, Desai PB. Induction of CYP3A4 by efavirenz in primary human hepatocytes: comparison with rifampin and phenobarbital. J Clin Pharmacol. 2004;44:1273–1281. doi: 10.1177/0091270004269142. [DOI] [PubMed] [Google Scholar]
- 15.Healan-Greenberg C, Waring JF, Kempf DJ, Blomme EA, Tirona RG, Kim RB. A human immunodeficiency virus protease inhibitor is a novel functional inhibitor of human pregnane X receptor. Drug Metab Dispos. 2008;36:500–507. doi: 10.1124/dmd.107.019547. [DOI] [PubMed] [Google Scholar]
- 16.Mouly S, et al. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin Pharmacol Ther. 2002;72:1–9. doi: 10.1067/mcp.2002.124519. [DOI] [PubMed] [Google Scholar]
- 17.Weiss J, Herzog M, König S, Storch CH, Ketabi-Kiyanvash N, Haefeli WE. Induction of multiple drug transporters by efavirenz. J Pharmacol Sci. 2009;109:242–250. doi: 10.1254/jphs.08209fp. [DOI] [PubMed] [Google Scholar]
- 18.Smith PF, DiCenzo R, Morse GD. Clinical pharmacokinetics of non-nucleoside reverse transcriptase inhibitors. Clin Pharmacokinet. 2001;40:893–905. doi: 10.2165/00003088-200140120-00002. [DOI] [PubMed] [Google Scholar]
- 19.Ngaimisi E, et al. Long-term efavirenz autoinduction and its effect on plasma exposure in HIV patients. Clin Pharmacol Ther. 2010;88:676–684. doi: 10.1038/clpt.2010.172. [DOI] [PubMed] [Google Scholar]
- 20.Bennett MT, Johns KW, Bondy GP. Ezetimibe is effective when added to maximally tolerated lipid lowering therapy in patients with HIV. Lipids Health Dis. 2007;6:15. doi: 10.1186/1476-511X-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berg-Wolf MV, Klibanov OM, Gaughan JP, Tedaldi EM. Ezetimibe combined with low-dose statin effectively lowers LDL in protease inhibitor treated patients. AIDS Patient Care STDs. 2008;22:483–488. doi: 10.1089/apc.2007.0206. [DOI] [PubMed] [Google Scholar]
- 22.Chow D, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS. 2009;23:2133–2141. doi: 10.1097/QAD.0b013e32833068e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coll B, Aragonés G, Parra S, Alonso-Villaverde C, Masana L. Ezetimibe effectively decreases LDL-cholesterol in HIV-infected patients. AIDS. 2006;20:1675–1677. doi: 10.1097/01.aids.0000238418.43937.3b. [DOI] [PubMed] [Google Scholar]
- 24.Negredo E, et al. Ezetimibe, a promising lipid-lowering agent for the treatment of dyslipidaemia in HIV-infected patients with poor response to statins. AIDS. 2006;20:2159–2164. doi: 10.1097/01.aids.0000247573.95880.db. [DOI] [PubMed] [Google Scholar]
- 25.Wohl DA, et al. Ezetimibe alone reduces low-density lipoprotein cholesterol in HIV-infected patients receiving combination antiretroviral therapy. Clin Infect Dis. 2008;47:1105–1108. doi: 10.1086/592116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosoglou T, Statkevich P, Johnson-Levonas AO, Paolini JF, Bergman AJ, Alton KB. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clin Pharmacokinet. 2005;44:467–494. doi: 10.2165/00003088-200544050-00002. [DOI] [PubMed] [Google Scholar]
- 27.Oswald S, et al. Intestinal expression of P-glycoprotein (ABCB1), multidrug resistance associated protein 2 (ABCC2), and uridine diphosphate-glucuronosyltransferase 1A1 predicts the disposition and modulates the effects of the cholesterol absorption inhibitor ezetimibe in humans. Clin Pharmacol Ther. 2006;79:206–217. doi: 10.1016/j.clpt.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Oswald S, et al. Disposition of ezetimibe is influenced by polymorphisms of the hepatic uptake carrier OATP1B1. Pharmacogenet Genomics. 2008;18:559–568. doi: 10.1097/FPC.0b013e3282fe9a2c. [DOI] [PubMed] [Google Scholar]
- 29.Storch CH, Theile D, Lindenmaier H, Haefeli WE, Weiss J. Comparison of the inhibitory activity of anti-HIV drugs on P-glycoprotein. Biochem Pharmacol. 2007;73:1573–1581. doi: 10.1016/j.bcp.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 30.Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J Clin Pharmacol. 2007;47:566–578. doi: 10.1177/0091270007299930. [DOI] [PubMed] [Google Scholar]
- 31.Weiss J, Theile D, Ketabi-Kiyanvash N, Lindenmaier H, Haefeli WE. Inhibition of MRP1/ABCC1, MRP2/ABCC2, and MRP3/ABCC3 by nucleoside, nucleotide, and non-nucleoside reverse transcriptase inhibitors. Drug Metab Dispos. 2007;35:340–344. doi: 10.1124/dmd.106.012765. [DOI] [PubMed] [Google Scholar]
- 32.Oscarson M, et al. Effects of rifampicin on global gene expression in human small intestine. Pharmacogenet Genomics. 2007;17:907–918. doi: 10.1097/FPC.0b013e3280143dfc. [DOI] [PubMed] [Google Scholar]
- 33.Hariparsad N, Carr BA, Evers R, Chu X. Comparison of immortalized Fa2N-4 cells and human hepatocytes as in vitro models for cytochrome P450 induction. Drug Metab Dispos. 2008;36:1046–1055. doi: 10.1124/dmd.108.020677. [DOI] [PubMed] [Google Scholar]
- 34.Josephson F, et al. CYP3A induction and inhibition by different antiretroviral regimens reflected by changes in plasma 4beta-hydroxycholesterol levels. Eur J Clin Pharmacol. 2008;64:775–781. doi: 10.1007/s00228-008-0492-8. [DOI] [PubMed] [Google Scholar]
- 35.Oswald S, et al. Disposition and sterol-lowering effect of ezetimibe are influenced by single-dose coadministration of rifampin, an inhibitor of multidrug transport proteins. Clin Pharmacol Ther. 2006;80:477–485. doi: 10.1016/j.clpt.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Bélanger AS, Caron P, Harvey M, Zimmerman PA, Mehlotra RK, Guillemette C. Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine. Drug Metab Dispos. 2009;37:1793–1796. doi: 10.1124/dmd.109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oswald S, et al. Pharmacokinetic and pharmacodynamic interactions between the immunosuppressant sirolimus and the lipid-lowering drug ezetimibe in healthy volunteers. Clin Pharmacol Ther. 2010;87:663–667. doi: 10.1038/clpt.2009.266. [DOI] [PubMed] [Google Scholar]
- 38.Oswald S, et al. Drug interactions between the immunosuppressant tacrolimus and the cholesterol absorption inhibitor ezetimibe in healthy volunteers. Clin Pharmacol Ther. 2011;89:524–528. doi: 10.1038/clpt.2011.4. [DOI] [PubMed] [Google Scholar]
- 39.Kastelein JJ, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–1443. doi: 10.1056/NEJMoa0800742. [DOI] [PubMed] [Google Scholar]
- 40.Rossebø AB, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 41.Sharp Collaborative Group. Study of Heart and Renal Protection (SHARP): randomized trial to assess the effects of lowering low-density lipoprotein cholesterol among 9,438 patients with chronic kidney disease. Am Heart J. 2010;160:785–794. doi: 10.1016/j.ahj.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 42.Ogburn ET, Jones DR, Masters AR, Xu C, Guo Y, Desta Z. Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab Dispos. 2010;38:1218–1229. doi: 10.1124/dmd.109.031393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oswald S, Scheuch E, Cascorbi I, Siegmund W. A LC-MS/MS method to quantify the novel cholesterol lowering drug ezetimibe in human serum, urine and feces in healthy subjects genotyped for SLCO1B1. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;830:143–150. doi: 10.1016/j.jchromb.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 44.Thelen KM, Laaksonen R, Päivä H, Lehtimäki T, Lütjohann D. High-dose statin treatment does not alter plasma marker for brain cholesterol metabolism in patients with moderately elevated plasma cholesterol levels. J Clin Pharmacol. 2006;46:812–816. doi: 10.1177/0091270006289851. [DOI] [PubMed] [Google Scholar]
- 45.Lütjohann D, et al. 4beta-hydroxycholesterol as a marker of CYP3A4 inhibition in vivo - effects of itraconazole in man. Int J Clin Pharmacol Ther. 2009;47:709–715. doi: 10.5414/cpp47709. [DOI] [PubMed] [Google Scholar]