

Abstract
We used large samples of expressed sequence tags to characterize the patterns of codon usage bias (CUB) in seven different Citrus species and to analyze their evolutionary effect on selection and base composition. We found that A- and T-ending codons are predominant in Citrus species. Next, we identified 21 codons for 18 different amino acids that were considered preferred codons in all seven species. We then performed correspondence analysis and constructed plots for the effective number of codons (ENCs) to analyze synonymous codon usage. Multiple regression analysis showed that gene expression in each species had a constant influence on the frequency of optional codons (FOP). Base composition differences between the proportions were large. Finally, positive selection was detected during the evolutionary process of the different Citrus species. Overall, our results suggest that codon usages were the result of positive selection. Codon usage variation among Citrus genes is influenced by translational selection, mutational bias, and gene length. CUB is strongly affected by selection pressure at the translational level, and gene length plays only a minor role. One possible explanation for this is that the selection-mediated codon bias is consistently strong in Citrus, which is one of the most widely cultivated fruit trees.
Keywords: citrus, codon usage, evolution
Background
The genetic code represents the set of rules by which information is encoded in DNA or mRNA sequences, and is subsequently translated into proteins by living cells. A three-nucleotide codon in a nucleic acid sequence specifies a single amino acid; a total of 61 codons specify only 20 different amino acids. Therefore, the majority of amino acids are represented by more than one codon and the genetic code is redundant. Most amino acids are encoded by two to six different codons. Different organisms show specific preferences for one of the several codons that encode the same amino acid, and hence the codons occur at different frequencies in genes.1–3
The mechanism by which these preferences arise, however, is not clearly understood. Mutation bias may play a role, but the selection driving evolution of codon usage remains unclear and may be species-specific. The quantification of codon usage bias (CUB), especially at the genomic scale, can increase our understanding of the evolution of living organisms. This phenomenon is now recognized as critical in shaping gene expression and cellular function through its effects on diverse processes ranging from RNA processing to protein translation and protein folding.
CUB is more complex in multicellular organisms than in unicellular organisms. Studies in various multicellular eukaryotic organisms have indicated that both mutational bias and selective forces impact codon usage.1 However, consensus on the relative contributions of these effects has yet to be reached. Selection on synonymous codon usage has been shown to occur in very diverse eukaryotes including plants, fungi, and invertebrates. In both Drosophila and Caenorhabditis, many studies have demonstrated that codon bias plays a major role in the selection of highly expressed genes. For example, optimal codons correspond to the most abundant tRNAs.4–7 These observations clearly support translation selection hypothesis that synonymous codon usage has been shaped by selection to improve the efficiency of translation. In vertebrates, human, and Xenopus,7 multivariate analyses reveal that the variability in codon usage is reflected essentially by a single major trend that is correlated strongly with the GC content at the third codon position (GC3s).8,9 In plants, there are a number of studies on Arabidopsis as a self-fertilizing model species for plant11–13 and Populus as a model species for trees.14–17Physcomitrella patens, a moss, is used as a model species to analyze codon usage, plant evolution, development, and physiology.18 In Arabidopsis thaliana, a clear correlation is observed between codon usage and gene expression levels and showed that this correlation is not due to a mutational bias.19 There is a study reported herein which shows that the expression pattern of Arabidopsis tissue-specific genes is an important factor in relation to their synonymous codon usage.1 Whittle et al20 have provided additional evidence of an association between codon bias and expression in plant reproductive organs. In this study, we quantified the relative importance of selective and neutral forces as causes of codon-usage bias within and between Citrus species.
Citrus species constitute one of the major tree fruit crops of the subtropical regions with great economic importance. Citrus is a large genus that includes several major cultivated species, including Citrus sinensis (sweet orange), Citrus reticulate (tangerine and mandarin), Citrus limon (lemon), Citrus grandis (pummelo), and Citrus paradise (grapefruit). It is not clear, however, how closely-related the citrus species are. Citrus taxonomy and phylogeny are very complicated and controversial, mainly due to sexual compatibility between citrus and related genera, the high frequency of bud mutations, and the long history of cultivation and wide dispersion.21 In the present study, more than a half million citrus Expressed Sequence Tags (ESTs) have been obtained and deposited to public databases in recent years.22 These sequences were obtained from various tissues of over 15 citrus accessions, related genera, and hybrids but about 85% of these sequences were derived from four major types: sweet orange (C. sinensis), clementine (C. clementina), mandarin (C. reticulata), and trifoliate orange (Poncirus trifoliata (L.) Raf.). We used these ESTs to characterize patterns of CUB in different Citrus species. We found that selection based on codon usage is widespread in commonly cultivated fruit tree species.
Methods
Data sources
All available ESTs for seven different species of Citrus were downloaded from PlantGDB): C. unshiu (19139 ESTs), C. trifoliata (62344), C. sinensis (203890), C. reticulata (55324), C. limonia (11045), C. aurantium (13668), and C. clementia (118353). The corresponding PlantGDB-assembled unique transcripts (PUTs) for C. unshiu, C. trifoliata, C. reticulata, C. limonia, and C. aurantium were also downloaded. These PUTs are unique transcripts assembled from all mRNA sequences for a given species available in public databases, and have been trimmed to remove bacterial contamination, repetitive sequences, and polyA tails. The PUTs were used as genes in this study, but do not represent full-length transcripts. To minimize sampling errors, only PUTS that are more than or equal to 100 codons and that have correct initial and termination codons were included in the dataset. We wrote C program to complete minimize sampling errors (supplementary file 1).
Codon usage analysis
The patterns of synonymous codon usage were analyzed in seven Citrus genomes. We combined the genes (PUTs) from seven genomes and calculated the relative synonymous codon usage (RSCU) for each gene. Several indices were analyzed using t-tests.
The most straight forward way to measure CUB is simply deviation from even usage. The RSCU23 statistics is calculated by dividing the observed usage of a codon by that expected if all codons were used equally frequently.24 Thus an RSCU of 1 indicates a codon is used as expected by random usage, RSCU > 1 indicates a codon used more frequently than expected randomly, and RSCU < 1 indicates a codon used less frequently than random.
The effective number of codons (ENC) was calculated to quantify the CUB of an open reading frame (ORF),25 which is the best estimator of absolute synonymous CUB.26 The larger the extent of codon preference in a gene, the smaller the ENC value. In an extremely biased gene where only one codon is used for each amino acid, this value would be 20; if all codons are used equally, it would be 61; and if the value of the ENC is greater than 40, the CUB was regarded as a low bias.28 The values of ENC were obtained by CodonW program.
The RSCU for all genes in each species was then calculated as well as the expression category separately using CodonW (J Peden, version 1.4.2 http://codonw.sourceforge.net/).27 The codons which are over-represented in highly expressed genes were then identified by comparing differences in RSCU (ΔRSCU) between high and low bias genes using t-tests using R.37 The major trend in codon usage is selection for optimum translation, and can be used to identify optimal codons. This is achieved by contrasting the codon usage of two groups of genes, composed of the genes that lie at either end of the principal trend (axis 1), the top and bottom 5% of genes (based on axis 1 ordination). Correspondence analysis (COA) of citrus genes generated a principal axis onto which the ordination of each gene was projected. The codon usage of 5% of the total number of genes from the extremes of the principal was pooled. The codon usage of both pools was compared using a two-way Chi squared contingency test to identify optimal codons. For the purposes of this test, the dataset with the lower ENC were putatively assigned as highly expressed. CodonW used ENC to partition genes. Finally, overall indices of codon usage for each of the five different species were calculated as the average of all positive ΔRSCU values (Table 1). Optimal codons have ΔRSCU > 0.3 at P < 0.05.
Table 1.
Differences in relative synonymous codon usage (RSCU) across codons between genes with high and low levels of expression.
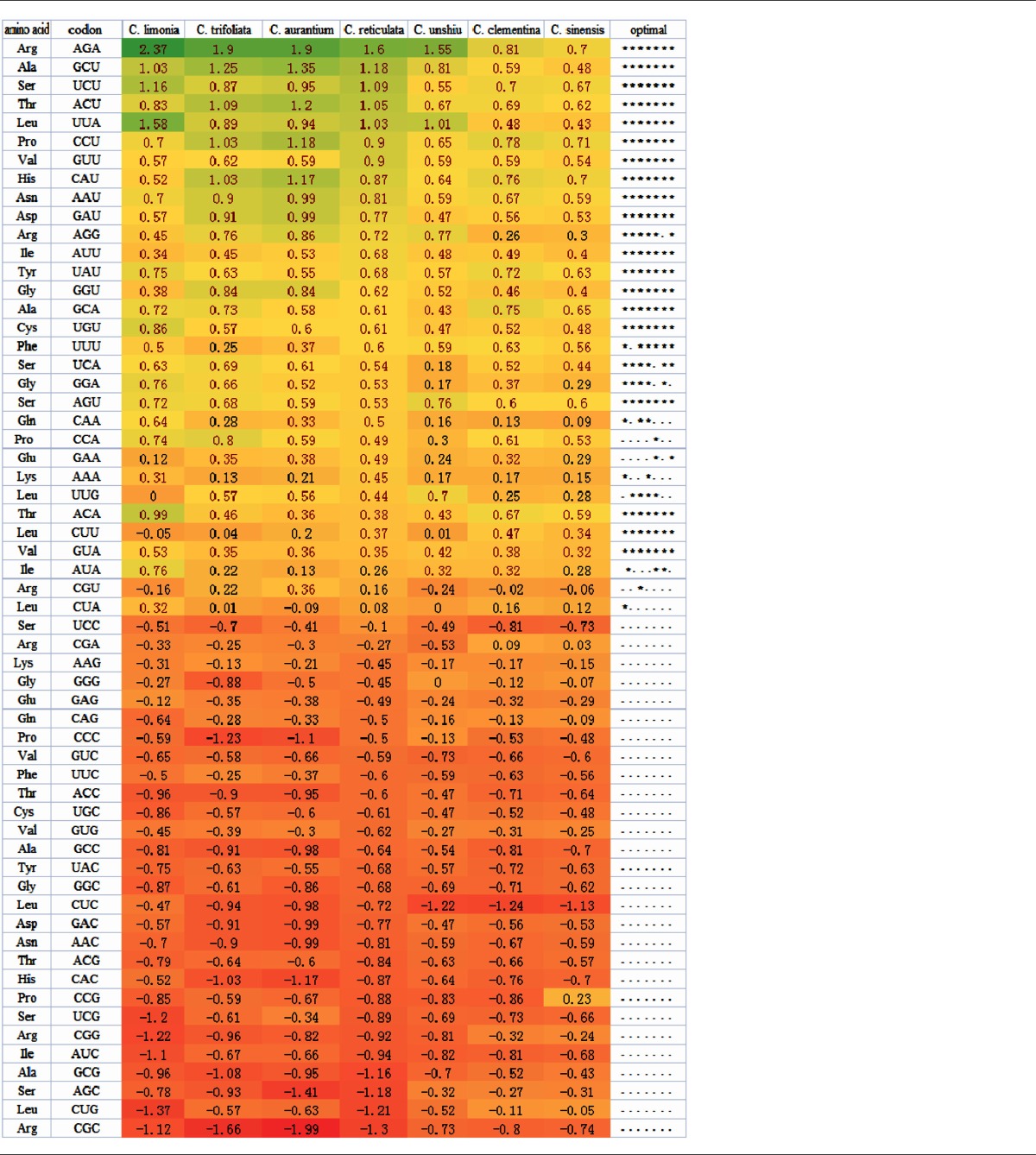
Notes: The right-most column shows codons with significantly increased usage in highly expressed genes, as determined by a t-test (P < 0.05). Each * represents a species for which the t-test was significant, in the order as they are listed in the figure. Codons above the horizontal dotted lines were used to design the optimal codons and were used to calculate frequencies of optimal codon usage (FOP) in all species. The color in the figure indicates the gradient of ΔRSCU values, from the most positive (green) to the most negative (orange).
Correspondence analysis (COA)
Correspondence analysis (COA) is an ordination technique that identifies the major trends in the variation of the data and distributes genes along continuous axes in accordance with these trends. It is conceptually similar to principal component analysis, but applies to categorical rather than continuous data. COA was performed by the values of RSCU in each gene, and was plotted in a 59-dimensional hyperspace according to their usage of the 59 sense codons (excluding Met, Trp, and termination codons). Major variation trends can be determined using these RSCU values and genes ordered according to their positions along the major axis, which can also be used to distinguish the major factors influencing the codon usage of a gene. Generally, the major trend influences codon usage variation among genes and occurs when the variability is more than 10%.30
This index measures the frequency of optimum codons (Fop) in a gene.31 It is a species-specific measure of bias towards particular codons that appear to be translational optimal in a species. It was a simple ratio between the frequency of optimal codons and the total number of synonymous codons.
The codon adaptation index (CAI; high values mean higher CUB and higher expressed level)32 and the frequency of GC at the third synonymously variable coding position, excluding Met, Trp, and termination codons (GC3s), were measured using the CodonW 1.4.2 program. Overall, indices of codon usage for each of the seven species were calculated as the average of all positive ΔRSCU values. We then performed correlation analysis using the Spearman’s rank correlation analysis in the multi-analysis software SPSS version 13.0. (http://codonw.sourceforge.net/). We identified codons based on ΔRSCU.
Orthologous genes
Orthologous groups were identified using the OrthoMCL program33 which can be used to infer orthologous families from multiple genomes. Among the identified families, only those with one-to-one orthology (defined as the core-set genes) relationships from the seven Citrus genomes were included for further analyses. To minimize sampling error, genes less than or equal to 100 codons or those containing internal stop codons were excluded. The core set, including 84 genes (supplementary Fig. 2) across the different species, was further analyzed.
Phylogenetic analysis
Groups of putatively orthologous sequences were aligned using different method (NJ, ME, ML, and MP) with Mega 4.0 software. The reliability of the tree was evaluated using the bootstrap method with 1,000 replications.
Substitution rate calculations
Comparative sequence data for 84 orthologous genes available from seven different species, dN/dS was also calculated for each gene using all available sequences and assuming a constant dN/dS over all branches of the phylogenetic tree (codeml runmode 0, model 1). To minimize sampling error, 33 orthologous gene (identity = 100, dN = 0 and dS = 0) were excluded. All identified 51 orthologous genes were also concatenated within species.
We compared the likelihood of different models of selection acting on different branches of a phylogenetic tree using the program codeml in the PAML package version 4.3. This program utilizes the codon substitution model and a maximum-likelihood method to calculate the likelihood of specified models. Twice, the difference in likelihoods of two models was then compared to a Chi-square distribution, with the degrees of freedom equal to the difference in the number of free parameters between the two models. Model M1 (neutral) assumed two classes of sites: the conserved sites at which ω = 0 and the neutral sites at which ω = 1. Model M2 (selection) added a third class of sites with ω as a free parameter, thus allowing for sites with ω > 1. Model M7 (beta) used a beta distribution B(p, q), which, depending on parameters p and q, can take various shapes (such as L, J, U, and inverted U shapes) in the interval (0, 1). Model M8 (beta and ω) adds an extra class of sites to the beta (M7) model, with the proportion and the ω ratio estimated from the data, thus allowing for sites with ω = 1. From these models, we constructed two likelihood ratio tests (LRTs; Table 4), which compared M1 (neutral) with M2 (selection), and M7 (beta) with M8 (beta and ω), respectively.
Table 4.
Likelihood ratio statistics.
| Comparison | 2Δℓ | df | P | χ21% |
|---|---|---|---|---|
| M1 (neutral) vs. M2 (selection) | 635.54 | 2 | 0.000E + 000 | 9.21 |
| M7 (beta) vs. M8 (beta and v) | 650.86 | 2 | 0.000E + 000 | 9.21 |
The significance of the LRT was usually calculated using the Chi-square approximation, which states that at the asymptote when there is a large amount of data, twice the difference in the log of maximum likelihood between the two models (the likelihood ratio statistic 2Δlogl) was then distributed as a Chi-square distribution with the degrees of freedom (df) given by the difference in the numbers of parameters in the two nested models. For the M1-M2 comparison, df = 2. For the M7-M8 comparison the use of df = 2 is expected to be conservative.
Results
CUB in seven Citrus species
We calculated the average ENC for each species set of complete codons (CDSs). We performed a bootstrap randomization test for CDSs with homolog candidates across all species to determine whether the ENCs for different species differed significantly (Fig. 1). Mean ENC is similar from 51.92 in C. unshiu to 53.75 in C. trifroliata. From this figure we can see the fact that GC is 0.3853 and GC3s is 0.4406. There is an obvious difference.
Figure 1.

Effective number of codons (ENC) as a measure of overall average codon usage bias (CUB) in seven Citrus species. The actual mean ENC, mean GC3s, and mean GC are shown below each bar. 95% confidence bars in standard error of mean are shown. A lower ENC represents greater bias.
Identification of optimal codons
Optimal codons were identified for all seven species based on ΔRSCU between genes with high and low bias (Table 1). The codon usage and RSCU of both datasets is shown (supplementary file 3). COA of seven citrus species generated a principal axis onto which the ordination of each gene was projected. The codon usage of 5% of the total number of genes from the extremes of the principal was pooled. The codon usage of both pools was compared using a two-way Chi squared contingency test. For the purposes of this test dataset with the lower ENC were putatively assigned as highly expressed.
Using this approach, we identified 19 codons with significant ΔRSCU values between genes with low and high levels of expression in all seven species (eg, AGA and GCU). An additional 4 codons showed ΔRSCU values that were significant in four or five species (eg, AGG and UCA). Some codons that did not show significant differences between high and low bias genes had positive ΔRSCU values in all species (eg, AUA and GUA). These are likely optimal codons (based on their positive ΔRSCU values), or their power may be too low to achieve statistical significance in one or a few species. On the other hand, some codons showed reversals of ΔRSCU between high and low bias genes (eg, CCG, UCG, and CGG). Whether these codons truly represent differences in codon preferences between species or simply represent statistical artifacts remains unclear. Based on the ΔRSCU analysis, we identified 21 codons for 19 different amino acids that were used to calculate the frequency of optimal codon usage in all genes across the species (Table 1).
ENC plot
Plotting ENC values against GC3s is one of the most effective ways to explore heterogeneity.25 In Figure 2, the ENC value of each gene is plotted against its corresponding GC3. Due to limited data, C. trifoliata was included in the analysis. The dark solid curve shows the expected position of genes whose codon usage was determined based on variation in GC3 content. If a particular gene is subject to GC compositional constraint to shape codon usage patterns, it will lie on a continuous curve, which represents random codon usage. If a gene is subject to selection for translational optimal codons, it will lie considerably below the expected curve. Although few genes in the seven Citrus species were on the expected curve, several points were under the solid curve. This suggests that these genes were not only subject to GC compositional constraints, but also natural selection. Based on Figure 2, the strength of selection was strong during the evolution of Citrus genome. Furthermore, as shown in Figure 3, the FOP was correlated with gene expression, gene length, and base composition. However, there were no similarities in the ratio of codon usage.
Figure 2.

The ENC plot of Citrus. The continuous curve represents the relationship between GC3s and ENC values under random codon usage.
Figure 3.

Proportion of variation in the frequency of optimal codon usage explained by gene expression, gene length, and base composition at synonymous sites.
Between-subject effects
Principal component analysis and tests of between-subject effects were used to investigate the dependent variable Fop in codon bias as a function of gene expression (CAI), base composition (GC3s), and sequence length. We use the software EVIEW6.0 to handle data in the seven files (supplementary file 4). First, we found the natural logarithm of four sequences (Fop, CAI, GC3S, and L_SYM), then the linear regression of FOP to the other three parameters was determined (supplementary file 5). The regression results can be found in supplementary file 6.
Figure 3 shows that in each species, expression level explained a significant amount of variation in codon bias. No obvious difference in gene expression was found among each species. For example, the radio of gene expression for C. limonia was 48.9117%, while it was 45.168% for C. aurantium. However, difference in GC3s was apparent. For C. sinensis, GC3s was found to be only 20.3695%, while that for C. trifoliata was 35.5777%. The influence of gene length was very low; the maximum was 5.0189% for C. limonia and the minimum was 0.1975% for C. sinensis. Thus, the influence of selection on the evolution of Citrus was significant.
Phylogeny of the seven species
The molecular phylogenetic trees were constructed by using the Nei-Gojobori method, Maximum Parsimony method, Maximum likelihood method, and Minimum Evolution method with Mega software. These trees were consistent across different substitution models and tree inference methods, all of which yielded the same trees, although the support for the tree topology in Figure 4 was not particularly strong without 100% bootstrap support for all branches when using the concatenated data. In addition, the tree in Figure 4 was also consistent with earlier phylogenetic study of the genus citrus using micro satellite (SSR) based markers.42
Figure 4.

Prediction of the ω value of each branch during evolutionary processes. Unrooted tree representing the phylogenetic relationship between the seven species. ML estimates non-synonymous (dN) and synonymous (dS) substitution rates, dN/dS ratios (in parentheses), and the maximum likelihood estimates of selection acting on preferred codons are shown above each branch and are calculated from the concatenated data set of 84 genes. Branch lengths are proportional to the synonymous substitution rate.
Because synonymous mutations were undetectable during natural selection, while non-synonymous mutations were under strong selective pressure, comparing the fixation rates of these two types of mutations is a powerful way to explore the effects of natural selection on the evolution of molecular sequences. Measurement of the non-synonymous/synonymous substitution rate ratio (ω = dN/dS), also known as the acceptance rate, is commonly used.28 This rate is an important indicator of selective pressure at the protein level, where ω = 1 represents neutral mutations, ω < 1 represents purifying selection, and ω > 1 represents diversifying positive selection. We used the codeml program of the PAML software to analyze ω (Fig. 4), which showed that most of the seven Citrus plants experienced positive selection. Using all available sequences and assuming constant dN/dS over all branches of the phylogenetic tree, we calculated average dN/dS values of 0.9479, 1.0257, 0.9123, 1.0151, 1.7285, 1.3131, and 0.6184 for C. aurantium, C. clementina, C. limonia, C. unshiu, C. sinensi, C. reticulate, and C. trifoliata, respectively. C. sinenesis in particular is the most common species used for citrus production globally. Its annual production accounts for two-thirds of total citrus production. Thus, in China, C. sinenesis is the most important artificially selected strain for citrus cultivation. However, C. aurantium development has a ω value of 0.9749, which represetns less cultivation, relatively. C. trifoliate, with a ω value of 0.61839, is rarely used for artificial selection as rootstock, and is more commonly used for purifying selection.
Table 3 lists parameter estimates and log-likelihood values under models of variable ω ratios among sites. Both two models that allow for the presence of positively selected sites (ie, M2 (selection) and M8 (beta and ω)) do suggest the presence of such sites (Table 3). Allowing for the presence of positively selected sites (with ω > 1) improves the fit of the models significantly. For example, the neutral model (M1) does not allow for sites with ω > 1. The selection model (M2) adds an additional site class, with the ω ratio estimated to be 4.514. The log-likelihood improvement was huge, as seen when 2Δℓ = 191.90 is compared with χ2 1% = 9.21 with df = 2 (Table 4). M8 involves more parameters than M7, and the LRT statistic 2Δℓ = 650.86 is much greater than the critical value χ2 1% = 9.21 with df = 2 (Table 2). The results suggest extreme variation in positive selection for 51 orthologous genes concatenated within species (see Methods). Table 3 lists sites inferred to be under positive selection under different models at the 95% cutting point.
Table 3.
Parameter estimates and log-likelihood values under models of variable ratios among sites.
| Model | P | LnL | Kappa | Estimates of parameters | Positively selected sites | |
|---|---|---|---|---|---|---|
| M1 | Nearly neutral | 1 | −64600.24 | 1.55928 | P0= 0.20008 | Not allowed |
| ω0= 0.19552 | ||||||
| P1= 0.79992 | ||||||
| ω1= 1.00000 | ||||||
| M2 | Positive selection | 3 | −64282.47 | 1.72982 | P0= 0.06407 | 1Q 5N 9L 15D 51E 632K 670R 685G 883R 886G 929S |
| ω0= 0.00000 | 1031W 1037A 1353E 1384R 1388S 1394H 1400A 1413D | |||||
| P1= 0.77815 | 1418L 1737E 1738R 1742C 1760V 1769S 1770R 1787V | |||||
| ω1= 1.00000 | 1973R 1976R 1992A 1995S 2266L 2272N 2280L 2283S | |||||
| P2= 0.15778 | 2285S 2298V 2304P 2305F 2308N 2352R 2362R 2363H | |||||
| ω2= 4.51408 | 2390T 2 2444S 2457L 2498L 2499F 2500F 2502K 2503N | |||||
| 2514S 2516C 2522Y 2535L 2570S 2587R 2660C 2662M | ||||||
| 2666L 2667K 2668A 2669M 2671T 2672S 2673S 2677L | ||||||
| 2678G 2679L 2680Q 2684K 2685P 2686F 2691H 2735W | ||||||
| 2743L 2748K 2756M 2818W 2821S 2822H 2823C 2827L | ||||||
| 2829G 2838A 2839V 2964K 3012L 3013R 3016S 3017N | ||||||
| 3018S 3019L 3023S 3027P 3032P 3034P 3036A 3037A | ||||||
| 3040F 3050F 3054L 3491P 3495V 3774I 3775P 3777Q | ||||||
| 3778R 3780Y 3793T 3808Y 3810Y 3816L 3830L 3832S | ||||||
| M7 | Beta | 2 | −64609.64 | 1.56036 | P = 0.45889 | Not allowed |
| q = 0.08925 | ||||||
| M8 | Beta and ω | 4 | −64284.21 | 1.71973 | P0= 0.84072 | 1Q 5N 9L 15D 51E 181M 632K 670R 685G 883R 886G |
| P = 0.27207 | 929S 1031W 1037A 1264T 1353E 1384R 1388S 1394H | |||||
| q = 0.03464 | 1400A 1413D 1418L 1737E 1738R 1742C 1760V 1769S | |||||
| (P1= 0.15928) | 1770R 1787V 1973R 1976R 1984R 1992A 1995S 2266L | |||||
| ω = 4.34756 | 2272N 2280L 2283S 2285S 2298V 2304P 2305F 2308N | |||||
| 2352R 2362R 2363H 2390T 2 2444S 2457L 2498L 2499F | ||||||
| 2500F 2502K 2503N 2514S 2516C 2522Y 2535L 2570S | ||||||
| 2587R 2660C 2662M 2666L 2667K 2668A 2669M 2671T | ||||||
| 2672S 2673S 2677L 2678G 2679L 2680Q 2684K 2685P | ||||||
| 2686F 2691H 2735W 2743L 2748K 2756M 2817T 2818W | ||||||
| 2819M 2821S 2822H 2823C 2827L 2829G 2838A 2839V | ||||||
| 2964K 3012L 3013R 3016S 3017N 3018S 3019L 3023S | ||||||
| 3027P 3032P 3034P 3036A 3037A 3040F 3050F 3054L | ||||||
| 3491P 3495V 3774I 3775P 3777Q 3778R 3780Y 3793T | ||||||
| 3808Y 3810Y 3816L 3830L 3832S |
Notes: P represents the number of free parameters in the ω-distribution. Sites inferred to be under positive selection at the 99% level are bold and those at the 95% level are in italic.
Table 2.
Optimal codon table in Citrus sinensis.
| Amino acid | Codon |
ΔRSCU
|
Frequency per thousand bases
|
|---|---|---|---|
| From EST | From CDS | ||
| Ala | GCU | 0.59 | 29.3 |
| GCA | 0.75 | 20.6 | |
| GCC | −0.81 | 15.9 | |
| GCG | −0.52 | 8.3 | |
| Arg | AGA | 0.81 | 14.9 |
| AGG | 0.26 | 14.9 | |
| CGU | −0.02 | 5.5 | |
| CGA | 0.09 | 4.8 | |
| CGG | −0.32 | 4.4 | |
| CGC | −0.8 | 4.6 | |
| Gly | GGU | 0.46 | 19.7 |
| GGA | 0.37 | 18.7 | |
| GGG | −0.12 | 14 | |
| GGC | −0.71 | 17.2 | |
| His | CAU | 0.76 | 12.4 |
| CAC | −0.76 | 10.6 | |
| Val | GUU | 0.59 | 27.6 |
| GUA | 0.38 | 8.3 | |
| GUC | −0.66 | 11.5 | |
| GUG | −0.31 | 21.2 | |
| Lys | AAA | 0.17 | 25.7 |
| AAG | −0.17 | 34 | |
| Phe | UUU | 0.63 | 23.3 |
| UUC | −0.63 | 21 | |
| Pro | CCU | 0.78 | 16.8 |
| CCC | −0.53 | 11.2 | |
| CCG | −0.86 | 7.3 | |
| CCA | 0.61 | 16 | |
| Thr | ACU | 0.69 | 18.5 |
| ACA | 0.67 | 15.1 | |
| Asn | AAU | 0.67 | 24.8 |
| AAC | −0.67 | 21.7 | |
| Asp | GAU | 0.56 | 33.8 |
| GAC | −0.56 | 18.6 | |
| Cys | UGU | 0.52 | 8.5 |
| UGC | −0.52 | 8.8 | |
| Gln | CAA | 0.13 | 18.7 |
| CAG | −0.13 | 16.5 | |
| Glu | GAA | 0.32 | 28.6 |
| GAG | −0.32 | 31.3 | |
| Ile | AUU | 0.49 | 24.2 |
| AUA | 0.32 | 12.6 | |
| AUC | −0.81 | 16.4 | |
| Leu | UUA | 0.48 | 12.3 |
| UUG | 0.25 | 22.4 | |
| CUU | 0.47 | 25.2 | |
| CUA | 0.16 | 8.4 | |
| CUC | −1.24 | 14.6 | |
| CUG | −0.11 | 13 | |
| Met | AUG | 0 | 0 |
| Tyr | UAU | 0.72 | 15.4 |
| UAC | −0.72 | 13.9 | |
| Ser | UCU | 0.7 | 17.7 |
| UCA | 0.52 | 17 | |
| AGU | 0.6 | 11.2 | |
| UCC | −0.81 | 9.8 | |
| UCG | −0.73 | 9.1 | |
| AGC | −0.27 | 12 | |
| Thr | ACC | −0.71 | 10.6 |
| ACG | −0.66 | 6.9 |
Notes: ΔRSCU: Relative synonymous codon usage in predicted genes with high and low gene expression levels based on the EST sequence in Citrus sinensis. Optimal codons (red box) were identified based on differences in relative synonymous codon usage. Frequency per thousand bases: use frequency per thousand bases in identified high-confidence coding sequences from full-length cDNA in Citrus sinensis. Optimal codons (green box) were identified based on different frequencies per thousand bases (P < 0.05).
Discussion
ESTs were used to estimate the accuracy of our data. Our CDS sequences obtained using EST electronic splicing was used for follow-up analysis. To verify the accuracy of our data, we used the known 165 full-length CDS sequence, including 47126 codons in the whole genome sequence of C. sinensis, to perform high-frequency codon analysis (http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=2711). As shown in Table 2, genes with high expression levels and significant increases in codon usage were common. Only a small number of optimal codons from the EST were present at the same frequency as the most common codons. These results are complicated because we used different data sets and different methods to acquire the data. However, our EST data analysis was reliable.
Only several plants have been completely sequenced, and more less plants are available for comparative analysis. Therefore, it is difficult to determine the total lengths of CDSs. In this study, we performed electronic splicing based on EST data to obtain CDS sequences for further analysis. However, the accuracy of this analysis was limited. Based on a comparison (T2) with C. sinensis, the codons of highly expressed genes based on full-length splicing and high usage codons (using full-length CDSs for direct calculation of the frequency) were similar.
As shown in Figure 2, biological selection played a major role (approximately 50%) in Citrus evolution. Compared to Populus15 and nematodes,34 the data for Citrus are highly ordered, and changes in gene expression, GC3s, and gene length had a lesser influence on the evolution of different species. In Populus, there were large differences among species in the amount of variation in codon usage explained by gene expression, ranging from 2.6% in P. trichocarpa to 16.9% in P. deltoids.15 We can see the consistently strong selection-mediated codon bias among Citrus species, possibly because Citrus have been cultivated over time. In addition, Codon W used ENC to partition genes rather than a more direct measure of gene expression in the case of COA. In some studies, optimal codons have been defined as those codons which occur more often (relative to their synonyms) in highly expressed genes, compared with lowly expressed genes.35 We used a modification of this definition, where optimal codons are defined as those levels.36 Significance is assessed by a two-way chi square contingency test with the criterion of P < 0.01. The advantage of this test is that differences in codon usage between highly and lowly expressed genes caused by random noise are suppressed.
Adding to the positive selection test (Table 3), we also found that there is evidence that ADP-ribosylation factor gene examined for positive selection have a class of sites with ω > 1 (evidence is not published), and ADP-ribosylation factor gene appears to contain some sites under putative positive selection. We speculate that it may be due to histone modification regulating citrus tree growth and response to environmental stimuli by the ADP-ribosylation (ADP-ribosylation) factor during evolutionary in citrus.
Codon usage is related to carrier genetic (DNA) and functional (protein) information. Thus, these unique coding strategies make studies on molecular evolution challenging.37 Variation in codon usage is represented by two major paradigms and is determined by either mutational bias or selection pressure. A unified theory for codon usage has not been determined as different species have different models. CUB in mammals and vertebrates is more strongly influenced by differential mutation pressure.24 The primary determinant of codon bias in human RNA viruses38 and plant viruses39 is mutational pressure, and not translational selection. On the other hand, in fast-growing organisms with large population sizes, such as Escherchia coli and Saccaromyces cerevisiae, codon usage is generally under selective pressure. Codon optimization results in more rapid translation rates and increased accuracy. As a result, translational selection is stronger in highly expressed genes, as is the case for the abovementioned organisms. In addition, codon usages in both Drosophila and Caenorhabditis are correlated with gene expression, with highly expressed genes having strongly biased codon usage. This is presumably due to increased selective pressure. In the present study, we found that Citrus experienced strong selective pressure via domestication of various species at several sites for extended periods of time. Naturally, Citrus may have originated as a bitter fruit plant, possibly in what is now the Malay Archipelago.40 The modern fruit species probably evolved in China, where there is a greater diversity of Citrus varieties and associated parasites than anywhere else in the world. The hybridization of pummelos and mandarin oranges in environments such as mixed Chinese gardens created both C. sinensis and C. aurantium. The multitude of natural hybrids and cultivated varieties, including spontaneous mutants, obscure the history of Citrus. However, as one of the world’s most widely cultivated fruit trees, selection pressure has played a role over an extended period of time.
Conclusion
Variation in codon usage among Citrus genes is influenced by translational selection, mutational bias, and gene length. CUB is strongly affected by selection pressure at the translational level. Base mutations also play an important role, while gene length has only a minor influence. It is possible that selection-mediated codon bias is consistently strong in Citrus, which is one of the most widely cultivated fruit trees.
Footnotes
Author Contributions
Produced and analyzed the data: CX, JD. Drafted the manuscript: CX, XG. Obtained funding: QZ. Designed the study: CT. Made critical revisions and approved the final version: QW. All authors reviewed and approved of the final manuscript.
Funding
This work was supported by the National Science Foundation of China (No. 31170561) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
Supplementary materials
Supplementary File 1
C program to minimize sampling errors
Supplementary File 2
The 84 orthologous genes of the core set used for further analysis
Supplementary File 3
The codon usage and RSCU of both datasets
Supplementary File 4
Result files from a COA created by CodonW
Supplementary File 5
Result files from EVIEW6.0 software
Supplementary File 6
The linear regression of FOP to the other parameters
References
- 1.Plotkin JB, Kudla G. Synonymous but not the same: The causes and consequences of codon bias. Nat Rev Genet. 2011;12(1):32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plotkin JB, Dushoff J, Desai MM, Fraser HB. Codon usage and selection on proteins. J Mol Evol. 2006;63(5):635–53. doi: 10.1007/s00239-005-0233-x. [DOI] [PubMed] [Google Scholar]
- 3.Camiolo S, Farina L, Porceddu A. The relation of codon bias to tissue-specific gene expression in Arabidopsis thaliana. Genetics. 2012;192(2):642–9. doi: 10.1534/genetics.112.143677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moriyama EN, Powell JR. Codon usage bias and tRNA abundance in Drosophila. J Mol Evol. 1997;45(5):514–23. doi: 10.1007/pl00006256. [DOI] [PubMed] [Google Scholar]
- 5.Duret L. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet. 2000;16(7):287–9. doi: 10.1016/s0168-9525(00)02041-2. [DOI] [PubMed] [Google Scholar]
- 6.Percudani R. Restricted wobble rules for eukaryotic genomes. Trends Genet. 2001;17(3):133–5. doi: 10.1016/s0168-9525(00)02208-3. [DOI] [PubMed] [Google Scholar]
- 7.Kanaya S, Yamada Y, Kinouchi M, Kudo Y, Ikemura T. Codon usage and tRNA genes in eukaryotes: correlation of codon usage diversity with translation efficiency and with CG-dinucleotide usage as assessed by multivariate analysis. J Mol Evol. 2001;53(4–5):290–8. doi: 10.1007/s002390010219. [DOI] [PubMed] [Google Scholar]
- 8.Scora RW. On the history and origin of Citrus. Bulletin of the Torrey Botanical Club. 1975;102(6):369–75. [Google Scholar]
- 9.Musto H, Cruveiller S, D’Onofrio G, Romero H, Bernardi G. Translational selection on codon usage in Xenopus laevis. Mol Biol Evol. 2001;18(3):1703–7. doi: 10.1093/oxfordjournals.molbev.a003958. [DOI] [PubMed] [Google Scholar]
- 10.Wright SI, Iorgovan G, Misra S, Mokhtari M. Neutral evolution of synonymous base composition in the Brassicaceae. J Mol Evol. 2007;64(1):136–41. doi: 10.1007/s00239-005-0274-1. [DOI] [PubMed] [Google Scholar]
- 11.Wright SI, Lauga B, Charlesworth D. Rates and patterns of molecular evolution in inbred and outbred Arabidopsis. Mol Biol Evol. 2002;19(9):1407–20. doi: 10.1093/oxfordjournals.molbev.a004204. [DOI] [PubMed] [Google Scholar]
- 12.Wright SI, Lauga B, Charlesworth D. Subdivision and haplotype structure in natural populations of Arabidopsis lyrata. Mol Ecol. 2003;12(5):1247–63. doi: 10.1046/j.1365-294x.2003.01743.x. [DOI] [PubMed] [Google Scholar]
- 13.Wright SI, Yau CB, Looseley M, Meyers BC. Effects of gene expression on molecular evolution in Arabidopsis thaliana and Arabidopsis lyrata. Mol Biol Evol. 2004;21(9):1719–26. doi: 10.1093/molbev/msh191. [DOI] [PubMed] [Google Scholar]
- 14.Zhou M, Tong CF, Shi JS. A preliminary analysis of synonymous codon usage in poplar species. Zhi Wu Sheng Li Yu Fen Zi Sheng Wu Xue Xue Bao. 2007;33(4):285–93. [PubMed] [Google Scholar]
- 15.Ingvarsson PK. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol Biol Evol. 2007;24(3):836–44. doi: 10.1093/molbev/msl212. [DOI] [PubMed] [Google Scholar]
- 16.Ingvarsson PK. Molecular evolution of synonymous codon usage in Populus. BMC Evol Biol. 2008;8:e307. doi: 10.1186/1471-2148-8-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingvarsson PK. Natural selection on synonymous and nonsynonymous mutations shapes patterns of polymorphism in Populus tremula. Mol Biol Evol. 2010;27(3):650–60. doi: 10.1093/molbev/msp255. [DOI] [PubMed] [Google Scholar]
- 18.Rensing SA, Fritzowsky D, Lang D, Reski R. Protein encoding genes in an ancient plant: analysis of codon usage, retained genes and splice sites in a moss, Physcomitrella patens. BMC Genomics. 2005;6:e43. doi: 10.1186/1471-2164-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci U S A. 1999;96(8):4482–7. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whittle CA, Malik MR, Krochko JE. Gender-specific selection on codon usage in plant genomes. BMC genomics. 2007;8:e169. doi: 10.1186/1471-2164-8-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlini DB, Stephan W. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics. 2003;163(1):239–43. doi: 10.1093/genetics/163.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delseny M, Han B, Hsing YI. High throughput DNA sequencing: the new sequencing revolution. Plant Sci. 2010;179(5):407–22. doi: 10.1016/j.plantsci.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 23.Sharp PM, Li WH. Codon usage in regulatory genes in Escherichia colidoes not reflect selection for ‘rare’ codon. Nucleic Acids Res. 1986;14(19):7737–49. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharp PM, Lloyd AT. In: An Atlas of Drosophila Genes. Maroni G, editor. New York, NY: Oxford University Press; 1993. pp. 378–97. [Google Scholar]
- 25.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–9. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 26.Comeron JM, Aguade M. An evaluation of measures of synonymous codon usage bias. J Mol Evol. 1998;47(3):268–74. doi: 10.1007/pl00006384. [DOI] [PubMed] [Google Scholar]
- 27.Peden JF. Analysis of codon usage. University of Nottingham; 2000. [Google Scholar]
- 28.Messier W, Stewart CB. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385(6612):151–4. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- 29.Anders FL. The ‘effective number of codons’ revisited. Biochem Biophys Res Commun. 2004;317(3):957–64. doi: 10.1016/j.bbrc.2004.03.138. [DOI] [PubMed] [Google Scholar]
- 30.Greenacre MJ. Theory and Applications Of Correspondence Analysis. Academic Press; London: 1984. [Google Scholar]
- 31.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 1981;151(3):389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 32.Sharp PM, Li WH. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–95. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cutter AD, Charlesworth B. Selection intensity on preferred codons correlates with overall codon usage bias in Caenorhabditis remanei. Curr Biol. 2006;16(20):2053–7. doi: 10.1016/j.cub.2006.08.067. [DOI] [PubMed] [Google Scholar]
- 35.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli system. J Mol Biol. 1981;151(3):389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 36.Lloyd AT, Sharp PM. Codon usage in Aspergillus nidulans. Mol Gen Genet. 1991;230(1–2):288–94. doi: 10.1007/BF00290679. [DOI] [PubMed] [Google Scholar]
- 37.Grantham R, Perrin P, Mouchiroud D. Patterns in codon usage of different kinds of species. Oxford Surveys in Evolutionary Biology. 1986;3:48–81. [Google Scholar]
- 38.Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003;92(1):1–7. doi: 10.1016/s0168-1702(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins GM, Pagel M, Gould EA, Zanotto PMA, Holmes EC. Evolution of base composition and codon usage bias in the genus Flavivirus. J Mol Evol. 2001;52(4):383–90. doi: 10.1007/s002390010168. [DOI] [PubMed] [Google Scholar]
- 40.McPhee J. Oranges. Farrar, Straus and Giroux; 1967. [Google Scholar]
- 41.Yang Z, Nielsen R, Hasegawa M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol Biol Evol. 1998;5(12):1600–11. doi: 10.1093/oxfordjournals.molbev.a025888. [DOI] [PubMed] [Google Scholar]
- 42.Jannati M, Fotouhi R, Abad AP. Genetic diversity analysis of Iranian citrus varieties using micro satellite (SSR) based markers. J Horticulture Forestry. 2009;1(7):120–5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1
C program to minimize sampling errors
Supplementary File 2
The 84 orthologous genes of the core set used for further analysis
Supplementary File 3
The codon usage and RSCU of both datasets
Supplementary File 4
Result files from a COA created by CodonW
Supplementary File 5
Result files from EVIEW6.0 software
Supplementary File 6
The linear regression of FOP to the other parameters