

Abstract
Neuropathology after traumatic brain injury (TBI) is the result of both the immediate impact injury and secondary injury mechanisms. Unresolved post-traumatic glial activation is a secondary injury mechanism that contributes to a chronic state of neuroinflammation in both animal models of TBI and human head injury patients. We recently demonstrated, using in vitro models, that p38α MAPK signaling in microglia is a key event in promoting cytokine production in response to diverse disease-relevant stressors and subsequent inflammatory neuronal dysfunction. From these findings, we hypothesized that the p38α signaling pathway in microglia could be contributing to the secondary neuropathologic sequelae after a diffuse TBI. Mice where microglia were p38α-deficient (p38α KO) were protected against TBI-induced motor deficits and synaptic protein loss. In wild-type (WT) mice, diffuse TBI produced microglia morphological activation that lasted for at least 7 d; however, p38α KO mice failed to activate this response. Unexpectedly, we found that the peak of the early, acute phase cytokine and chemokine levels was increased in injured p38α KO mice compared with injured WT mice. The increased cytokine levels in the p38α KO mice could not be accounted for by more infiltration of macrophages or neutrophils, or increased astrogliosis. By 7 d after injury, the cytokine and chemokine levels remained elevated in injured WT mice but not in p38α KO mice. Together, these data suggest that p38α balances the inflammatory response by acutely attenuating the early proinflammatory cytokine surge while perpetuating the chronic microglia activation after TBI.
Introduction
Traumatic brain injury (TBI) represents a spectrum of pathological conditions, which range from focal to diffuse, and from mild to severe (Saatman et al., 2008). Regardless of the cause of the injury, fundamental to the survival of the organism is a repair process that is remarkably conserved among different tissues and in response to different injuries (Gurtner et al., 2008). Inflammation is central to the classical described repair process and, as such, is critical for health of the organism. However, inflammation that is inefficient, excessive, or prolonged can result in infection, edema, or reactive oxygen species and produce collateral damage, which persists after the primary injury. Therefore, understanding the role of inflammation in the multicellular repair process after a brain injury is of great medical and biological importance. The consequences of even a mild brain injury can be long-lasting and increase the risk of dementia (Uryu et al., 2007; Gavett et al., 2010; Johnson et al., 2012). Associated with the secondary disease process after a TBI is persistent microglia activation lasting for more than a decade (Ramlackhansingh et al., 2011; Giunta et al., 2012). Microglia, the resident CNS macrophage, respond within 30 min to a brain injury (Davalos et al., 2005; Nimmerjahn et al., 2005). Once the microglia respond to chemoattractants and damage-associated molecular patterns, the microglia release inflammatory mediators (e.g., IL-1β, TNFα, and nitric oxide). The inflammatory mediators and damage-associated molecular patterns act on cytokine receptors and conserved pattern recognition receptors (e.g., Toll-like receptors) to initiate an acute phase response (Morganti-Kossmann et al., 2007; Chen and Nunez, 2010). Phenotypic subpopulations of activated microglia, based on a molecular signature of gene expression, are characterized as a classical proinflammatory state (M1 microglia) or an alternatively activated state (M2 microglia) associated with anti-inflammatory and wound healing functions (Colton, 2009; David and Kroner, 2011).
Central to linking the inflammatory stimuli in microglia to the cellular response is the mitogen-activated protein kinase (MAPK) p38α. The expression of p38α is ubiquitous; however, recent studies have shown that p38α has unique functions depending on cell, tissue, and disease stimulus. In experimental autoimmune encephalomyelitis, p38α in dendritic cells, but not T cells or macrophages, is necessary for the disease progression by regulating a TH17 response (Huang et al., 2012). In microglia, p38 has been shown to be important for phagocytosis of β-amyloid and axon debris (Reed-Geaghan et al., 2009; Hosmane et al., 2012) as well as production of IL-1β and TNFα (Bachstetter et al., 2011). However, the role of p38α in the repair process after a brain injury has not been defined. We used a diffuse brain injury model without contusion, cavitation, or tissue destruction, but with profound microglia activation (Cao et al., 2012). We report here that p38α regulates the microglia morphological activation and limits the acute phase cytokine response to the diffuse TBI.
Materials and Methods
Animals.
The Institutional Animal Care and Use Committee of the University of Kentucky approved the use of animals in this study, which was conducted in accordance with the principles of animal care and experimentation in the Guide for the Care and Use of Laboratory Animals. To generate p38α conditional knock-out mice, the first exon of the p38α gene (MAPK14) was flanked by two loxP sites and then crossed with mice that contain Cre driven by the lysozyme promoter, as previously described (Kang et al., 2008; Bachstetter et al., 2011; Xing et al., 2011). The restricted cell-type expression of the lysozyme promoter (Clarke et al., 1996; Clausen et al., 1999) results in cell-specific deletion of p38α MAPK in myeloid cells, including microglia, as previously validated (Bachstetter et al., 2011; Xing et al., 2011). The mice (all backcrossed to C57 Bl/6) were maintained and bred as follows: p38αfl/fl wild-type and not carrying the Cre allele (p38α WT) were bred with Lys-Cre hemizygotes. This generates litters where ∼50% of the mice are microglia/macrophage conditional p38α KO and ∼50% are p38αfl/fl littermates (used as WT controls). All experiments with p38α mice used both sexes of mice at ∼50:50 ratio. Genotyping was performed by Transnetyx. Male C57BL/6J mice were obtained from The Jackson Laboratory.
Midline fluid percussion injury (mFPI).
The animals were subjected to mFPI as previously described (Lifshitz, 2008). Briefly, 3- to 4-month-old male and female mice were used for the experiment. To prepare for the mFPI, mice were anesthetized with 5% isoflurane before stabilizing the head using ear bars in a stereotaxic frame (David Kopf Instruments). Anesthesia was maintained with continuous inhalation of isoflurane (3.5%, 1 L/min). A midline incision was made in the scalp to expose the skull. A 3.0-mm-diameter craniotomy was made using a trephine along the sagittal suture, midway between bregma and lambda. Mice were excluded from the experiment if dura was breached during the craniotomy. The top portion of the Luer-Loc hub of a 20-gauge needle (BD Biosciences) was cut away, beveled, scored, and affixed over the craniotomy site using cyanoacrylate. Dental acrylic was applied around the hub to provide stability during the injury induction. After the dental acrylic hardened, a Luer-Loc cap was applied to the hub and sutures were applied where necessary to close the skin around the dental acrylic head stage. Mice were given 16–24 h to recover from the craniotomy before injury.
Before injury, mice were anesthetized for 5 min with 5% isoflurane in a small induction chamber. Then, the Luer-loc cap was removed, and the hub was filled with normal saline and attached to the male end of the fluid percussion device, ensuring that no air bubbles were introduced into the system. Once a normal breathing pattern resumed, before sensitivity to stimulation, a pressure pulse was generated by the fluid percussion device (Custom Design and Fabrication). An injury of moderate severity (1.2 ± 0.05 atm) was administered by releasing the pendulum onto the fluid filled piston, moments before the reflexive responses returned. After injury, the hub assembly was removed en bloc. The injury site was inspected, and the incision was sutured. The time to right was recorded. The mice were placed in a cage with a warming pad and monitored until normal movement and grooming resumed. Sham mice were subjected to all the experimental and surgical conditions described above, except that the fluid percussion injury was not administered.
Rotarod.
Personnel blinded to animal groups measured the distance mice traveled using the Rotor-Rod system from San Diego Instruments. Animals were pretrained (60 s at 4 rpm for 3 trials) on the rotarod for two consecutive days before the craniotomy. Testing began at 24 h after injury and was repeated for the next 6 d. For the test, the animal was placed on the rod with the starting speed set to 4 rpm, and the speed was then continuously accelerated up to 25 rpm over 5 min. When the animal fell off the rod breaking the photo beam, the instrument recorded latency to fall and total distance traveled. Two trials were run daily for each animal, and the mean of the two trials was plotted.
Death and brain tissue harvesting.
Mice were killed by sodium pentobarbital overdose and transcardially perfused with ice-cold PBS for 5 min. The brains were rapidly removed. On ice, the brain was bisected along the sagittal plane with one hemisphere further dissected and flash frozen in liquid nitrogen and stored at −80°C for subsequent biochemical evaluation. The other hemisphere was immersion fixed in 4% paraformaldehyde for 14–16 h, before cryoprotection in a 30% sucrose/PBS solution.
Biochemical endpoint analyses.
For protein measurements, a PBS fraction and a detergent fraction were generated from neocortical tissue using a high shear homogenizer (Omni TH115). The PBS fraction was prepared by homogenizing the cortical tissue in a 1:10 (weight/volume) of ice-cold freshly prepared lysis buffer consisting of 1 μg/ml Leupeptin, 1 mm PMSF, and 1 mm ethylenediaminetetraacetic acid. The cortical homogenate was centrifuged at 12,000 × g for 20 min at 4°C in a Beckman Microfuge 18, and supernatants collected for measurement of cytokines. A detergent-solubilized fraction was prepared by resuspending and rehomogenizing the pellet in a detergent-containing lysis buffer (tissue protein extraction reagent with Halt protease and phosphatase inhibitor mixture; Thermo Scientific) at the same volume used for first homogenization. Homogenates were cleared by centrifugation at 12,000 × g for 20 min at 4°C, and supernatants were used for measurement of synaptic proteins.
Cytokine levels were measured as previously described (Bachstetter et al., 2011; Bachstetter et al., 2012) using Meso Scale Discovery (MSD) custom multiplex high-sensitivity ELISA kits according to the manufacturer's instructions with minor modifications. Briefly, 25 μl of the PBS fraction supernatant was loaded per well of the MSD plate (∼100 μg of protein). The sample was incubated at 4°C for ∼14 h. All incubation steps were done using an Eppendorf MixMate at 1000 rpm. Cytokine levels were normalized to the total amount of protein in the sample loaded as determined by BCA Protein Assay (Thermo Scientific).
Synaptic proteins levels were measured by immunoblots as previously described (Bachstetter et al., 2012). Blots were probed using reagents and manufacturer recommendations for Odyssey Infrared imaging system (LI-COR Biosciences). The detergent-solubilized fraction (1 μg) was prepared in SDS buffer (LI-COR), separated on a 10% NuPAGE Novex Bis-Tris Midi Gel (Invitrogen), and transferred to nitrocellulose membrane using the iBlot system (Invitrogen). Blots were probed with the following primary antibodies: rabbit anti-PSD95 (1:2000, Cell Signaling Technology, catalog #3450); mouse anti-synaptophysin (clone SY38, 1:30,000, Millipore, catalog #MAB5258); rabbit anti-syntaxin 1 (1:100,000, Millipore, catalog #ab5820), mouse anti-SNAP 25 (clone 20) (1:10,000, BD Biosciences, catalog #610366). Rabbit anti-GAPDH (clone 14C10) (1:10,000, Cell Signaling Technology, catalog #2118) was used as a loading control. The Odyssey Infrared imaging system (LI-COR Biosciences) and integrated intensity values were used in statistics.
For gene expression, dissected neocortical tissues stored at −80°C were used for RNA isolation using RNeasy mini-columns (QIAGEN, catalog #74104) with on-column DNase treatment (QIAGEN, catalog #79254) according to the manufacturer's protocol. RNA quantity and quality were determined using A260/A280 readings by NanoDrop (Thermo Scientific). Reverse transcription (RT) was done following the manufacturer's protocol using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, catalog #4368814). A no template and a no RT control were conducted to control for contamination. Real-time PCR was performed using the TaqMan Gene Expression assay kit (Applied Biosystems, catalog #4444964) according to the manufacturer's instructions on a ViiA 7 Real-Time PCR System (Applied Biosystems). The following TaqMan probes (Applied Biosystems) were used: IL-1β (Mm00434228_m1), IL-6 (Mm00446190_m1), TNFα (Mm00443258_m1), CCL2 (Mm00441242_m1), CCL3 (Mm00441258_m1), CD45 (Mm01293575_m1), CD68 (Mm00839636_g1), MHCII (Mm00439216_m1), arg1 (Mm00475988_m1), YM1 (Mm00657889_mH), GLAST (Mm00600697_m1), GLT-1 (Mm00441457_m1), GFAP (Mm00546086_m1), S100B (Mm00485897_m1), VIM (Mm01333430_m1), and 18S (Hs99999901_s1). Relative gene expression was calculated by the 2−ΔΔCT method.
Immunohistochemistry (IHC).
Using a sliding microtome with a freezing stage, we collected serial coronal sections (30 μm) of the hemi-brain through the entire hemisphere and stored the sections in cryoprotectant at −20°C. Staining procedures were conducted on free-floating sections using every 12th section through the entire hemisphere. Primary and secondary antibodies were diluted in 3% normal goat serum (LAMPIRE Biological Laboratories, catalog #7332500) with 0.2% Triton X-100. Endogenous peroxidase activity was quenched with 3% H2O2 in methanol, before the tissue blocking in 10% normal goat serum with 0.2% Triton X-100. Primary antibodies used included the following: rabbit anti-GFAP (1:10,000, Dako, catalog #Z0334); rabbit anti-IBA1 (1:10,000, Wako, catalog #019-19741); rat anti-CD45 (YW62.3) (1:10,000, Thermo Scientific, catalog #MA1447081); rat anti-CD68 (1:5000, Serotec catalog #MCA1957T); rat anti-Ly6G/C (GR1) (1:800, BD Biosciences PharMingen catalog #550291); and rabbit anti-p-p38 (1:1000, Cell Signaling Technology, catalog #9211). For the detection of GFAP and IBA1, an HRP-conjugated goat anti-rabbit IgG was used. For all other primary antibodies, a biotinylated secondary antibody was amplified in avidin-biotin substrate (ABC kit, Vector Laboratories). All sections were developed in 0.5 mg/ml 3,3-diaminobenzidine tetrahydrochloride solution (Sigma, catalog #D5637). The tissue sections were dehydrated through gradients of ethyl alcohol and finally xylene. The sections were then coverslipped with Permount Mounting Medium (Fisher Scientific).
Quantitative image analysis of IHC.
As previously described (Bachstetter et al., 2012), the Aperio ScanScope XT digital slidescanner was used to image the entire stained slide at 20× magnification to create a single high-resolution digital image. The neocortex was outlined using the Aperio ImageScope software. The Aperio-positive pixel count algorithm (version 9) was used to quantify the amount of specific staining in the region. The number of positive pixels was normalized to the number of total pixels (positive and negative) to account for variations in the size of the region sampled. Color and intensity thresholds were established to detect the immunostaining as positive pixels and background staining as negative pixels. To quantify the number of IBA1+ clusters of microglia, the Aperio nuclear algorithm (version 9). We operationally defined microglia clusters as groups of cells where the processes of the cells were overlapping, and with a total area covered by cells of >1000 μm. To this end, a threshold was set in the nuclear algorithm, so only clusters of microglia >1000 μm, regardless of curvature or roundness, would be quantified. We empirically found that these settings would detect multicellular groups of microglia (>4) while not detecting individual microglia. Once conditions were established for an immunohistochemical stain, the entire batch of slides was analyzed with the same parameters. The resulting color markup of the analysis was confirmed for each slide. Because of the limited number of GR1+ and p-p38+ cells, the Aperio ImageScope software was not used. GR1+ cells were manually counted in the cortex, and the number of cells was then averaged by the number of sections. For p-p38 quantification, three or four randomly selected fields of the neocortex (−1.5 to −2.5 mm bregma) were collected for each animal using a Nikon Eclipse 90i upright microscope equipped with a Nikon DS-Ri1 digital camera, and the number of positive cells were assessed using the Nikon Elements BR software package, according to previously described methods (Sudduth et al., 2012). Personnel blind to the experimental conditions performed all quantifications.
Statistics.
Statistical analysis was conducted using GraphPad prism software, version 5 (GraphPad Software). Groups of two were compared by unpaired t test. One-way ANOVA followed by Bonferroni's multiple-comparison test was used for comparisons among three or more groups, unless otherwise indicated. Statistical significance was defined as p < 0.05. Values are expressed as mean ± SEM.
Results
Myeloid-specific deletion of p38α reduces TBI-induced motor deficits
We have previously reported using in vitro assays that microglia p38α is critical for the production of proinflammatory cytokines and microglia-mediated neuronal death (Xing et al., 2011); however, the relative in vivo contribution of microglia p38α to a brain injury is unknown. To test the role of microglia p38α in vivo, we used a model of diffuse TBI, which causes a traumatic axonal injury throughout the mediodorsal neocortex, hippocampus, and dorsolateral thalamus, without the formation of compression, contusion, or tissue loss (Kelley et al., 2006; Kelley et al., 2007). There is leakage of the blood–brain barrier in the model (Kelley et al., 2007), but the lack of a focal necrotic core allowed us to address the role of microglia response to trauma with only a limited influx of circulating adaptive and innate immune cells. We compared the injury-induced responses in WT mice and in p38α conditional KO mice where p38α is selectively deficient in cells of the myeloid linage: neutrophils/macrophages/microglia (p38α KO mice) (Kang et al., 2008; Bachstetter et al., 2011; Hume, 2011; Xing et al., 2011).
To assess motor function, we used the rotarod task, which is highly sensitive to vestibulomotor deficits associated with brain injury (Hamm, 2001). Adult mice were trained for 2 d on the rotarod task until all mice met the criterion of 1 min at 4 rpm. From day 1 to day 7 after the diffuse brain injury, mice were tested on an accelerating rotarod (0–25 rpm over 5 min). As shown in Figure 1A, injured WT mice showed a significant motor impairment after injury compared with the uninjured sham mice, whereas the p38α KO mice did not exhibit any significant motor impairment after injury.
Figure 1.
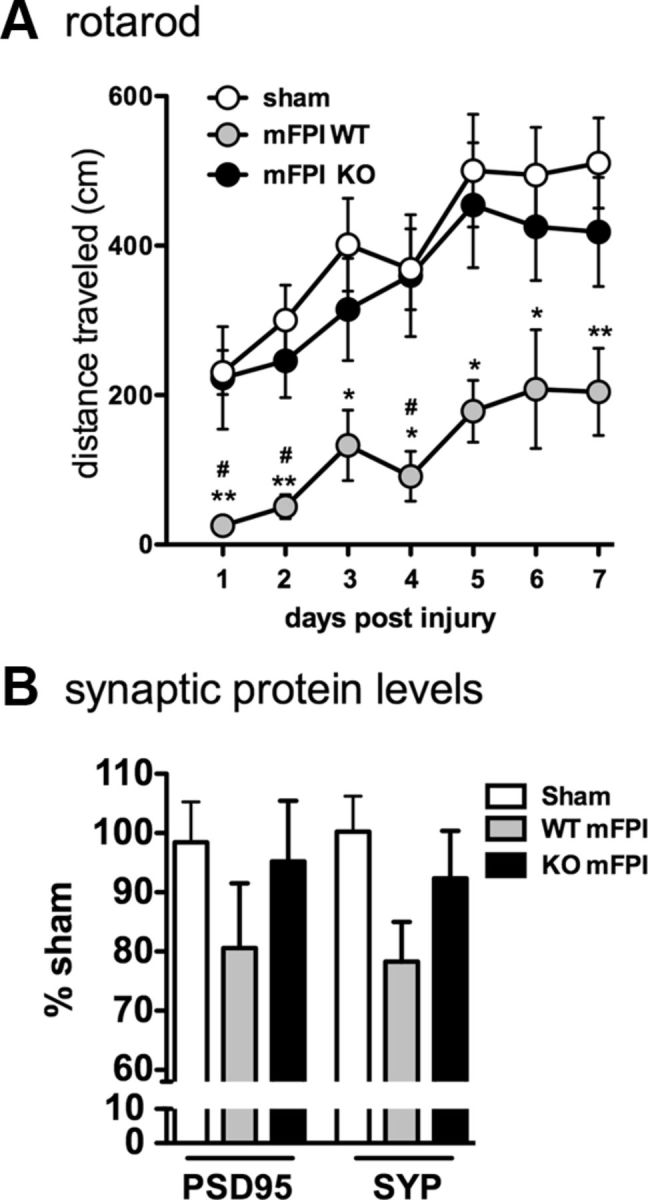
Myeloid-specific deletion of p38α protects mice from diffuse brain injury-induced vestibulomotor impairments and synaptic protein loss. A, Injured WT mice (mFPI WT) demonstrated significant impairments in rotarod performance compared with sham-injured mice and mFPI p38α KO mice. The injured KO mice were not significantly different from the sham mice. B, Although the differences did not reach statistical significance, at 7 d after injury, levels of the synaptic proteins PSD95 and synaptophysin (SYP) were decreased in the WT mice compared with sham, whereas the injury-induced decrease was not seen in the KO mice; n = 8–14 per group. *p < 0.05, sham versus mFPI WT. **p < 0.01, sham versus mFPI WT. #p < 0.05, mFPI WT versus mFPI KO.
Synaptic protein levels in the cortex 7 d after injury were quantified to determine whether the observed motor deficit was also associated with synaptic loss. We found that there was a decrease in PSD95 and synaptophysin protein levels after the diffuse brain injury in the WT mice, although the decrease in protein levels did not reach statistical significance as determined by a one-way ANOVA (Fig. 1B). Interestingly, the injured microglia p38α KO mice showed no decrease in synaptic protein levels and were indistinguishable from sham controls (Fig. 1B).
Inflammatory cytokine and chemokine response peaks during the first 9 h after diffuse brain injury
To determine the temporal changes in the inflammatory response after diffuse brain injury, we subjected male C57BL/6J mice to mFPI and then measured by real-time PCR a panel of cytokine, chemokine, microglia, and astrocyte markers in the cortex at select time points after the injury (Fig. 2). Cytokines and chemokines were found to peak between 3 h and 9 h after the injury (Fig. 2A). Two markers associated with the alternative activation state (M2) microglia phenotype, arg1 and YM1, were found to be strongly increased by 9 h; however, by 7 d, the expression of the M2 markers was below the sham level (Fig. 2B). The diffuse brain injury had a limited effect on astrocyte markers, with the exception of GFAP and vimentin (VIM) (Fig. 2C).
Figure 2.
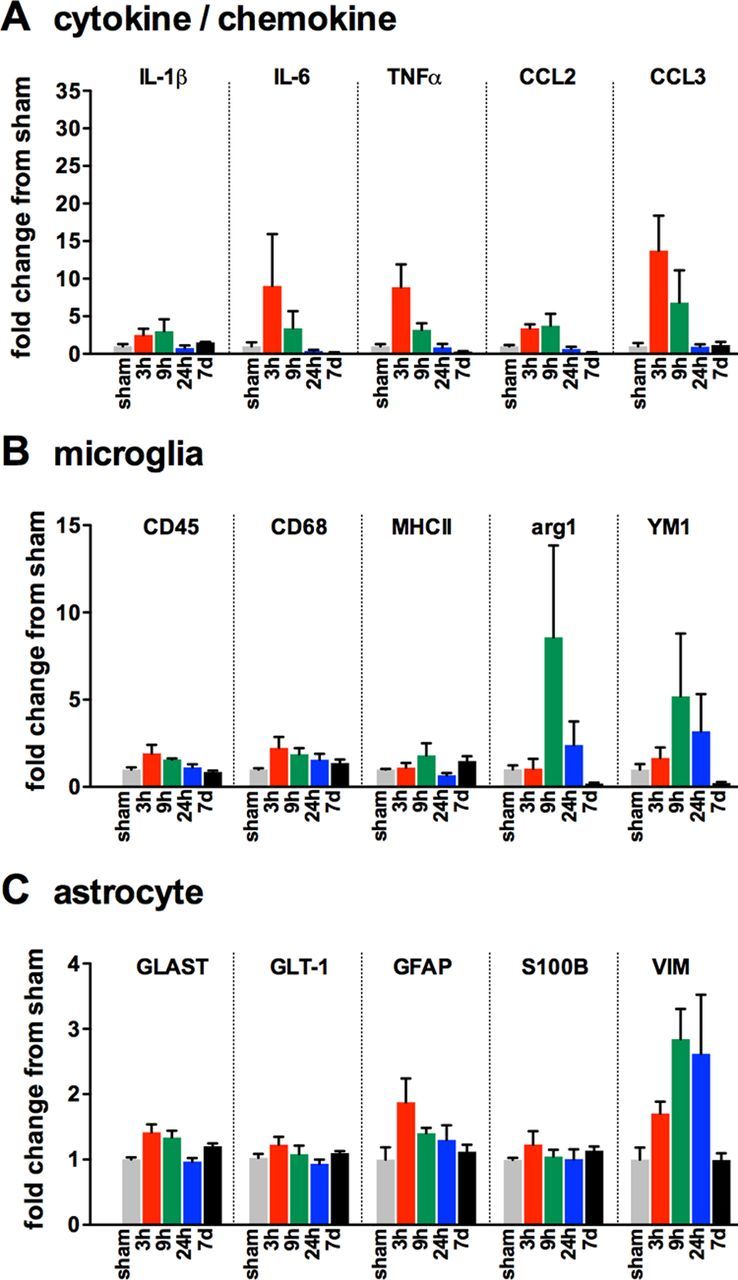
Changes in gene expression associated with glial activation after diffuse brain injury. Temporal changes in cytokine/chemokine (A), microglia (B), and astrocyte markers (C) were measured in the cortex of male C57BL/6J mice subjected to sham conditions (gray), or at 3 h (red), 9 h (green), 24 h (blue), or 7 d (black) after mFPI; n = 3 per time point.
Microglia continue to express p-p38α for at least 7 d after a diffuse brain injury
Our previous work strongly implicates p38α in the IL-1β and TNFα response from microglia evoked by inflammatory stimuli (Bachstetter et al., 2011). As the peak cytokine response was found to occur between 3 to 9 h after the diffuse brain injury, we predicted that, at ∼6 h after the injury, we would see elevated p-p38α in the injured brain. Figure 3A shows the result of p-p38 IHC in mice 6 h and 7 d after the mFPI. In the WT mFPI mice, p-p38+ cells were found throughout the injured cortex at 6 h, with a similar number of p-p38+ cells found at 7 d after the diffuse TBI. In contrast, p-p38+ cells were nearly absent in the p38α KO mFPI mice (Fig. 3B). IHC for p-p38 is not specific for cell type or p38 isotype. However, the observation that the p38α KO mice show essentially no p-p38 staining further validates the KO and provides support that, after a diffuse TBI, it is myeloid cells, presumably microglia, and the α isoform of p38 that is responsible for the p-p38 increase seen after injury.
Figure 3.

The increase in p-p38 after diffuse brain injury is associated with myeloid cells. A, p-p38 IHC was seen in the WT mFPI mice at 6 h and 7 d post-injury (p.i.) but was nearly absent in the sham-injured mice and the p38α KO mFPI mice. B, Quantification shows a significant increase in the number of p-p38+ cells in the injured WT mice at 6 h, which persists to 7 d after injury. Injured p38α KO mice show little to no p-p38 immunoreactivity and are not different from sham-injured mice at either time point (n = 6–12 per group). *p < 0.05. **p < 0.01.
Microglia activation in response to a diffuse TBI is dependent on p38α MAPK
Microglia, the resident tissue immune effector cells of the CNS, rapidly respond to tissue trauma by altering their morphology and moving to the site of injury (Davalos et al., 2005). The intracellular signal transduction that regulates this response is not fully known. We have previously reported in rats that the mFPI model induces a classical activation of microglia using IBA1 (Cao et al., 2012). This activation of microglia after mFPI without tissue loss provided us with the opportunity to probe the intracellular pathways involved in the microglia morphological response using an in vivo disease relevant injury model. Figure 4A shows the result of IBA1 IHC in mice 6 h and 7 d after the mFPI. In the sham-injured mice, a uniform spatial distribution of the IBA1+ microglia, characteristic of quiescent phenotype, was seen in the sham mice at both time points. After the brain injury in WT mice, there was a strong morphological activation of the IBA1+ cells, including large multicellular groups, or clusters, of microglia. Quantification of the intensity of the IBA1 immunoreactivity throughout the neocortex demonstrated a significant increase in IBA1 staining in the injured WT mice compared with the injured p38α KO mice (Fig. 4B). IHC for CD68, a macrophage marker associated with phagocytic cells (da Silva and Gordon, 1999; Kurushima et al., 2000), also clearly showed an upregulation at 6 h and 7 d after injury in WT mice (Fig. 4C). In contrast, the p38α KO mice failed to show increased CD68+ staining after the injury (Fig. 4D).
Figure 4.

p38α KO microglia fail to respond morphologically to a diffuse brain injury. A, IBA1 IHC shows a strong increase in staining and morphological alterations in the WT mFPI mice post-injury (p.i.), which were absent in the KO mFPI mice 6 h after injury and remained diminished in the KO mice 7 d after injury. Arrows indicate clusters of microglia around what appears to be a blood vessel. B, Quantification shows a significant increase in IBA1 staining in the injured WT mice at 6 h, which persists to 7 d after injury. Microglia in the KO mice demonstrated a failure to activate during the acute (6 h) or chronic phase (7 d) after the injury. C, CD68 IHC further supports the lack of a microglia morphological response to the diffuse brain injury in the absence of p38α, both at acute time points and during the chronic phase after the injury. D, Quantification shows a significant increase in CD68 staining in the WT-injured mice at 6 h that persisted to 7 d after injury, with minimal response in the KO mice. Data are presented as percentage of sham; n = 6–8 per group. *p < 0.05. **p < 0.01.
As shown in Figure 5, by 6 h after injury in the WT mice, a strong increase in the IBA1 immunoreactivity was found, the uniform microglia distribution was lost, and the cells formed clusters where the individual cell processes become indistinguishable by light microscopy. In some instances, the morphology of the microglia took on a rod shape perpendicular to the cortex (Fig. 5A,B) (Cao et al., 2012; Ziebell et al., 2012). At times, the clusters of microglia appeared to take on the shape of a layer-V pyramidal neuron (Fig. 5C). Microglia in the WT mice at 6 h after injury were also found to have processes with large swellings (Fig. 5D). In the p38α KO mice, at 6 h after injury, these unusual microglia morphologies were not observed (Fig. 5E,F). Instead, microglia in the injured p38α KO mice were found to have thin highly ramified processes similar to the sham mice (Fig. 5G), and in stark contrast to the injured WT mice. Using the Aperio nuclear algorithm, we quantified the number of microglia clusters 6 h after injury and found that the p38α was necessary for the microglia to form multicellular clusters (Fig. 5H–J).
Figure 5.
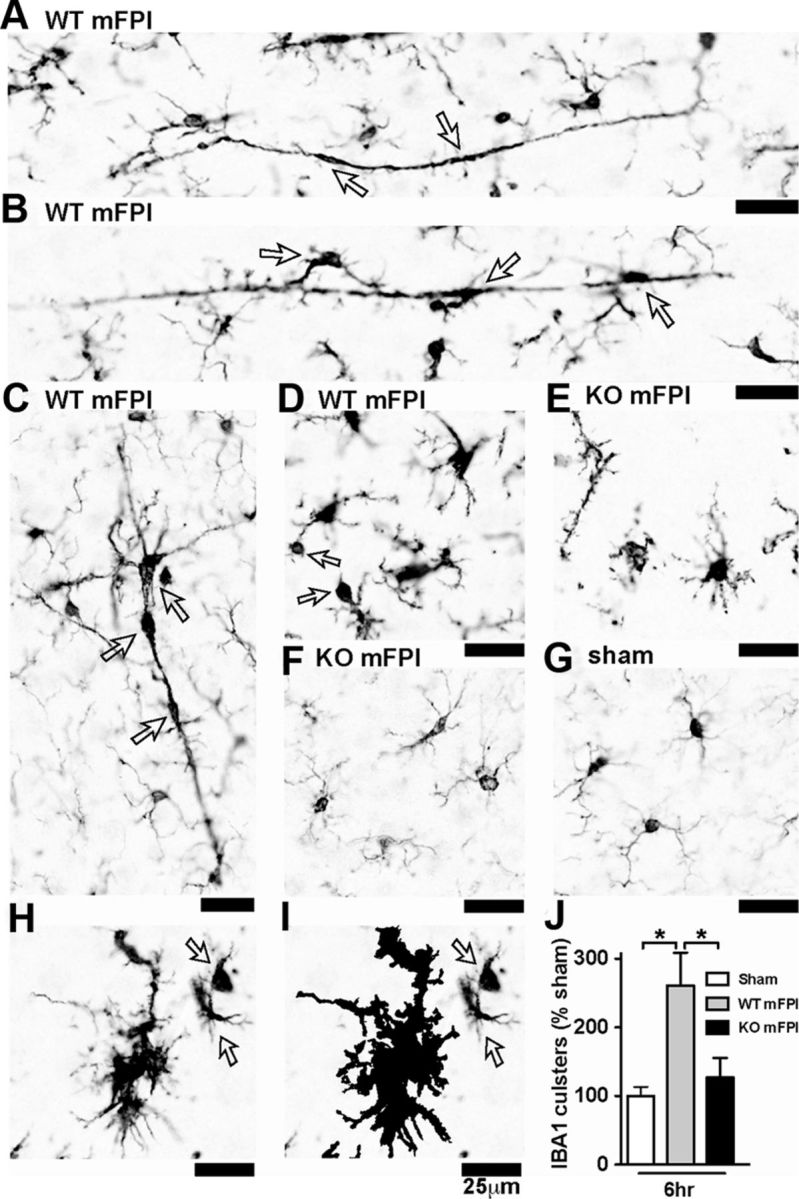
Microglia clustering is dependent on p38α KO after a diffuse TBI. A–D, IBA1 IHC shows the spectrum of microglia activation seen in the WT mice 6 h after the diffuse brain injury. Note the long, rod-shaped microglia (A), trains of multiple microglia in plane with the axonal columns in the cortex (B), and multiple microglia in the shape of a layer-V pyramidal cell (C), as well as IBA1+ cells with retracted processes with large swellings (D). In contrast, the p38α KO mice exhibited thin ramified microglia processes (E) or looked morphologically similar (F) to sham-injured mice (G). A cluster of microglia from a WT mFPI mouse shows the IBA1 staining (H), and the Aperio nuclear algorithm generated markup shows the reliable detection of the microglia clusters (demonstrated here as the black markup) and not individual cells (arrows) (I). Quantification shows a significant increase in IBA1 clusters in the WT-injured mice at 6 h after injury (J). Data are presented as percentage of sham; n = 6–8 per group. *p < 0.05.
Suppression of the acute proinflammatory response after a diffuse TBI requires p38α in myeloid cells
Previously, we have found, in a diffuse TBI model, that a surge of proinflammatory cytokines occurs within the first 12 h, which is important for subsequent cognitive impairments (Lloyd et al., 2008). Using in vitro assays, we have also reported that p38α is a key intracellular signaling kinase in microglia involved in the production of proinflammatory cytokines (Bachstetter et al., 2011). Therefore, we hypothesized that microglia p38α is critical for the cytokine surge after a diffuse brain injury. However, unexpectedly and contrary to our hypothesis, we found elevated levels of the proinflammatory cytokines IL-1β, IL-6, and TNFα (Fig. 6A) and the chemokines CCL2, CCL5, and CXCL1 (Fig. 6B) in the cortex of p38α KO mice 6 h after the injury compared with the sham-injured and WT mFPI mice. Interestingly, by 7 d after the injury, the levels of IL-1β remained significantly elevated in the WT mFPI mice but were back to basal in the p38α KO mFPI mice (Fig. 6A). The levels of the other cytokines and chemokines were also low to undetected by 7 d after injury in WT and p38α KO mice.
Figure 6.

Cytokine and chemokine levels are acutely enhanced in the p38α KO mice after a diffuse brain injury. The protein levels of a panel of inflammatory cytokines (A) and chemokines (B) were measured in the neocortex by MSD multiplex immunoassay. The injured p38α KO mice were found to have significantly enhanced cytokine response at 6 h; but by 7 d after injury, the cytokine response in the KO mice was below (IL-1β) or equal to (IL-6) the response of the WT-injured mice. The chemokines were also significantly increased at 6 h in the p38α KO mice; and by 7 d, the levels were near baseline; n = 6–12 per group. *p < 0.05. **p < 0.01. ***p < 0.001. N.D., Not detected.
Gene expression changes reveal increased M1 cytokine/chemokine and heightened M2 response in the p38α KO mice at 6 h after injury
To begin to explore the mechanism by which the acute proinflammatory response is increased in the KO mice, we measured a panel of cytokine, chemokine, microglia, and astrocyte markers by real-time PCR. As shown in Figure 7A, a significant increase was seen in the expression of proinflammatory cytokines IL-1β, TNFα, and the chemokines CCL2 and CCL3 in the cortex of p38α KO mice 6 h after the injury compared with the injured WT and sham-injured mice, replicating the changes in cytokines and chemokines seen at the protein level. By 7 d after the injury, the levels of IL-1β expression in the KO mice were found to be significantly less than that of the WT mice (Fig. 7A). Interestingly, we also found that, at 6 h after the injury, KO mice had a more robust M2 response, as shown by elevated expression of the M2 markers arg1 and YM1, although the increase did reach statistical significance as determined by a one-way ANOVA (Fig. 7B). Whether these effects are solely from microglia remains to be shown. Few changes were seen in the expression of astrocyte markers after the diffuse brain injury at either time point. However, we did find that, at 6 h after the injury, the expression of GFAP and VIM was significantly increased in the KO mice compared with the WT mice (Fig. 7C).
Figure 7.
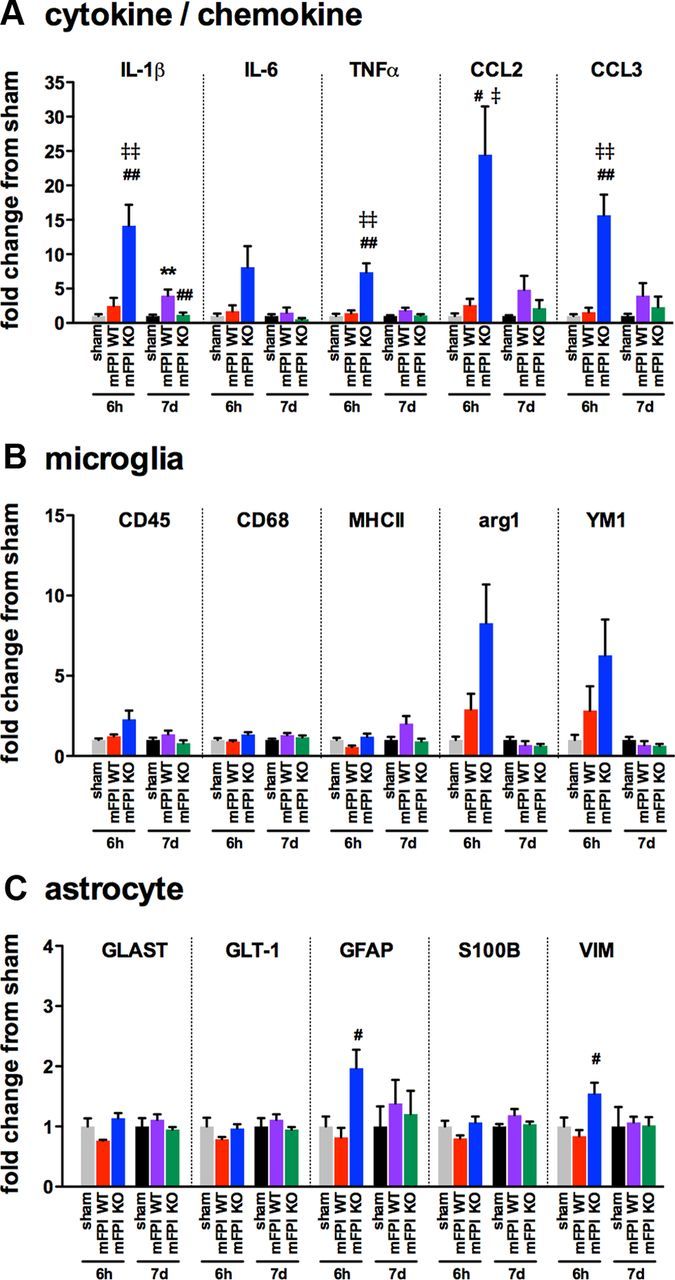
Enhanced cytokine and chemokine gene expression is seen in the p38α KO mice at 6 h after diffuse brain injury. Temporal changes in cytokine/chemokine (A), microglia (B), and astrocyte markers (C) were measured from the cortex harvested from sham mice (gray represents 6 h; black, 7 d), injured WT mice (red, 6 h; purple, 7 d), or injured p38α KO mice (blue, 6 h; green, 7 d); n = 4–10 per group. **p < 0.01, sham versus mFPI WT. #p < 0.05, mFPI WT versus mFPI KO. ##p < 0.01, mFPI WT versus mFPI KO. ‡p < 0.05, sham versus mFPI KO. ‡‡p < 0.01, sham versus mFPI KO.
Astrogliosis remains intact in p38α KO mice after a diffuse TBI
As we found increased expression of GFAP and VIM in the p38α KO mice, we sought to determine whether there might be an increasing astrogliosis that may account for the acute increase in cytokines seen in the p38α KO mice. Therefore, we performed IHC for GFAP, a marker of astrocytes, which in the mouse is normally found expressed at low levels in the cortex. As shown in Figure 8, in the sham-injured mice, very little GFAP staining was seen in the cortex. The diffuse TBI caused a strong astrogliosis, which was most evident at 7 d after injury when injured mice showed a robust increase in GFAP staining regardless of genotype (Fig. 8A). Quantification of staining in the cortex demonstrated that the p38α KO mice had significantly less GFAP staining at 6 h after injury compared with the injured WT mice. By 7 d after injury, there was no detectable difference in GFAP staining between the injured WT or p38α KO mice (Fig. 8A,B).
Figure 8.
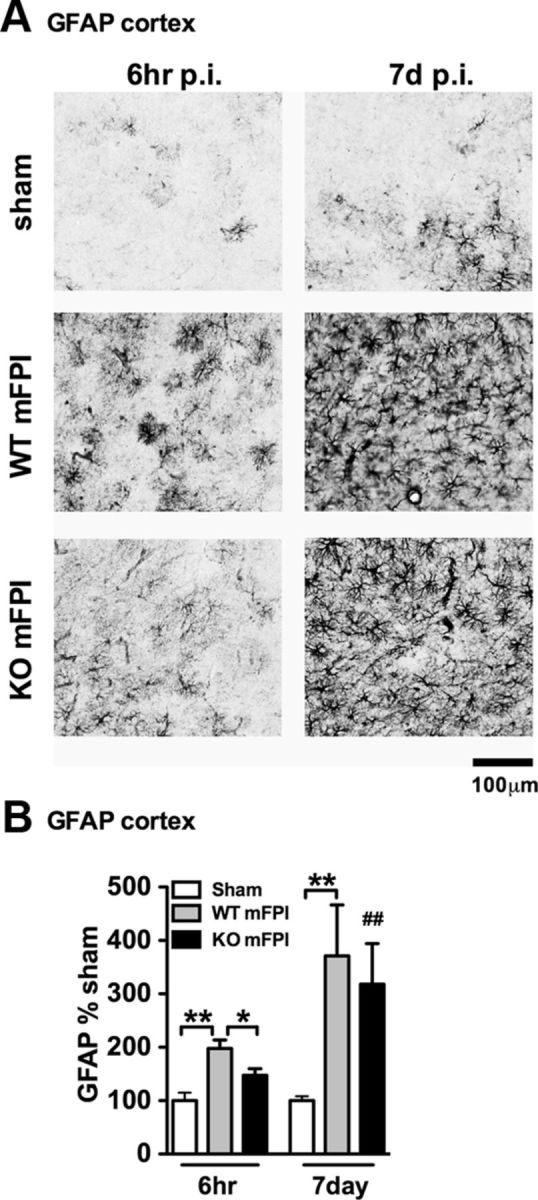
There is intact astrogliosis in myeloid-deficient p38α KO mice after injury. A, GFAP IHC shows a strong increase in staining and morphological alterations in the WT mFPI mice 6 h after injury, which was not as prominent in the p38α KO mFPI mice. By 7 d after injury, WT and KO mice showed similar GFAP staining. B, Quantification shows a significant increase in GFAP at 6 h after injury in WT mice but not KO mice. At 7 d, there was no difference in astrogliosis between the WT and KO injured mice. Data are presented as percentage of sham (n = 6–12 per group). *p < 0.05. **p < 0.01. ##p < 0.01, sham versus mFPI KO (t test).
The number of infiltrating innate immune cells do not account for the increase in cytokines seen in the KO mice after a diffuse TBI
CD45 is a transmembrane protein tyrosine phosphatase that is expressed on all nucleated hematopoietic cells (Penninger et al., 2001). The expression of CD45 on resident microglia is low; but upon stimulation, the expression of CD45 on microglia is upregulated (Sedgwick et al., 1991), and high expression of CD45 as determined by flow cytometry is associated with infiltrating macrophages (Mizutani et al., 2012). Figure 9A shows representative photomicrographs of the CD45 IHC. A significant increase in CD45 staining was seen in the cortex of injured WT mice, but a much more limited increase in CD45 staining in the p38α KO mice (Fig. 9B).
Figure 9.

Myeloid-deficient p38α KO mice have a limited increase in CD45, but similar changes in GR1+ monocytes/neutrophils, compared with WT mice. A, Representative photomicrographs of CD45+ IHC in the cortex 6 h post-injury (p.i.). B, A significant increase in CD45+ staining was found in the cortex of WT mFPI mice compared with sham-injured mice, whereas p38α KO mice showed very little increase in CD45+ staining. C, The number of GR1+ cells were counted in the cortex and presented as the average number of GR1+ cells per coronal tissue section. WT and KO mice showed similar numbers of GR1+ cells after injury (n = 6–12 per group). *p < 0.05. **p < 0.01. ***p < 0.001.
Previous studies have suggested that macrophages are necessary to limit the number of neutrophils after trauma (Lucas et al., 2010), by regulating neutrophil recruitment and clearance (Soehnlein and Lindbom, 2010). As a myeloid cell, neutrophils would also be deficient in p38α in the KO mouse model; therefore, we quantified the number of GR1+ cells in the cortex to determine whether there was an increase in the number of neutrophils. We found a significant increase (p < 0.05) in the number of GR1+ cells in the cortex after diffuse brain injury in WT mice; however, there was no difference between the injured WT or p38α KO mice in the number of GR1+ cells (Fig. 9C). This suggests that neutrophil recruitment after the diffuse brain injury is independent of p38α, as we do not see a difference in the number of GR1+ cells. In contrast, the activation and/or recruitment of macrophage/microglia after the diffuse brain injury is p38α-dependent, as there is less CD45 staining in the KO mice. Our data clearly show that the numbers of CD45+ cells and GR1+ cells are either decreased or the same in the p38α KO mice; therefore, an increased number of infiltrating immune cells is not the mechanism for the increase in cytokines seen in the KO mice. However, it should be noted that determining the number of CD45+ cells and GR1+ cells alone does not rule out the possibility that the activation state and/or ratio of the infiltrating monocytes/macrophages versus brain-resident microglia may differ in the p38 WT or KO mice and that there may be qualitative differences in the responsiveness caused by the lack of p38α in the cells that do infiltrate.
Discussion
In the present study, we have identified p38α as a critical component in the intracellular signaling mechanisms by which a diffuse brain injury leads to activation of microglia. Moreover, we found that p38α has an important role in perpetuating injury-induced microglia activation, despite the fact that acutely p38α suppresses the peak cytokine response to diffuse brain injury. We propose a model (Fig. 10) where the hypertrophy and directional chemotaxis of microglia after a brain injury are dependent on p38α. In the absence of p38α, there is a failure of microglia priming such that the enhanced level of cytokines seen in the p38α KO mice does not produce microglia activation or chronically elevated cytokines. Therefore, microglia priming by p38α is important to preserve a state of chronic neuroinflammation after a diffuse brain injury; inhibition or elimination of p38α can improve outcome.
Figure 10.
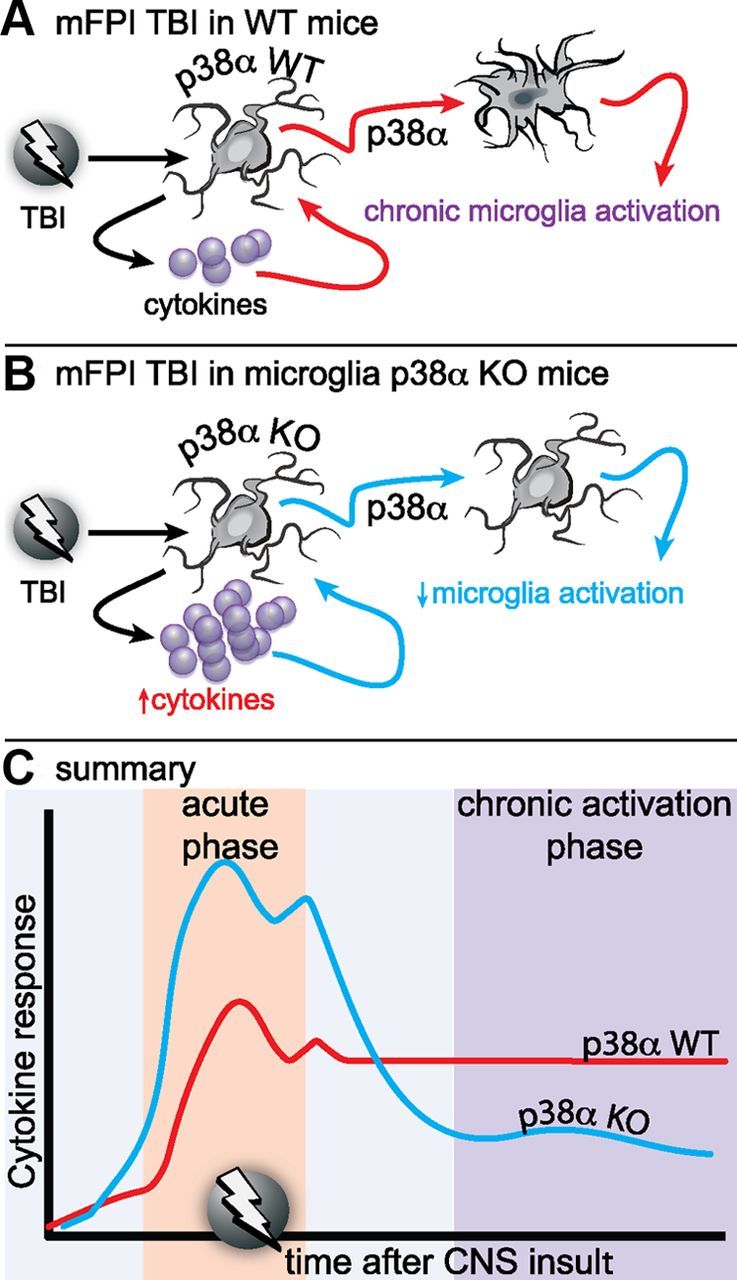
Proposed model of the role of p38α in the microglia response to diffuse brain injury. A, In WT mice, a mild to moderate diffuse brain injury induces a reactive glia response associated with elevated inflammatory cytokines and chemokines, morphological hypertrophy, and loss of microglia spatial domains as early as 6 h after injury. The reactive glia response, which is exemplified by an “M1” proinflammatory phenotype, persists for at least 7 d after the injury. B, In the p38α KO mice, the acute phase cytokine response is elevated compared with the WT mice. However, despite the higher cytokine response, there is a failure of the microglia deficient in p38α to exhibit an acute or chronic phase morphological activation response. C, Our results suggest that p38α is a critical kinase in the detrimental inflammatory feedforward loop responsible for neuromotor deficits.
Microglia are known to respond rapidly (30 min) and migrate to the source of injury (Davalos et al., 2005; Nimmerjahn et al., 2005). The signal transduction mechanisms in microglia that lead to this response are poorly understood, yet there is strong support for the involvement of p38 in chemotaxis (Heit et al., 2002, 2008), with chemoattractant selectivity (i.e., complement C5a) not being p38α-dependent (Isfort et al., 2011), whereas IL-1β is p38-dependent (Ferreira et al., 2012). Dead, dying, and injured cells release “find me” and “eat me” signals that attract microglia and macrophages to isolate the damage and remove the dead cells (Ravichandran, 2011). Purinergic signaling is the most well-described chemoattractant in the CNS (Davalos et al., 2005; Nimmerjahn et al., 2005; Haynes et al., 2006). ATP has been shown to increase levels of phospho-p38 in microglia (Trang et al., 2009). However, the role of p38 in chemotaxis to ATP is not clear, as chemotaxis to ATP has been reported to be both dependent (Heit et al., 2008) and independent of p38 (Isfort et al., 2011). This may be the result of differences in chemoattractant signals that activate microglia distally (chemokines) versus chemoattractant signals at the end target and most likely involves an integration of these signals. In complex gradients of chemoattractants, p38 and PI3K can have opposing roles in the distal versus end target movement, with p38 important for movement toward end target (Heit et al., 2008). Our data suggest that p38 may participate in the integration of the chemoattractant signals after the injury. At 6 h after the diffuse brain injury, we found a striking activation of microglia, with a loss of the normal spatial distribution, clustering of multiple cells around presumably injured neurons and blood vessels, and the appearance of processes with swollen inclusions. In injured mice where p38α is deficient in microglia, we found only a limited increase in microglia reactivity, at both 6 h and 7 d after the injury, and failure of microglia to form multicellular clusters. We also found very limited CD45 staining in the injured p38α KO mice, which could represent a decreased level of macrophage infiltration.
Microglia are known to phagocytose dying cells (Katayama et al., 2012), myelin and axonal debris (Tanaka et al., 2009; Ghasemlou et al., 2010), misfolded proteins (Reed-Geaghan et al., 2009), and synapses in response to injury such as ischemia (Wake et al., 2009), or as an activity-dependent synaptic pruning process (Schafer et al., 2012). In many instances, this process has been shown to be p38-dependent (Reed-Geaghan et al., 2009; Tanaka et al., 2009; Ghasemlou et al., 2010; Katayama et al., 2012). Therefore, the failure of microglia to activate and form multicellular clusters may reflect failure to activate a p38-dependent phagocytosis response, which is supported by the diminished CD68 response seen in the injured p38α KO mice.
In the diffuse brain injury model in this study, previous reports demonstrate axonal injury, but lack of neuronal death. Moreover, the injured neurons in this model show signs of axonal regeneration and recovery with the peak of the neuron injury response occurring during the first 7 d and partial recovery over the subsequent month (Singleton et al., 2002; Greer et al., 2011; Greer et al., 2012), suggesting that, although these neurons are endangered, they are salvageable. Our data suggest that limiting the microglia response to the injury is potentially beneficial, as shown by the lack of motor impairments in the p38α KO mice. The clearance of axonal debris, which in vitro is p38-dependent, is beneficial for the promotion of axonal growth (Hosmane et al., 2012). However, if the axon was damaged, but not beyond repair, clearance of this axon would be detrimental. Therefore, regulating the microglia response and/or p38 signaling in a mild brain injury may produce a significantly different outcome than in a TBI with pronounced neuronal death or blood–brain barrier damage.
Evidence from clinical studies and preclinical animal models suggest the key role of proinflammatory cytokine overproduction from activated glia as a potential driving force for pathology progression in TBI (Schmidt et al., 2005; Kelley et al., 2006; Lloyd et al., 2008; Cao et al., 2012). One of the most striking findings of our study was the demonstration that the inflammatory cytokine response in the CNS, which dogmatically has been associated with the activation of microglia, can be clearly disconnected from the microglia activation via a p38α-dependent mechanism after a diffuse brain injury. The seemingly divergent reaction to the diffuse brain injury (i.e., a robust increase in acute cytokine response with a lack of microglia activation in the p38α conditional KO mice) distinctly shows that microglia activation, as determined morphologically, does not necessitate the production of cytokines, and vice versa. Nevertheless, our data suggest that, if microglia are incapable of responding to the acute phase cytokine response, because of a lack of p38α, then the increase in cytokines does not lead to chronic or enhanced neuroinflammation and subsequent synaptic and behavioral impairments.
Although p38α is undoubtedly important in the production of proinflammatory cytokines (Bachstetter et al., 2011), it is becoming apparent that p38α also activates negative feedback pathways that limit the production of proinflammatory cytokines (for review, see Bachstetter and Van Eldik, 2010). The evolutionary importance of a tightly regulated immune response is exemplified by the numerous known ways and potential yet undiscovered ways that the inflammatory response is self-limiting. It has previously been demonstrated in a variety of cell types that p38 can antagonize JNK, ERK, and NFκB signaling (Zhang et al., 2001; Mathur et al., 2004; Gorska et al., 2007; Heinrichsdorff et al., 2008; Kim et al., 2008). One well-described mechanism of negative feedback is the dual-specificity protein phosphatase 1, which dephosphorylates both phosphothreonine and phosphotyrosine residues to inactivate p38 and JNK (Liu et al., 2007). The expression of dual-specificity protein phosphatase 1 is induced by p38 and ERK via a CREB-binding site in the promoter of dual-specificity protein phosphatase 1 (Sommer et al., 2000). In addition, p38α has been shown to block the phosphorylation of ERK by a direct interaction (Zhang et al., 2001). Therefore, the increase in proinflammatory cytokines we see at 6 h in the p38α KO mice could be a result of prolonged JNK activation; more work is necessary to test this hypothesis.
Previous studies report increased levels of p38 after TBI and spinal cord injury (SCI) (Mori et al., 2002; Xu et al., 2006; Stirling et al., 2008; Ghasemlou et al., 2010) However, preclinical experiments using small-molecule p38 inhibitors have demonstrated mixed results, with a lack of neuroprotection after a focal TBI (Mori et al., 2002), or moderate SCI (Stirling et al., 2008), while showing protection in a mild SCI (Xu et al., 2006). A recent study using mice deficient in MK2, a direct downstream substrate of p38, demonstrated reduced inflammation and improved locomotor recovery after SCI (Ghasemlou et al., 2010). These results are in agreement with our current study, which implicates p38 as important for regulating inflammation as part of the repair process to injury. However, inflammation is also critical to the repair process; and depending on the type and severity of the injury, inhibiting the p38-MK2 pathway can slow wound healing (Thuraisingam et al., 2010). Nevertheless, in a sterile environment, such as the CNS, suppressing the secondary injury response leading to the chronic inflammatory sequelae via the p38 pathway can be neuroprotective as demonstrated in preclinical models of ischemia (Barone et al., 2001; Legos et al., 2001; Wang et al., 2002), Alzheimer's disease (Culbert et al., 2006; Munoz et al., 2007), and Parkinson's disease (Thomas et al., 2008). Although the p38 pathway appears to be a promising clinical target for the treatment of CNS disorders with dysregulated neuroinflammatory responses, more studies are still necessary to determine therapeutic window, dosing, and long-term efficacy and to improve the pharmacodynamics and selectivity of small-molecule p38 inhibitors. In summation, our study demonstrates that p38α is a critical intracellular signaling molecule in microglia regulation of the inflammatory repair process after a diffuse TBI.
Footnotes
This work was supported in part by the Alzheimer's Association Zenith award to L.J.V.E. and National Institutes of Health Grant R01 NS064247 to L.J.V.E., Grants R01 NS065052, R21 NS072611, and R01 NS065052-S to J.L., and Grant F32 AG037280 to A.D.B. We thank Dr. Huiping Jiang (Boehringer Ingelheim Pharmaceuticals) and Dr. Jiahuai Han (Scripps Research Institute) for the kind gifts of the knock-out mice; and Edgardo Dimayuga, Jordan Harrison, Janna Neltner, and Bin Xing for assistance with various aspects of this work.
The authors declare no competing financial interests.
References
- Bachstetter AD, Van Eldik LJ. The p38 MAP kinase family as regulators of proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. 2010;1:199–211. [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ. Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine upregulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta) J Neuroinflammation. 2011;8:79. doi: 10.1186/1742-2094-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Norris CM, Sompol P, Wilcock DM, Goulding D, Neltner JH, St Clair D, Watterson DM, Van Eldik LJ. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer's disease-related pathology. J Neurosci. 2012;32:10201–10210. doi: 10.1523/JNEUROSCI.1496-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF, McVey MJ, Legos JJ, Erhardt JA, Nelson AH, Ohlstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001;296:312–321. [PubMed] [Google Scholar]
- Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012;225:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S, Greaves DR, Chung LP, Tree P, Gordon S. The human lysozyme promoter directs reporter gene expression to activated myelomonocytic cells in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:1434–1438. doi: 10.1073/pnas.93.4.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert AA, Skaper SD, Howlett DR, Evans NA, Facci L, Soden PE, Seymour ZM, Guillot F, Gaestel M, Richardson JC. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity: relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2006;281:23658–23667. doi: 10.1074/jbc.M513646200. [DOI] [PubMed] [Google Scholar]
- da Silva RP, Gordon S. Phagocytosis stimulates alternative glycosylation of macrosialin (mouse CD68), a macrophage-specific endosomal protein. Biochem J. 1999;338:687–694. [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Santos T, Cortes L, Cochaud S, Agasse F, Silva AP, Xapelli S, Malva JO. Neuropeptide Y inhibits interleukin-1 β-induced microglia motility. J Neurochem. 2012;120:93–105. doi: 10.1111/j.1471-4159.2011.07541.x. [DOI] [PubMed] [Google Scholar]
- Gavett BE, Stern RA, Cantu RC, Nowinski CJ, McKee AC. Mild traumatic brain injury: a risk factor for neurodegeneration. Alzheimers Res Ther. 2010;2:18. doi: 10.1186/alzrt42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemlou N, Lopez-Vales R, Lachance C, Thuraisingam T, Gaestel M, Radzioch D, David S. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) contributes to secondary damage after spinal cord injury. J Neurosci. 2010;30:13750–13759. doi: 10.1523/JNEUROSCI.2998-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: a prime target for Alzheimer's disease prevention. J Neuroinflammation. 2012;9:185. doi: 10.1186/1742-2094-9-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorska MM, Liang Q, Stafford SJ, Goplen N, Dharajiya N, Guo L, Sur S, Gaestel M, Alam R. MK2 controls the level of negative feedback in the NF-κB pathway and is essential for vascular permeability and airway inflammation. J Exp Med. 2007;204:1637–1652. doi: 10.1084/jem.20062621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JE, McGinn MJ, Povlishock JT. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci. 2011;31:5089–5105. doi: 10.1523/JNEUROSCI.5103-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JE, Povlishock JT, Jacobs KM. Electrophysiological abnormalities in both axotomized and nonaxotomized pyramidal neurons following mild traumatic brain injury. J Neurosci. 2012;32:6682–6687. doi: 10.1523/JNEUROSCI.0881-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Hamm RJ. Neurobehavioral assessment of outcome following traumatic brain injury in rats: an evaluation of selected measures. J Neurotrauma. 2001;18:1207–1216. doi: 10.1089/089771501317095241. [DOI] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y(12) receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Heinrichsdorff J, Luedde T, Perdiguero E, Nebreda AR, Pasparakis M. p38 α MAPK inhibits JNK activation and collaborates with IκB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep. 2008;9:1048–1054. doi: 10.1038/embor.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol. 2008;9:743–752. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- Hosmane S, Tegenge MA, Rajbhandari L, Uapinyoying P, Kumar NG, Thakor N, Venkatesan A. Toll/interleukin-1 receptor domain-containing adapter inducing interferon-β mediates microglial phagocytosis of degenerating axons. J Neurosci. 2012;32:7745–7757. doi: 10.1523/JNEUROSCI.0203-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Wang Y, Vogel P, Kanneganti TD, Otsu K, Chi H. Signaling via the kinase p38alpha programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat Immunol. 2012;13:152–161. doi: 10.1038/ni.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol. 2011;89:525–538. doi: 10.1189/jlb.0810472. [DOI] [PubMed] [Google Scholar]
- Isfort K, Ebert F, Bornhorst J, Sargin S, Kardakaris R, Pasparakis M, Bähler M, Schwerdtle T, Schwab A, Hanley PJ. Real-time imaging reveals that P2Y2 and P2Y12 receptor agonists are not chemoattractants and macrophage chemotaxis to complement C5a is phosphatidylinositol 3-kinase (PI3K)- and p38 mitogen-activated protein kinase (MAPK)-independent. J Biol Chem. 2011;286:44776–44787. doi: 10.1074/jbc.M111.289793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Chen J, Otsuka M, Mols J, Ren S, Wang Y, Han J. Macrophage deletion of p38α partially impairs lipopolysaccharide-induced cellular activation. J Immunol. 2008;180:5075–5082. doi: 10.4049/jimmunol.180.7.5075. [DOI] [PubMed] [Google Scholar]
- Katayama T, Kobayashi H, Okamura T, Yamasaki-Katayama Y, Kibayashi T, Kimura H, Ohsawa K, Kohsaka S, Minami M. Accumulating microglia phagocytose injured neurons in hippocampal slice cultures: involvement of p38 MAP kinase. PLoS One. 2012;7:e40813. doi: 10.1371/journal.pone.0040813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BJ, Farkas O, Lifshitz J, Povlishock JT. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp Neurol. 2006;198:350–360. doi: 10.1016/j.expneurol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, Lawrence T, Otsu K, Brissette JL, Arthur JS, Park JM. The kinase p38 α serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008;9:1019–1027. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurushima H, Ramprasad M, Kondratenko N, Foster DM, Quehenberger O, Steinberg D. Surface expression and rapid internalization of macrosialin (mouse CD68) on elicited mouse peritoneal macrophages. J Leukoc Biol. 2000;67:104–108. doi: 10.1002/jlb.67.1.104. [DOI] [PubMed] [Google Scholar]
- Legos JJ, Erhardt JA, White RF, Lenhard SC, Chandra S, Parsons AA, Tuma RF, Barone FC. SB 239063, a novel p38 inhibitor, attenuates early neuronal injury following ischemia. Brain Res. 2001;892:70–77. doi: 10.1016/s0006-8993(00)03228-5. [DOI] [PubMed] [Google Scholar]
- Lifshitz J. Fluid percussion injury. In: Chen J, editor. Animal models of acute neurological injuries. Totowa, NJ: Humana; 2008. pp. 369–384. [Google Scholar]
- Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases: regulating the immune response. Nat Rev Immunol. 2007;7:202–212. doi: 10.1038/nri2035. [DOI] [PubMed] [Google Scholar]
- Lloyd E, Somera-Molina K, Van Eldik LJ, Watterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small-molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, Roers A, Eming SA. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- Mathur RK, Awasthi A, Wadhone P, Ramanamurthy B, Saha B. Reciprocal CD40 signals through p38MAPK and ERK-1/2 induce counteracting immune responses. Nat Med. 2004;10:540–544. doi: 10.1038/nm1045. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol. 2012;188:29–36. doi: 10.4049/jimmunol.1100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Mori T, Wang X, Jung JC, Sumii T, Singhal AB, Fini ME, Dixon CE, Alessandrini A, Lo EH. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Metab. 2002;22:444–452. doi: 10.1097/00004647-200204000-00008. [DOI] [PubMed] [Google Scholar]
- Munoz L, Ralay Ranaivo H, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterson DM. A novel p38 α MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Penninger JM, Irie-Sasaki J, Sasaki T, Oliveira-dos-Santos AJ. CD45: new jobs for an old acquaintance. Nat Immunol. 2001;2:389–396. doi: 10.1038/87687. [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–455. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury: an inflammatory disease? Brain Res Brain Res Rev. 2005;48:388–399. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton RH, Zhu J, Stone JR, Povlishock JT. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791–802. doi: 10.1523/JNEUROSCI.22-03-00791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- Sommer A, Burkhardt H, Keyse SM, Lüscher B. Synergistic activation of the mkp-1 gene by protein kinase A signaling and USF, but not c-Myc. FEBS Lett. 2000;474:146–150. doi: 10.1016/s0014-5793(00)01566-0. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Liu J, Plunet W, Steeves JD, Tetzlaff W. SB203580, a p38 mitogen-activated protein kinase inhibitor, fails to improve functional outcome following a moderate spinal cord injury in rat. Neuroscience. 2008;155:128–137. doi: 10.1016/j.neuroscience.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Sudduth TL, Wilson JG, Everhart A, Colton CA, Wilcock DM. Lithium treatment of APPSwDI/NOS2−/− mice leads to reduced hyperphosphorylated τ, increased amyloid deposition and altered inflammatory phenotype. PLoS One. 2012;7:e31993. doi: 10.1371/journal.pone.0031993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Ueno M, Yamashita T. Engulfment of axon debris by microglia requires p38 MAPK activity. J Biol Chem. 2009;284:21626–21636. doi: 10.1074/jbc.M109.005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Timmer M, Cesnulevicius K, Hitti E, Kotlyarov A, Gaestel M. MAPKAP kinase 2 deficiency prevents neurons from cell death by reducing neuroinflammation: relevance in a mouse model of Parkinson's disease. J Neurochem. 2008;105:2039–2052. doi: 10.1111/j.1471-4159.2008.05310.x. [DOI] [PubMed] [Google Scholar]
- Thuraisingam T, Xu YZ, Eadie K, Heravi M, Guiot MC, Greemberg R, Gaestel M, Radzioch D. MAPKAPK-2 signaling is critical for cutaneous wound healing. J Invest Dermatol. 2010;130:278–286. doi: 10.1038/jid.2009.209. [DOI] [PubMed] [Google Scholar]
- Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29:3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xu L, Wang H, Young PR, Gaestel M, Feuerstein GZ. Mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 deficiency protects brain from ischemic injury in mice. J Biol Chem. 2002;277:43968–43972. doi: 10.1074/jbc.M206837200. [DOI] [PubMed] [Google Scholar]
- Xing B, Bachstetter AD, Van Eldik LJ. Microglial p38α MAPK is critical for LPS-induced neuron degeneration, through a mechanism involving TNFα. Mol Neurodegener. 2011;6:84. doi: 10.1186/1750-1326-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wang BR, Wang X, Kuang F, Duan XL, Jiao XY, Ju G. ERK1/2 and p38 mitogen-activated protein kinase mediate iNOS-induced spinal neuron degeneration after acute traumatic spinal cord injury. Life Sci. 2006;79:1895–1905. doi: 10.1016/j.lfs.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Zhang H, Shi X, Hampong M, Blanis L, Pelech S. Stress-induced inhibition of ERK1 and ERK2 by direct interaction with p38 MAP kinase. J Biol Chem. 2001;276:6905–6908. doi: 10.1074/jbc.C000917200. [DOI] [PubMed] [Google Scholar]
- Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J. Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation. 2012;9:247. doi: 10.1186/1742-2094-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]