

GRK-mediated Ser-129 phosphorylation of membrane-associated α-synuclein enhances dopamine uptake without altering cell surface expression of dopamine transporter. The data suggest a role of Ser-129–phosphorylated α-synuclein in modulating intracellular dopamine levels and damaging dopaminergic neurons in Parkinson disease.
Abstract
Most α-synuclein (α-syn) deposited in Lewy bodies, the pathological hallmark of Parkinson disease (PD), is phosphorylated at Ser-129. However, the physiological and pathological roles of this modification are unclear. Here we investigate the effects of Ser-129 phosphorylation on dopamine (DA) uptake in dopaminergic SH-SY5Y cells expressing α-syn. Subcellular fractionation of small interfering RNA (siRNA)–treated cells shows that G protein–coupled receptor kinase 3 (GRK3), GRK5, GRK6, and casein kinase 2 (CK2) contribute to Ser-129 phosphorylation of membrane-associated α-syn, whereas cytosolic α-syn is phosphorylated exclusively by CK2. Expression of wild-type α-syn increases DA uptake, and this effect is diminished by introducing the S129A mutation into α-syn. However, wild-type and S129A α-syn equally increase the cell surface expression of dopamine transporter (DAT) in SH-SY5Y cells and nonneuronal HEK293 cells. In addition, siRNA-mediated knockdown of GRK5 or GRK6 significantly attenuates DA uptake without altering DAT cell surface expression, whereas knockdown of CK2 has no effect on uptake. Taken together, our results demonstrate that membrane-associated α-syn enhances DA uptake capacity of DAT by GRKs-mediated Ser-129 phosphorylation, suggesting that α-syn modulates intracellular DA levels with no functional redundancy in Ser-129 phosphorylation between GRKs and CK2.
INTRODUCTION
Parkinson disease (PD) is the most common movement disorder. The pathological hallmarks of PD are loss of dopaminergic neurons in the substantia nigra pars compacta and the appearance of fibrillar aggregates of α-synuclein (α-syn), called Lewy bodies (LBs) and Lewy neurites, in surviving nigral neurons (Spillantini et al., 1997; Eriksen et al., 2003; Lee and Trojanowski, 2006). Three different point mutations in the SNCA gene, which encodes α-syn, cause rare autosomal dominantly inherited forms of PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004). Multiplication of SNCA is also associated with familial PD, indicating that increased expression per se can lead to dopaminergic neurodegeneration (Singleton et al., 2003). Accumulating evidence indicates that prefibrillar intermediates, such as soluble oligomers and protofibrils, are highly toxic to neurons, whereas mature fibrils are less toxic (Volles et al., 2001; Xu et al., 2002; Sharon et al., 2003; Chen et al., 2009). However, the mechanisms of α-syn neurotoxicity are unclear. Furthermore, it is unknown how this widely distributed protein preferentially damages dopaminergic neurons. Direct measurement of cytosolic dopamine (DA) levels in l-3,4-dihydroxyphenylalanine–treated primary dopaminergic neuronal cultures provided important insights into the problem by showing that elevation of cytosolic DA levels was required to evoke selective death of dopaminergic neurons and that toxity of cytosolic DA depended on the presence of a-syn (Mosharov et al., 2009). The data suggest that the pathogenic effects of α-syn are downstream of DA synthesis. In support of the data, earlier studies showed that DA stabilized α-syn as the form of toxic protofibrils (Conway et al., 2001), and DA-modified α-syn inhibited chaperone-mediated autophagy (Martinez-Vicente et al., 2008). The increase in cytosolic DA levels and subsequent interaction of cytosolic DA with α-syn seem to play a key role in preferential degeneration of dopaminergic neurons in PD.
Approximately 90% of α-syn deposited in LBs is phosphorylated at Ser-129 (Fujiwara et al., 2002; Anderson et al., 2006). In contrast, only 4% or less of total α-syn is phosphorylated at this residue in normal brain (Fujiwara et al., 2002; Anderson et al., 2006). This suggests that levels of Ser-129–phosphorylated α-syn are tightly regulated by kinases, phosphatases, and degradation pathways (Machiya et al., 2010) and that accumulation of Ser-129–phosphorylated α-syn above a certain threshold leads to the formation of LBs and dopaminergic neurodegeneration (Fujiwara et al., 2002). Indeed, coexpression of α-syn and a Drosophila homologue of G protein–coupled receptor kinase 2 (Gprk2) in a Drosophila model of PD yielded Ser-129–phosphorylated α-syn and enhanced α-syn toxicity (Chen and Feany, 2005). In addition, in a rat recombinant adeno-associated virus (AAV)–based model, coexpression of A53T α-syn and human G protein–coupled receptor kinase 6 (GRK6) accelerated α-syn–induced degeneration of dopaminergic neurons (Sato et al., 2011). However, the role of increased Ser-129 phosphorylation in dopaminergic neurodegeneration in PD is unknown. In this study, we investigate the effects of Ser-129 phosphorylation on DA uptake in human dopaminergic SH-SY5Y cells. We report that GRKs-mediated phosphorylation of membrane-associated α-syn increases DA uptake without enhancing cell surface expression of dopamine transporter (DAT).
RESULTS
Contribution of endogenous GRKs and casein kinase 2 to Ser-129 phosphorylation of α-syn in SH-SY5Y cells
Multiple kinases, including GRK2, GRK3, GRK5, GRK6 (Pronin et al., 2000; Sakamoto et al., 2009), casein kinase 1 (CK1), CK2 (Okochi et al., 2000), Polo-like kinase 2 (PLK2), and PLK3 (Inglis et al., 2009; Mbefo et al., 2010) phosphorylate α-syn at Ser-129 in vitro. In this study, we focused on the roles of GRKs and CK2 on Ser-129 phosphorylation of α-syn in human dopaminergic SH-SY5Y cells. We did not examine CK1 or PLKs because the multiple CK1 isoforms made it difficult to assess their effects and commercial antibodies that recognize endogenous PLK proteins were unavailable. We examined the effects of kinases on Ser-129 phosphorylation of α-syn by using a cell line stably expressing wild-type α-syn (wt-aS/SH) because expression of endogenous α-syn was not high enough to allow detection of the Ser-129–phosphorylated form by Western blotting (see discussion of Figure 4C; Machiya et al., 2010).
FIGURE 4:

Ser-129 phosphorylation state of α-syn affects DA uptake. (A) DA uptake through endogenous DAT in wt-aS/SH cells. The cells were incubated in buffer containing 20 nM [3H]DA and 5 μM cold DA for 10 min in the absence or presence of either 10 μM GBR12909 or 10 μM nomifensine. Incorporated radioactivity was measured. (B) Effects of Ser-129 phosphorylation of α-syn on DA uptake through endogenous DAT. Top, DA uptake kinetics in parental cells, wt-aS/SH cells, and S129A-aS/SH cells. All these cell lines were transfected with control siRNA to ensure comparability with the following kinase knockdowns. The assays were performed with the mixture of [3H]DA and various concentrations of cold DA in the absence or presence of GBR12909. Middle, Eadie–Hofstee plot of the kinetics data. Bottom, summary of kinetic parameters. (C) Expression levels of wild-type and S129A α-syn in each stable cell line. Cell lysates (20 μg/lane) were analyzed by WB with antibodies recognizing β-actin, Ser-129–phosphorylated α-syn or total α-syn. (D) Effects of Ser-129 phosphorylation of α-syn on DA uptake in SH-SY5Y cells stably coexpressing DAT and GFP (GFP+DAT/SH), α-syn (wt-aS+DAT/SH), or S129A mutant α-syn (S129A-aS+DAT/SH). Top, DA uptake kinetics. Bottom, summary of kinetic parameters. Data represent means ± SD, and p values were estimated by one-way ANOVA with a Bonferroni correction (*p < 0.05; **p < 0.01).
We first investigated the contribution of endogenous GRKs to Ser-129 phosphorylation of α-syn in wt-aS/SH cells using small interfering RNA (siRNA)–mediated knockdown. Knockdowns of GRK2 and GRK6 suppressed endogenous expression by ∼50% compared with control siRNA (Figure 1A). Knockdowns of GRK3 and GRK5 suppressed endogenous expression by ∼70% (Figure 1A). The siRNAs did not alter the expression of nontarget GRKs. Knockdowns of GRK3, GRK5, and GRK6 significantly decreased levels of Ser-129–phosphorylated α-syn by 34 ± 14% (mean ± SD, p = 0.015, n = 4), 27 ± 14% (p = 0.028, n = 5), and 19 ± 11% (p = 0.048, n = 5), respectively (Figure 1B). GRK2 knockdown did not alter the phosphorylation level (11 ± 34% increase, p = 0.548, n = 5; Figure 1B).
FIGURE 1:

The contribution of endogenous GRKs and CK2 to Ser-129 phosphorylation of α-syn in SH-SY5Y cells stably expressing wild-type α-syn (wt-aS/SH). Cell lysates (20 μg/lane) were analyzed by Western blotting (WB) with the indicated antibodies. For loading control, the same amounts of samples were blotted with β-actin antibody. (A) The levels of GRKs in the cells transfected with siRNAs against individual GRKs. The graphs show the target/β-actin ratios in GRK siRNAs relative to control siRNA. (B) Effects of siRNA-mediated knockdowns of GRKs on Ser-129 phosphorylation of α-syn. Cell lysates were loaded along with recombinant α-syn proteins and Ser-129–phosphorylated α-syn proteins for standards. Bands of total α-syn, including phosphorylated and nonphosphorylated forms, were detected by Syn-1 antibody. Relative band intensities of Ser-129–phosphorylated α-syn and total α-syn were corrected by plotting them on the standard curves and then normalized to the intensities of β-actin. The graph shows the Ser-129–phosphorylated α-syn/total α-syn ratios in GRK siRNAs relative to control siRNA. (C) The levels of CK2 in the cells transfected with siRNAs against individual CK2 α-subunits. The graphs show the target/β-actin ratios in CK2 siRNAs relative to control siRNA. (D) Effects of siRNA-mediated knockdowns of CK2 α-subunits on Ser-129 phosphorylation of α-syn. The graph shows the Ser-129–phosphorylated α-syn/total α-syn ratios in CK2 siRNAs relative to control siRNA. Data represent means ± SD, and p values were estimated by unpaired Student's t test (*p < 0.05).
We next examined the effects of siRNA-mediated knockdowns of the catalytic α subunits (α and α′) of CK2 on Ser-129 phosphorylation of α-syn. Knockdowns of CK2 α and α′ subunits suppressed endogenous expression by ∼60 and ∼90%, respectively (Figure 1C). CK2 α′ subunit knockdown significantly decreased the level of Ser-129–phosphorylated α-syn (30 ± 24% decrease, p = 0.042, n = 4). Although CK2 α subunit knockdown increased the level of Ser-129–phosphorylated α-syn, this effect was not statistically significant (41 ± 61% increase, p = 0.226, n = 5; Figure 1D).
Subcellular distribution of GRKs, CK2, and α-syn in SH-SY5Y cells
We examined the subcellular distribution of GRKs, CK2, and α-syn in SH-SY5Y cells. Although α-syn is principally a cytosolic protein, a portion of it is known to be reversibly associated with membrane components (Davidson et al., 1998; McLean et al., 2000; Perrin et al., 2000). In addition, GRK2 and GRK3 rapidly translocate from the cytoplasm to the plasma membrane in response to G protein–coupled receptor (GPCR) activation (Boekhoff et al., 1994; Premont et al., 1995). To minimize artificial dissociation of membrane-associated proteins from membrane components during homogenization, we treated the living cells with a membrane-permeable and thiol-cleavable cross-linker, dithiobis(succinimidyl propionate) (DSP; spacer arm length 12 Å) before subcellular fractionation. To qualitatively evaluate this fractionation technique, we examined the distribution of transmembrane and cytosolic proteins. Type I transmembrane proteins amyloid precursor protein and nicastrin (NCT) were exclusively recovered in the membrane fractions, whereas a cytosolic protein, Cu/Zn superoxide dismutase, was primarily recovered in the cytosolic fractions (Figure 2A). DSP treatment did not alter the distribution of these proteins (Figure 2A). To further assess whether DSP treatment influences fractionation, we examined the subcellular distribution of mutant α-syn, which lacks amino acids 20–60 for lipid binding (Δ20–60 α-syn), in cells transiently transfected with the mutant cDNA (Perrin et al., 2000; Karube et al., 2008). This deletion mutant was abundant in the cytosolic fractions, and its subcellular distribution was unaffected by DSP treatment, indicating that DSP did not cross-link non–membrane-associated proteins with membrane components (Figure 2B). Taken together, these results suggested that this fractionation technique was able to separate integral membrane and cytosolic proteins.
FIGURE 2:

Subcellular distribution of GRKs, CK2, and α-syn proteins in SH-SY5Y cells. Cells were treated with or without 1 mM DSP at 37°C for 30 min, followed by separation into the cytosol and membrane fractions by ultracentrifugation. Equal aliquots of the cytosol and membrane fractions were loaded on SDS–PAGE along with total RIPA lysates (20 μg/lane) and analyzed by WB. (A) Subcellular distribution of Cu/Zn superoxide dismutase (SOD1), amyloid precursor protein (APP), and nicastrin (NCT). Samples were analyzed by WB with each antibody. (B) Effect of chemical cross-linking by DSP on subcellular fractionation of α-syn. Cells transiently transfected with wild-type or deletion mutant α-syn (Δ20–60 α-syn) cDNA were analyzed by WB with Syn-1 antibody. SOD1, APP, and NCT were used as fractionation markers. (C) Subcellular distribution of GRKs, CK2, and α-syn proteins. Wt-aS/SH cells were analyzed by WB with the indicated antibodies. Representative blots are shown. In the graphs of A–C, subcellular localizations are expressed as percentages of the levels in the membrane fractions to the total levels in the cytosol and membrane fractions. Data represent means ± SD, and p values were estimated by unpaired Student's t test (*p < 0.05).
When we examined the cell line stably expressing wild-type α-syn (wt-aS/SH), GRKs including GRK3, GRK5, and GRK6 were more abundant in the membrane fractions than in the cytosolic fractions in the presence of DSP (Figure 2C). GRK5 and GRK6 were predominant in the membrane fractions, irrespective of DSP treatment, whereas DSP treatment significantly increased the amounts of GRK3 recovered in the membrane fractions from 29.5 ± 11.2 to 69.8 ± 10.8% (p = 0.002, n = 4). GRK2 was distributed nearly equally in the cytosolic and membrane fractions in the presence of DSP. CK2 containing α′ subunit was slightly abundant in the cytosolic fractions, even in the presence of DSP (Figure 2C). CK2 containing α subunit was distributed almost equally in the cytosolic and membrane fractions in the presence of DSP. α-Syn was abundantly present in the cytosolic fractions, irrespective of DSP treatment (Figure 2C). However, DSP treatment significantly increased the amounts of α-syn in the membrane fractions from 12.3 ± 4.6 to 29.5 ± 9.5% (p = 0.017, n = 4). In addition, DSP treatment significantly increased the proportion of Ser-129–phosphorylated α-syn in the membrane fractions from 15.4 ± 7.0 to 24.9 ± 2.4% (p = 0.042, n = 4; Figure 2C). When we compared the subcellular distribution of wild-type α-syn between Figure 2B and Figure 2C, DSP treatment yielded larger amounts of membrane-associated α-syn in transiently transfected cells than in the stable cell line. Because DSP treatment was performed in the same conditions, these findings suggested that the subcellular distribution of α-syn was affected by the gene expression system in cells. To assess whether target proteins were excessively cross-linked in the membrane fractions during DSP treatment at 37°C, we investigated their subcellular distribution by DSP treatment at 4°C. The amounts of GRK2, GRK3, GRK6, total α-syn, and phosphorylated α-syn in the membrane fractions significantly increased, whereas the subcellular distribution of GRK5 and CK2 α′ subunit was not altered by DSP (Supplemental Figure S1). There was no obvious difference in the effects of DSP on the subcellular distribution of these proteins between 4 and 37°C incubations.
GRKs and CK2-mediated Ser-129 phosphorylation of cytosolic and membrane-associated α-syn
To identify the cellular compartments where α-syn undergoes Ser-129 phosphorylation by GRKs and CK2, we performed subcellular fractionation of cells transfected with siRNAs against individual GRKs or CK2. The cells were treated with DSP, immediately followed by the fractionation. Data were compared with those for control siRNA. Knockdowns of GRK3, GRK5, and GRK6 significantly decreased the amount of Ser-129–phosphorylated α-syn in the membrane fractions by 30 ± 17% (p = 0.017, n = 5), 43 ± 36% (p = 0.031, n = 5), and 30 ± 19% (p = 0.026, n = 5), respectively (Figure 3A). However, knockdowns of these kinases did not alter the amounts of Ser-129–phosphorylated α-syn in the cytosolic fractions (Figure 3A). In contrast, GRK2 knockdown increased the amounts of Ser-129–phosphorylated α-syn in the cytosolic fractions (31 ± 23% increase, p = 0.037, n = 5). CK2 α′ subunit knockdown significantly decreased the amounts of Ser-129–phosphorylated α-syn in both the cytosolic (46 ± 28% decrease, p = 0.021, n = 5) and membrane fractions (57 ± 37% decrease, p = 0.027, n = 5; Figure 3B). In contrast, CK2 α subunit knockdown significantly increased the amounts of Ser-129–phosphorylated α-syn in the membrane fractions (135 ± 85% increase, p = 0.024, n = 5; Figure 3B). These results demonstrated that GRK3, GRK5, and GRK6 contributed to the phosphorylation of membrane-associated α-syn, whereas CK2 containing α′ subunit contributed to the phosphorylation of both cytosolic and membrane-associated α-syn. These results also raised the possibility that GRK2 and CK2 containing α subunit inhibited the phosphorylation of cytosolic and membrane-associated α-syn, respectively.
FIGURE 3:

The contribution of endogenous GRKs and CK2 to Ser-129 phosphorylation of cytosolic and membrane-associated α-syn in SH-SY5Y cells. Wt-aS/SH cells were transfected with siRNAs against individual GRKs or CK2 α-subunits. The cells were treated with DSP, followed by subcellular fractionation. Equal aliquots of the cytosol and membrane fractions were loaded on SDS–PAGE and analyzed by WB with the indicated antibodies. Effects of siRNA-mediated knockdowns of GRKs (A) and CK2 α-subunits (B) on the phosphorylation are shown. SOD1, APP, and NCT are used as fractionation markers. Relative band intensities of Ser-129–phosphorylated α-syn and total α-syn were normalized to those of β-actin. All graphs show the Ser-129–phosphorylated α-syn/total α-syn ratios in target siRNAs relative to control siRNA. Data represent means ± SD, and p values were estimated by unpaired Student's t test (*p < 0.05).
Effects of Ser-129 phosphorylation of α-syn on DA uptake
α-Syn has been reported to bind to the C-terminus of DAT, and overexpression of α-syn increases DA uptake by enhancing the cell surface expression of the transporter (Lee et al., 2001). To test whether Ser-129 phosphorylation affects α-syn function, we investigated the effects of Ser-129–phosphorylated α-syn on DA uptake through endogenous DAT. In wt-aS/SH cells, treatment with DA reuptake inhibitors GBR12909 and nomifensine effectively decreased DA uptake by 85.2 ± 2.0 and 80.4 ± 1.7%, respectively (Figure 4A). When we analyzed DA uptake kinetics in wt-aS/SH cells, Vmax for DA uptake was increased 3.9-fold compared with parental cells (p < 0.001, n = 3; Figure 4B). Vmax was also 1.8-fold higher than that in S129A-aS/SH cells, which stably expressed the phosphorylation-incompetent S129A α-syn mutant (p < 0.001, n = 3; Figure 4B). The expression levels of α-syn proteins in these stable cell lines were comparable (the intensity of the α-syn/β-actin ratio in wt-aS/SH cells relative to S129A-aS/SH cells was 1.05; Figure 4C). Km values for DA uptake showed that the affinity of DAT for DA in wt-aS/SH cells was lower than in parental (p = 0.049, n = 3) or S129A-aS/SH (p = 0.004, n = 3; Figure 4B) cells. To further clarify the effects of Ser-129–phosphorylated α-syn on DA uptake, we assessed DA uptake kinetics in cell lines coexpressing DAT and α-syn (aS+DAT/SH cells). Vmax for DA uptake in wt-aS+DAT/SH cells was significantly higher than in S129A-aS+DAT/SH cells (p = 0.003, n = 3; Figure 4D). Vmax in control GFP+DAT/SH cells was the lowest among the cell lines (p < 0.001, n = 3). Km for DA uptake in wt-aS+DAT/SH cells was higher than in S129A-aS+DAT/SH or GFP+DAT/SH cells, although the differences were not statistically significant (Figure 4D). These findings indicated that the enhanced DA uptake in wt-aS/SH and wt-aS+DAT/SH cells was not due to higher affinity of DAT for DA.
We next investigated the cell surface expression of DAT. The cell surface expression of endogenous DAT in wt-aS/SH cells was measured by binding of a 3H-labeled DAT-specific ligand, 2β-carbomethoxy-3β-(4-fluorophenyl)tropane (CFT), because endogenous DAT in SH-SY5Y cells was undetectable by Western blotting. The cell surface expression of endogenous DAT in wt-aS/SH cells (341 ± 18 fmol/mg protein, n = 3) was ∼1.7-fold higher than in parental cells (204.3 ± 12.9 fmol/mg protein, n = 3; p < 0.001; Figure 5A). However, the increase in wt-aS/SH cells was similar to that observed in S129A-aS/SH cells (356 ± 36.4 fmol/mg protein, n = 3; Figure 5A). Next we assessed the cell surface expression of exogenous DAT in wt-aS+DAT/SH cells by surface biotinylation. The cell surface expression of DAT in cells coexpressing the transporter and wild-type α-syn was ∼1.5-fold higher than in cells coexpressing the transporter and green fluorescent protein (GFP; p < 0.001, n = 3; Figure 5B). However, the increase in DAT expression in cells coexpressing wild-type α-syn was similar to that observed in cells coexpressing S129A α-syn (1.51 ± 0.13–fold increase compared with cells coexpressing GFP, n = 3; Figure 5B). The glycosylated, mature form of NCT was found exclusively in avidin-precipitated complexes, suggesting that extracellular regions of membrane proteins were selectively biotinylated (Arawaka et al., 2002).
FIGURE 5:
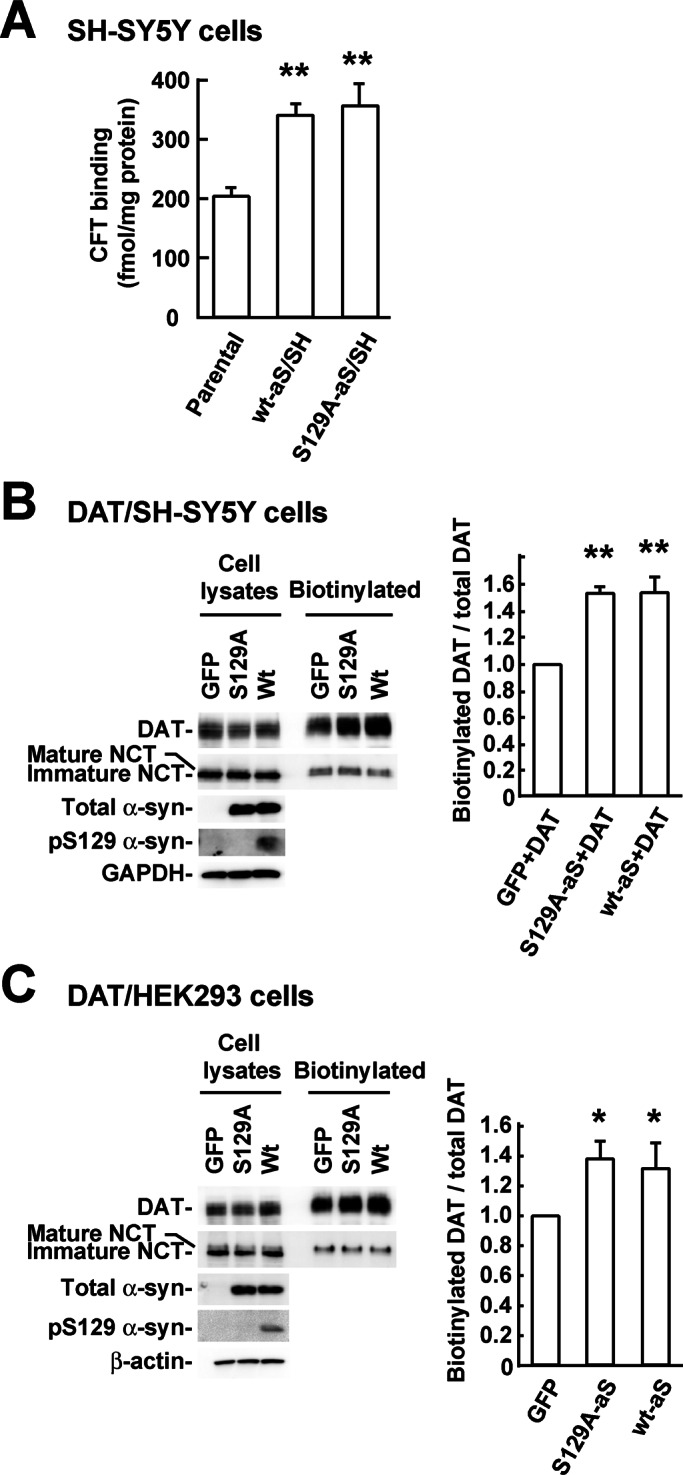
Effects of Ser-129 phosphorylation of α-syn on the cell surface expression of DAT. (A) The cell surface expressions of endogenous DAT in parental SH-SY5Y cells, wt-aS/SH cells, and S129A-aS/SH cells were measured by [3H]CFT binding assay. (B) The cell surface expressions of DAT in GFP+DAT/SH, wt-aS+DAT/SH, and S129A-aS+DAT/SH cells were assessed by surface biotinylation. Avidin-precipitated complexes were analyzed by blots with antibodies against DAT or NCT. Total cell lysates (20 μg/lane) were blotted with antibodies recognizing DAT, NCT, total α-syn, Ser-129–phosphorylated α-syn, or glyceraldehyde-3-phosphate dehydrogenase. The graph shows the biotinylated DAT/total DAT ratios in α-syn–coexpressed cells relative to GFP-coexpressed cells. (C) The cell surface expressions of DAT in nonneuronal HEK293 cells stably expressing the transporter (DAT/HEK). DAT/HEK cells were transiently transfected with GFP, wild-type α-syn, or S129A mutant α-syn cDNA and then treated with surface biotinylation. Avidin-precipitated complexes were analyzed by blots with antibodies against DAT or NCT. Total cell lysates were blotted with the indicated antibodies. The graph shows the biotinylated DAT/total DAT ratios in α-syn–expressed cells relative to GFP-expressed cells. Data represent means ± SD, and p values were estimated by one-way ANOVA with a Bonferroni correction (*p < 0.05; **p < 0.01).
To assess whether α-syn elevates DAT cell surface expression in nonneuronal cells, we examined the effects of α-syn expression in human embryonic kidney 293 (HEK293) cells stably expressing DAT (DAT/HEK). DAT/HEK cells were transiently transfected with wild-type α-syn, S129A α-syn, or GFP. DAT cell surface expression in cells expressing wild-type α-syn was ∼1.3-fold higher than in cells expressing GFP (p = 0.016, n = 4; Figure 5C). The magnitude of the increase was comparable with that in cells expressing S129A α-syn (1.37 ± 0.13–fold increase compared with cells expressing GFP, n = 4; Figure 5C).
Differential effects of GRKs and CK2-mediated Ser-129 phosphorylation of α-syn on DA uptake
To elucidate the effect of Ser-129 phosphorylation of α-syn on DA uptake, we examined the DA uptake kinetics in wt-aS/SH cells transfected with siRNA against GRK5, GRK6, or CK2 α′ subunit. Data were compared with those for control siRNA. Vmax for DA uptake in cells transfected with GRK6 siRNA was significantly decreased by ∼38% (p < 0.001, n = 3; Figure 6, A and C). GRK5 knockdown also decreased DA uptake but to a lesser extent (∼12% decrease, p = 0.007, n = 3; Figure 6, A and C). There was no significant change in Km values for DA uptake among the GRK5, GRK6, and control siRNA-mediated knockdowns (Figure 6, B and C). CK2 α′ subunit knockdown did not alter Vmax and Km values for DA uptake (Figure 6, A–C). In addition, the GRK5 and GRK6 knockdowns did not affect cell surface expression of DAT in wt-aS/SH cells, as measured with the [3H]CFT binding assay (Figure 6D). Because knockdown of GRK5 or GRK6 did not reduce the expression levels of total α-syn, as shown in Figure 1B, these findings suggested that knockdown of GRK5 or GRK6 modulated DA uptake through Ser-129 phosphorylation of α-syn without changing cell surface expression of DAT. To further assess whether knockdowns of GRKs attenuate DA uptake through α-syn, we investigated uptake in S129A-aS/SH cells transfected with siRNA against GRK6. Western blotting data showed that GRK6 siRNA reduced the expression levels of endogenous GRK6, whereas total α-syn (S129A α-syn) levels were comparable with control siRNA (Supplemental Figure S2A). Ser-129–phosphorylated α-syn was undetectable. When we compared DA uptake with control siRNA, GRK6 knockdown did not alter Vmax (15.4 ± 0.9 vs. 15.2 ± 0.9 pmol/mg protein/min in control and GRK6 siRNA, respectively; p = 0.419, n = 3, unpaired Student's t test) and Km values (0.30 ± 0.07 vs. 0.33 ± 0.05 μM in control and GRK6 siRNA, respectively; p = 0.649, n = 3) for DA uptake (Supplemental Figure S2B). We then investigated whether the reduced expression of α-syn affected DA uptake by using parental SH-SY5Y cells transfected with siRNA against α-syn. Recent articles demonstrated that the detection of endogenous α-syn monomers in Western blotting was improved by treating the blotted membranes with 0.4% paraformaldehyde (Lee and Kamitani, 2011; Dettmer et al., 2013). This enabled us to reproducibly assess the expression of endogenous α-syn in parental SH-SY5Y cells, although endogenous Ser-129–phosphorylated α-syn was undetectable (Figure 6E). α-Syn knockdown significantly suppressed endogenous expression by 86.7 ± 3.2% (Figure 6E). α-Syn knockdown significantly decreased Vmax for DA uptake by ∼38.1% (9.3 ± 0.3 vs. 5.5 ± 0.5 pmol/mg protein/min in control and α-syn siRNA, respectively; p < 0.001, n = 3, unpaired Student's t test), whereas Km values were comparable between control and α-syn siRNAs (0.36 ± 0.04 vs. 0.35 ± 0.06 μM in control and α-syn siRNA, respectively; p = 0.772, n = 3; Figure 6E).
FIGURE 6:

siRNA-mediated knockdown of GRK5 or GRK6 decreases DA uptake in SH-SY5Y cells. (A) The graph shows the effects of siRNA-mediated knockdowns of GRKs or CK2 on the DA uptake kinetics. Wt-aS/SH cells were transfected with 1 nM nonsilencing control siRNA or 1 nM siRNA targeting GRK5, GRK6, or CK2 α′ subunit. (B) The graph shows the Eadie–Hofstee plot of the kinetics data. (C) Summary of kinetic parameters. Data represent means ± SD, and p values were estimated by one-way ANOVA with a Bonferroni correction (**p < 0.01). (D) Effects of siRNA knockdown of GRK5, GRK6, or CK2 α′ subunit on the cell surface expressions of endogenous DAT in wt-aS/SH cells. The assays were performed with [3H]CFT. (E) Effects of siRNA-mediated knockdown of α-syn on DA uptake in parental SH-SY5Y cells. Left, Western blotting (WB) data of triplicate samples of control and α-syn siRNA cells. Cells were transfected with 10 nM nonsilencing control siRNA or 10 nM siRNA targeting α-syn. Cell lysates (5 μg/lane) were analyzed by WB with the indicated antibodies. Note that endogenous α-syn could be detected by treating the blotted membranes with 0.4% paraformaldehyde solution for 10 min before incubation in blocking buffer. α-Syn siRNA reduced the expression levels of endogenous α-syn. Right, DA uptake kinetics of control and α-syn siRNAs. Data represent means ± SD.
DISCUSSION
Our results demonstrate that in dopaminergic SH-SY5Y cells, GRK3, GRK5, GRK6, and CK2 containing α′ subunit contribute to Ser-129 phosphorylation of membrane-associated α-syn, whereas Ser-129 phosphorylation of cytosolic α-syn is mediated exclusively by CK2. The expression of α-syn increased DA uptake and cell surface expression of DAT. Experiments using cells stably expressing wild-type α-syn or phosphorylation-incompetent S129A mutant α-syn showed that Ser-129 phosphorylation enhanced the ability of α-syn to increase DA uptake, although phosphorylation did not affect cell surface expression of DAT. The Ser-129 phosphorylation–independent increase in DAT cell surface expression was also observed in nonneuronal HEK293 cells. The increase in DA uptake was significantly attenuated by siRNA knockdown of GRK5 or GRK6, which did not alter cell surface expression of DAT. In contrast, knockdown of CK2 α′ subunit failed to decrease DA uptake. Collectively these results indicate that GRKs-mediated Ser-129 phosphorylation of membrane-associated α-syn enhances DA uptake capacity of DAT.
These effects of α-syn were consistent with previous experiments showing that siRNA-mediated knockdown of α-syn resulted in significant decreases in Vmax for DA uptake and the surface density of DAT in SH-SY5Y cells (Fountaine and Wade-Martins, 2007). We also observed that siRNA knockdown of endogenous α-syn significantly decreased DA uptake in SH-SY5Y cells. An earlier report showed that the formation of α-syn–DAT complexes facilitated DA uptake and membrane clustering of DAT in nonneuronal Ltk− cells and HEK293 cells (Lee et al., 2001). The importance of the α-syn–DAT interaction is further supported by the in vivo findings that α-syn–knockout mice, as well as α- and β-syn double-knockout mice, exhibited a small but statistically significant reduction of DA uptake (Chandra et al., 2004; Chadchankar et al., 2011), and α-syn transgenic mice showed a modest increase in the DAT expression (Richfield et al., 2002). However, nonneuronal cell cultures overexpressing α-syn (Oaks and Sidhu, 2011) and α-syn–knockout mice with the different genetic background (Dauer et al., 2002) yielded conflicting results (Venda et al., 2010). There has been a lack of consistency regarding the effects of α-syn on the function and cell surface expression of DAT. It should be noted that toxic effects of overexpressed α-syn may lose the function and trafficking of DAT (Yamakado et al., 2012). Recent study showed that the earliest change of dopaminergic neurodegeneration was a marked reduction of DA reuptake in a rat PD model using AAV-mediated overexpression of α-syn (Lundblad et al., 2012). In addition, our data suggested that GRKs-mediated Ser-129 phosphorylation of α-syn was one of the factors for observing the relevant interactions.
We found that the contribution of GRKs and CK2 to Ser-129 phosphorylation differed for cytosolic and membrane-associated α-syn. The present experiments using DSP-treated cells showed that ∼70% of endogenous GRK3, GRK5, and GRK6 proteins was distributed in the membrane fractions, whereas CK2 containing α′ subunit in the membrane fractions was ∼40%. The difference in the subcellular distribution among GRKs and CK2 may be insufficient to explain how these GRKs contribute only to phosphorylation of membrane-associated α-syn. GRK5 and GRK6 are subdivided into the same group in the GRK family, and this group is known to exhibit a constitutive association with cellular membranes (Reiter and Lefkowitz, 2006). GRK5 binding to the cytoplasmic face of the plasma membrane is mediated by electrostatic interactions involving the amphipathic α-helix (Thiyagarajan et al., 2004). GRK6 attaches to the plasma membrane by amphipathic α-helix and/or the C-terminal palmitoyl moieties (Stoffel et al., 1994; Jiang et al., 2007). In GRK5 and GRK6, the predominance of membrane-associated proteins (∼70% of total fractions) seems to reflect their constitutive association with cellular membranes. Of importance, earlier studies revealed that GRK5 and GRK6 mutants, which were defective in plasma membrane localization, decreased ability to phosphorylate the soluble substrates, as well as membrane-bound ones (Stoffel et al., 1998; Thiyagarajan et al., 2004). In addition, our experiments showed that DSP treatment significantly increased the amounts of GRK3 in the membrane fractions by ∼70% of total fractions. This finding suggested that the majority of GRK3 was localized close to membrane components. GRK3, which constitutes the GRK2 subgroup, undergoes stimulus-dependent translocation from the cytosol to the plasma membrane (Boekhoff et al., 1994). The translocation to the plasma membrane is mediated by the C-terminal pleckstrin homology domain, which binds to both acidic membrane phospholipids and free G-protein βγ subunits (Boekhoff et al., 1994; Premont et al., 1995). Binding of the membrane lipids modulates the activity of GRKs containing the pleckstrin homology domain (DebBurman et al., 1995). Previous in vitro experiments revealed that GRK2 and GRK5 could phosphorylate α-syn at Ser-129, and this phosphorylation was enhanced by phospholipids (Pronin et al., 2000). Thus it seems possible that the interaction of GRK3, GRK5, and GRK6 with cellular membranes regulates contribution to phosphorylation of α-syn in addition to their predominant distribution in the membrane fractions. In addition, our data showed that siRNA knockdown of GRK2 elevated the amounts of Ser-129–phosphorylated α-syn in the cytosol, suggesting that GRK2 acts as an inhibitory factor on phosphorylation of cytosolic α-syn. Although the mechanism by which GRK2 does not exert its catalytic activity to cytosolic α-syn is unknown, GRK2 may functionally block phosphorylation of α-syn by other GRKs in the cytosol. It would be important to elucidate why GRK3, GRK5, and GRK6 fail to exert their basal activities to phosphorylate α-syn in the cytosol.
The present experiments using siRNA knockdowns against individual GRKs or CK2 α′ subunit demonstrated that multiple kinases contributed to Ser-129 phosphorylation of membrane-associated α-syn, suggesting redundancy of these kinases in the phosphorylation. The findings raise the question of how different kinases modulate the phosphorylation. We did not find obvious evidence that remaining kinases compensated for the reduced expression or activity of target kinase in the phosphorylation, because siRNA knockdown of each kinase significantly decreased Ser-129 phosphorylation of membrane-associated α-syn. However, the present data do not exclude the possibility that other kinases require a longer time or complete deletion of target kinase to exert the compensatory effects. In addition, our siRNA knockdown data showed that the total sum of reduced effects on Ser-129 phosphorylation of membrane-associated α-syn was estimated as >100%. One may speculate that DSP treatment at 37°C excessively trapped phosphorylated α-syn in the membrane fractions. However, there was no significant difference in the subcellular distribution between DSP treatment conditions of 37 and 4°C. Alternatively, siRNA knockdowns of these kinases may induce the interaction with a protein that inhibits Ser-129 phosphorylation of membrane-associated α-syn. Among the kinases, siRNA knockdown of CK2 α′ subunit had the greatest effect on phosphorylation of membrane-associated α-syn, whereas siRNA knockdown of CK2 α subunit increased the phosphorylation. The decreased expression of α′ subunit may yield replacement with CK2 containing α subunit that is inactive for phosphorylation of membrane-associated α-syn, resulting in further reduction of the phosphorylation.
We also found that GRKs and CK2 had distinct effects on DAT through Ser-129 phosphorylation of membrane-associated α-syn. Although knockdown of CK2 α′ subunit suppressed Ser-129 phosphorylation of membrane-associated α-syn more effectively than that of individual GRKs, modulation of DAT function was seen by knockdown of GRK5 or GRK6 but not of CK2. The exact reason for this is unknown from the present study. α-Syn is known to interact with the C-terminal tail of DAT (Lee et al., 2001). DAT is reported to directly interact with DA receptor, a GPCR, which can be desensitized by GRKs (Ito et al., 1999; Kim et al., 2001; Lee et al., 2007). These findings raise the possibility that GRKs may be localized with membrane-associated α-syn–DAT complexes more closely than CK2. In addition, a previous article showed that Sept4 physically associated with α-syn and DAT and functioned as a component of the presynaptic scaffold for the machinery of DA uptake (Ihara et al., 2007). That article further demonstrated that Sept4 partially interfered with Ser-129 phosphorylation of α-syn by CK2 in vitro. There may be a scaffold protein that affects interaction between kinases and membrane-associated α-syn–DAT complexes. Alternatively, DAT-bound α-syn may change more easily accessible conformation to GRKs than CK2. Further studies using a cellular model in which GRK levels are varied would help to clarify whether the Ser-129 phosphorylation state affects the interaction between α-syn and DAT. In addition, phosphorylated α-syn is observed in cortical LBs (Fujiwara et al., 2002) and rat primary cortical neurons (Machiya et al., 2010). It is unclear what role phosphorylated α-syn plays in cerebral cortical neurons. α-Syn is reported to interact with norepinephrine and serotonin transporters in a manner similar to DAT (Oaks and Sidhu, 2011). This may suggest that Ser-129 phosphorylation of α-syn has effects on the function of these transporters in cerebral cortical neurons.
In in vivo experiments using recombinant AAV, coexpression of A53T α-syn and GRK6 yielded authentically phosphorylated α-syn at Ser-129 and accelerated α-syn–induced degeneration of neurons (Sato et al., 2011). The accelerating effect was transient and did not affect the formation of α-syn aggregates, suggesting that the toxicity mediated by Ser-129 phosphorylation is independent of α-syn aggregation. GRKs-mediated Ser-129-phoshorylation of membrane-associated α-syn may be involved in dopaminergic neurodegeneration in PD by elevating the intracellular concentration of DA. Additional research on the role of Ser-129 phosphorylation would further our understanding of the mechanisms that cause α-syn toxicity and facilitate the development of therapeutic strategies for PD.
MATERIALS AND METHODS
Plasmid cDNAs and reagents
Human wild-type α-syn and phosphorylation-incompetent mutant S129A α-syn cDNAs were subcloned into the pcDNA3.1(+) vector (Invitrogen, Carlsbad, CA) as previously described (Machiya et al., 2010). Human DAT cDNA (IMAGE clone ID #40146999) was obtained from Open Biosystems (Huntsville, AL) and subcloned into the pcDNA3.1/Zeo vector (Invitrogen). Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Cell culture and transfection
Human dopaminergic neuroblastoma SH-SY5Y cells (ECACC #94030304) were maintained in Ham-12/Eagle's minimum essential medium supplemented with 15% fetal bovine serum (Invitrogen), 2 mM l-glutamine (Invitrogen), and 1× nonessential amino acids. SH-SY5Y cell lines stably expressing either wild-type α-syn (wt-aS/SH #4) or S129A mutant α-syn (S129A-aS/SH #17) were selected against 1 mg/ml G418 (Invitrogen). For making SH-SY5Y cell lines stably coexpressing DAT and GFP (GFP+DAT/SH), wild-type α-syn (wt-aS+DAT/SH), or S129A α-syn (S129A-aS+DAT/SH), cell lines stably expressing DAT were first generated in selection medium containing 500 μg/ml Zeocin, and then double stable cell lines were obtained in selection medium containing 1 mg/ml G418. HEK293 cells were maintained in Eagle's minimum essential medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum and 2 mM l-glutamine. HEK293 cells stably expressing DAT (DAT/HEK) were selected against 500 μg/ml Zeocin. For the transient overexpression, the cells were transfected with cDNA using Lipofectamine Plus Reagents (Invitrogen) according to the manufacturer's protocol and harvested after 48 h of transfection.
siRNA transfection
Approximately 30% confluent cells in six-well plates were transfected with siRNA oligonucleotides (final concentration at 1 nM, stealth RNA interference [RNAi]; Invitrogen) using RNAiMAX reagent (Invitrogen) according to the manufacturer's protocol. We used the 25-nucleotide-long siRNAs for the knockdown of GRK2 (5′-GGAAGCGUGACACAGGCAAGAUGUA-3′), GRK3 (5′-CAAGAAACAAGUGACAUCAACUCUU-3′), GRK5 (5′-GACCACACAGACGACGACUUCUACU-3′), GRK6 (5′-GCCGACUACCUCGACAGCAUCUACU-3′), CK2α (5′-ACCAGACGUUAACAGACUAUGAUAU-3′), or CK2α′ (5′-GCAUGAUCUUUCGAAGGGAACCAUU-3′). As a nonsilencing control for stealth siRNAs, the stealth RNAi–negative control medium GC (Invitrogen) was used. At 72 h after transfection, the medium was discarded, and the cells were retransfected with siRNAs. At 48 h after a second transfection, the cells were harvested for experiments. For the α-syn knockdown, we transfected cells with 10 nM siRNA oligonucleotides (5′-GACCAAAGAGCAAGUGACAAAUGUU-3′).
Chemical cross-linking and subcellular fractionation
For chemical cross-linking, the cells in 100 mm-dishes were quickly rinsed twice with ice-cold phosphate-buffered saline (PBS; pH 7.4; 2.97 mM Na2HPO4⋅7H2O, 1.06 mM KH2PO4, 155 mM NaCl) and incubated in 6 ml of PBS containing 1 mM of membrane permeable cross-linker DSP (Thermo Scientific, Waltham, MA) for 30 min at 37°C. Then the buffer was removed and the cells were rinsed twice with Tris-buffered saline (TBS; 25 mM Tris-HCl, pH 7.4, 137 mM NaCl, 2.7 mM KCl) to quench the cross-linking reaction.
For subcellular fractionation, the cross-linked cells were suspended in 5 vol/wt of homogenization buffer (TBS plus 2 mM EDTA, 1× protease inhibitor cocktail [Roche, Indianapolis, IN], and 1× PhosSTOP [Roche]) and disrupted by a Teflon Potter homogenizer. The homogenate was centrifuged at 600 × g for 10 min and the supernatant was centrifuged at 100,000 × g for 30 min at 4°C. The resultant supernatant was recovered as the cytosol fraction. The post–100,000 × g pellet was disrupted by sonication in the same volume of radioimmunoprecipitation assay (RIPA) buffer (homogenization buffer plus 1% Triton-X 100, 0.5% deoxycholic acid, and 0.1% SDS), and the homogenate was incubated on ice for 30 min. After centrifugation at 12,000 × g for 30 min, the supernatant was recovered as the membrane fraction.
Primary antibodies
The following antibodies were used: anti–α-syn (Syn-1; it recognizes total α-syn, including nonphosphorylated and phosphorylated forms; BD Transduction Laboratories, Lexington, KY), anti–Ser-129-phosphorylated α-syn (psyn#64; Wako, Osaka, Japan), anti-GRK2 (#sc-562; Santa Cruz Biotechnology, Santa Cruz, CA), anti-GRK3 (#sc-563; Santa Cruz Biotechnology), anti-GRK6 (#sc-566; Santa Cruz Biotechnology), anti-GRK6 (#EPR2046(2), rabbit monoclonal; Abcam, Cambridge, MA), anti-CK2α (#sc-6479; Santa Cruz Biotechnology), anti-CK2α′ (#sc-6481; Santa Cruz Biotechnology), anti–β-actin (AC-15; Sigma-Aldrich), anti-Cu/Zn superoxide dismutase (SOD1-100; Stressgen, San Diego, CA), anti–amyloid precursor protein (#A8717; Sigma-Aldrich), anti-NCT (#16420; BD Transduction Laboratories), anti–glyceraldehyde-3-phosphate dehydrogenase (#6C5, Abcam), and anti-DAT (MAB369; Millipore, Billerica, MA). Hamster anti-GRK5 monoclonal antibody (#139) was described previously (Sakamoto et al., 2009).
Western blotting
For cell lysis, the cells were suspended in homogenization buffer containing 1% Nonidet P-40 and 10% glycerol and kept on ice for 30 min. After centrifugation at 12,000 × g for 30 min, the resultant supernatant was collected and stored at −80°C until use. Samples were denatured by boiling for 5 min in Laemmli's sample buffer containing 2.5% 2-mercaptoethanol. To detect DAT, samples were incubated at 37°C for 30 min in Laemmli's sample buffer containing 0.2 M dithiothreitol. Equal amounts of denatured samples were subjected to 12.5% polyacrylamide gel. Western blotting was performed by the methods described previously (Machiya et al., 2010). Signals were detected using a charge-coupled device camera, VersaDog 5000 (Bio-Rad, Hercules, CA). When we detected phosphorylated α-syn, we incubated the membrane in buffer containing 50 mM NaF. For detection of endogenous α-syn, membranes were incubated with 0.4% paraformaldehyde/PBS for 10 min at room temperature, rinsed three times with TBS containing 0.05% (vol/vol) Tween 20 (TBS-T), and blocked in 5% skim milk in TBS-T (Lee and Kamitani, 2011; Dettmer et al., 2013). For estimating the expression levels of total α-syn and Ser-129–phosphorylated α-syn, we used purified recombinant α-syn proteins and Ser-129–phosphorylated α-syn proteins as standards, respectively (Machiya et al., 2010). A set of diluted standards was subjected to SDS–PAGE along with samples. After quantifying band intensities of samples with Quantity One software (Bio-Rad), we corrected their relative intensities by plotting them on the standard curve. Statistical comparisons were made by unpaired Student's t test.
[3H]DA uptake assay
DA uptake kinetics in SH-SY5Y cells was measured using [3H]DA as described previously (Jiang et al., 2004). Briefly, cells were plated at a density of (1–5) × 105 cells per well on 12-well plates 1 d before assays. Medium was removed, and cells were rinsed with uptake buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, 130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 10 mM glucose, 1 mM ascorbic acid). Assays were initiated by the addition of uptake buffer containing 20 nM [3H]DA ([7,8-3H]DA, 50.0 Ci/mmol; GE Healthcare, Piscataway, NJ) along with 0.125–10 or 0.5–60 μM nonlabeling DA. After incubation for 10 min at 37°C, the cells were immediately rinsed three times with ice-cold uptake buffer without ascorbic acid and then solubilized with 0.5 ml of 1% SDS. Incorporated radioactivity was measured by using a liquid scintillation counter (TRI-CARB 2300TR; PerkinElmer, Waltham, MA). Nonspecific [3H]DA uptake was measured in the presence of DAT specific antagonist 10 μM GBR12909 and was subtracted from total counts. The protein concentrations were measured by bicinchoninic acid methods (Thermo Scientific). Statistical comparisons were performed using one-way analysis of variance (ANOVA) with a Bonferroni correction.
[3H]CFT binding assay
We assessed the cell surface expression of endogenous DAT by performing [3H]CFT binding assay on the cells as described previously (Jiang et al., 2004). The cells were plated at a density of (1–5) × 105 cells per well on 12-well plates 1 d before assays. Medium was removed, and cells were rinsed with binding buffer (10 mM phosphate, pH 7.4, 0.32 M sucrose). Assays were initiated by the addition of binding buffer containing 3H-labeled 4 nM CFT (85.9 Ci/mmol; PerkinElmer). After incubation for 2 h at 4°C, the cells were immediately rinsed twice with ice-cold binding buffer and then solubilized with 1% SDS. Incorporated radioactivity was measured by using a liquid scintillation counter. Nonspecific binding was determined in the presence of DA reuptake inhibitor 10 μM nomifensine and was subtracted from total counts. Statistical comparisons were performed using one-way ANOVA with a Bonferroni correction.
Cell surface biotinylation
The cells were washed three times with PBS+ (PBS, 0.1 mM CaCl2, 1 mM MgCl2) and then incubated in PBS+ containing 1 mg/ml SulfoNHS-SS-biotin (Thermo Scientific) with gentle agitation for 30 min at 4°C. The reaction was quenched by incubating the cells with PBS+ containing 0.1 M glycine for 20 min. The cells were washed three times with PBS+ and lysed with RIPA buffer containing 10% glycerol on ice for 30 min. Samples was divided into two aliquots. One was used for isolation of biotinylated proteins with UltraLink immobilized NeutrAvidin beads (Thermo Scientific), and the other was used to determine total DAT. Samples were analyzed by Western blotting with anti-DAT antibody.
Supplementary Material
Acknowledgments
This work was supported in part by a Grant-in-Aid from the Global Center of Excellence Program (F03) of the Japan Society for the Promotion of Science (T.K.) and a Grant-in-Aid for Scientific Research (C) (No. 23591230) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (S.A.).
Abbreviations used:
- α-syn
α-synuclein
- AAV
adeno-associated virus
- CFT
2β-carbomethoxy-3β-(4-fluorophenyl)tropane
- CK
casein kinase
- DA
dopamine
- DAT
dopamine transporter
- DSP
dithiobis(succinimidyl propionate)
- GRK
G protein–coupled receptor kinase
- LB
Lewy body
- NCT
nicastrin
- PD
Parkinson disease
- siRNA
small interfering RNA
- TBS
Tris-buffered saline
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0903) on April 10, 2013.
We declare no conflict of interest.
REFERENCES
- Anderson JP, et al. Phosphorylation of Ser-129 is the dominant pathological modification ofα-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Arawaka S, et al. The levels of mature glycosylated nicastrin are regulated and correlate with γ-secretase processing of amyloid β-precursor protein. J Neurochem. 2002;83:1065–1071. doi: 10.1046/j.1471-4159.2002.01207.x. [DOI] [PubMed] [Google Scholar]
- Boekhoff I, Inglese J, Schleicher S, Koch WJ, Lefkowitz RJ, Breer H. Olfactory desensitization requires membrane targeting of receptor kinase mediated by beta gamma-subunits of heterotrimeric G proteins. J Biol Chem. 1994;269:37–40. [PubMed] [Google Scholar]
- Chadchankar H, Ihalainen J, Tanila H, Yavich L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res. 2011;1382:37–44. doi: 10.1016/j.brainres.2011.01.064. [DOI] [PubMed] [Google Scholar]
- Chandra S, et al. Double-knockout mice for α- and β-synucleins: effect on synaptic functions. Proc Natl Acad Sci USA. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Feany MB. α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Chen L, Periquet M, Wang X, Negro A, McLean PJ, Hyman BT, Feany MB. Tyrosine and serine phosphorylation of α-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J Clin Invest. 2009;119:3257–3265. doi: 10.1172/JCI39088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT Jr. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Dauer W, et al. Resistance of α-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- DebBurman SK, Ptasienski J, Boetticher E, Lomasney JW, Benovic JL, Hosey MM. Lipid-mediated regulation of G protein-coupled receptor kinases 2 and 3. J Biol Chem. 1995;270:5742–5747. doi: 10.1074/jbc.270.11.5742. [DOI] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D. In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J Biol Chem. 2013;288:6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Dawson TM, Dickson DW, Petrucelli L. Caught in the act: α-synuclein is the culprit in Parkinson's disease. Neuron. 2003;40:453–456. doi: 10.1016/s0896-6273(03)00684-6. [DOI] [PubMed] [Google Scholar]
- Fountaine TM, Wade-Martins R. RNA interference-mediated knockdown of α-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J Neurosci Res. 2007;85:351–363. doi: 10.1002/jnr.21125. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Ihara M, et al. Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of α-synuclein neurotoxicity. Neuron. 2007;53:519–533. doi: 10.1016/j.neuron.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Inglis KJ, et al. Polo-like kinase 2 (PLK2) phosphorylates α-synuclein at serine 129 in central nervous system. J Biol Chem. 2009;284:2598–2602. doi: 10.1074/jbc.C800206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Haga T, Lameh J, Sadee W. Sequestration of dopamine D2 receptors depends on coexpression of G-protein-coupled receptor kinases 2 or 5. Eur J Biochem. 1999;260:112–119. doi: 10.1046/j.1432-1327.1999.00125.x. [DOI] [PubMed] [Google Scholar]
- Jiang H, Jiang Q, Feng J. Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J Biol Chem. 2004;279:54380–54386. doi: 10.1074/jbc.M409282200. [DOI] [PubMed] [Google Scholar]
- Jiang X, Benovic JL, Wedegaertner PB. Plasma membrane and nuclear localization of G protein-coupled receptor kinase 6A. Mol Biol Cell. 2007;18:2960–2969. doi: 10.1091/mbc.E07-01-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karube H, et al. N-terminal region of α-synuclein is essential for the fatty acid-induced oligomerization of the molecules. FEBS Lett. 2008;582:3693–3700. doi: 10.1016/j.febslet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem. 2001;276:37409–37414. doi: 10.1074/jbc.M106728200. [DOI] [PubMed] [Google Scholar]
- Kruger R, et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Lee BR, Kamitani T. Improved immunodetection of endogenous α-synuclein. PLoS One. 2011;6:e23939. doi: 10.1371/journal.pone.0023939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of α-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15:916–926. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Pei L, Moszczynska A, Vukusic B, Fletcher PJ, Liu F. Dopamine transporter cell surface localization facilitated by a direct interaction with the dopamine D2 receptor. EMBO J. 2007;26:2127–2136. doi: 10.1038/sj.emboj.7601656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological α-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc Natl Acad Sci USA. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M, Koyama S, Kato T. Phosphorylated α-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J Biol Chem. 2010;285:40732–40744. doi: 10.1074/jbc.M110.141952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbefo MK, et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem. 2010;285:2807–2822. doi: 10.1074/jbc.M109.081950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Ribich S, Hyman BT. Membrane association and protein conformation of α-synuclein in intact neurons. Effect of Parkinson's disease-linked mutations. J Biol Chem. 2000;275:8812–8816. doi: 10.1074/jbc.275.12.8812. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, et al. Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oaks AW, Sidhu A. Synuclein modulation of monoamine transporters. FEBS Lett. 2011;585:1001–1006. doi: 10.1016/j.febslet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson's disease associated α-synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Woods WS, Clayton DF, George JM. Interaction of human α-synuclein and Parkinson's disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem. 2000;275:34393–34398. doi: 10.1074/jbc.M004851200. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Premont RT, Inglese J, Lefkowitz RJ. Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J. 1995;9:175–182. doi: 10.1096/fasebj.9.2.7781920. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Morris AJ, Surguchov A, Benovic JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000;275:26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ. Behavioral and neurochemical effects of wild-type and mutated human α-synuclein in transgenic mice. Exp Neurol. 2002;175:35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- Sakamoto M, et al. Contribution of endogenous G-protein-coupled receptor kinases to Ser129 phosphorylation of α-synuclein in HEK293 cells. Biochem Biophys Res Commun. 2009;384:378–382. doi: 10.1016/j.bbrc.2009.04.130. [DOI] [PubMed] [Google Scholar]
- Sato H, Arawaka S, Hara S, Fukushima S, Koga K, Koyama S, Kato T. Authentically phosphorylated α-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson's disease. J Neurosci. 2011;31:16884–16894. doi: 10.1523/JNEUROSCI.3967-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of α-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Singleton AB, et al. α-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stoffel RH, Inglese J, Macrae AD, Lefkowitz RJ, Premont RT. Palmitoylation increases the kinase activity of the G protein-coupled receptor kinase, GRK6. Biochemistry. 1998;37:16053–16059. doi: 10.1021/bi981432d. [DOI] [PubMed] [Google Scholar]
- Stoffel RH, Randall RR, Premont RT, Lefkowitz RJ, Inglese J. Palmitoylation of G protein-coupled receptor kinase, GRK6. Lipid modification diversity in the GRK family. J Biol Chem. 1994;269:27791–27794. [PubMed] [Google Scholar]
- Thiyagarajan MM, Stracquatanio RP, Pronin AN, Evanko DS, Benovic JL, Wedegaertner PB. A predicted amphipathic helix mediates plasma membrane localization of GRK5. J Biol Chem. 2004;279:17989–17995. doi: 10.1074/jbc.M310738200. [DOI] [PubMed] [Google Scholar]
- Venda LL, Cragg SJ, Buchman VL, Wade-Martins R. α-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci. 2010;33:559–568. doi: 10.1016/j.tins.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT Jr. Vesicle permeabilization by protofibrillar α-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of α-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- Yamakado H, Moriwaki Y, Yamasaki N, Miyakawa T, Kurisu J, Uemura K, Inoue H, Takahashi M, Takahashi R. α-Synuclein BAC transgenic mice as a model for Parkinson's disease manifested decreased anxiety-like behavior and hyperlocomotion. Neurosci Res. 2012;73:173–177. doi: 10.1016/j.neures.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.