

β1 integrin is a major regulator of invadopodium maturation. Studies reveal that β1 integrin–mediated adhesion is a key upstream switch that induces Arg-dependent cortactin phosphorylation, actin polymerization, and MMP recruitment to invadopodia for extracellular matrix degradation.
Abstract
β1 integrin has been shown to promote metastasis in a number of tumor models, including breast, ovarian, pancreatic, and skin cancer; however, the mechanism by which it does so is poorly understood. Invasive membrane protrusions called invadopodia are believed to facilitate extracellular matrix degradation and intravasation during metastasis. Previous work showed that β1 integrin localizes to invadopodia, but its role in regulating invadopodial function has not been well characterized. We find that β1 integrin is required for the formation of mature, degradation-competent invadopodia in both two- and three-dimensional matrices but is dispensable for invadopodium precursor formation in metastatic human breast cancer cells. β1 integrin is activated during invadopodium precursor maturation, and forced β1 integrin activation enhances the rate of invadopodial matrix proteolysis. Furthermore, β1 integrin interacts with the tyrosine kinase Arg and stimulates Arg-dependent phosphorylation of cortactin on tyrosine 421. Silencing β1 integrin with small interfering RNA completely abrogates Arg-dependent cortactin phosphorylation and cofilin-dependent barbed-end formation at invadopodia, leading to a significant decrease in the number and stability of mature invadopodia. These results describe a fundamental role for β1 integrin in controlling actin polymerization–dependent invadopodial maturation and matrix degradation in metastatic tumor cells.
INTRODUCTION
Although significant advances have been made in the screening and treatment of primary cancers, metastasis remains the major cause of cancer-related death in these patients. For cells to escape from the primary tumor, actin-based invasive protrusions called invadopodia are believed to facilitate tumor cell basement membrane degradation, migration through the stroma, and intravasation (Eckert et al., 2011; Huttenlocher and Horwitz, 2011; Linder et al., 2011; Magalhaes et al., 2011). Invadopodia are enriched in actin regulators, such as N‑WASp, cortactin, Arp2/3, and cofilin, as well as in proteinases (e.g., membrane type 1 matrix metalloprotease [MT1-MMP], MMP-2, MMP‑9, and seprase; Mueller et al., 1999; Oser et al., 2009). In contrast to podosomes formed by hematopoietic cells, focal adhesion proteins (e.g., vinculin, paxillin, and focal adhesion kinase) are not concentrated in invadopodial protrusions (cores), with the exception of β1 integrin and talin (Mueller et al., 1999; Chan et al., 2009; Huttenlocher and Horwitz, 2011; Linder et al., 2011). Although it is well established that podosomes are adhesive structures, it is unclear whether the invadopodium core adheres to the extracellular matrix (ECM) and whether local adhesion regulates invadopodial function in metastatic tumor cells (Linder et al., 2011).
Integrins belong to a family of heterodimeric transmembrane adhesion proteins consisting of 18 α and 8 β subunits that can assemble into 24 distinct heterodimers (Takada et al., 2007). In addition to facilitating cell adhesion to the ECM, integrins have also been implicated in nearly every stage of tumor development, including tumor induction, epithelial–mesenchymal transition, local invasion, migration, intravasation, extravasation, and dissemination (Huttenlocher et al., 1998; Felding-Habermann et al., 2001; Guo and Giancotti, 2004; White et al., 2004; Maschler et al., 2005; Park et al., 2006; Sameni et al., 2008; Huck et al., 2010). β1 integrin is up-regulated in highly invasive breast carcinoma cells in vivo and promotes metastasis in a number of tumor models, including breast, ovarian, pancreatic, and skin cancer (Trikha et al., 1994; Wang et al., 2004; Huck et al., 2010; Grzesiak et al., 2011; Lahlou and Muller, 2011; Mitra et al., 2011). Consistent with a role in tumor cell invasion, β1 integrin has been shown to promote degradation of type IV collagen at the whole-cell level and localize to invadopodia; Chen and coworkers demonstrated that α3β1 and α6β1 integrins localize to the invadopodium core in highly metastatic LOX melanoma cells (Mueller et al., 1999; Sameni et al., 2008). Although it is known that β1 integrin activates p190RhoGAP and recruits seprase to the invadopodium core (Nakahara et al., 1998; Mueller et al., 1999), the detailed mechanism by which β1 integrin regulates invadopodia is poorly understood (Buccione et al., 2009; Linder et al., 2011; Murphy and Courtneidge, 2011).
The stages of invadopodium formation and maturation have been characterized (Artym et al., 2006; Oser et al., 2009). Briefly, growth factors (e.g., epidermal growth factor [EGF]) stimulate invadopodium precursor formation through the assembly of a core structure comprising cortactin, N-WASp, cofilin, and Tks5 (Artym et al., 2006; Oser et al., 2009). The activated EGF receptor (EGFR) is believed to recruit the tyrosine kinase Src by its SH2 domain, leading to Src-mediated activation of the Abl-family kinase Arg (Mader et al., 2011). Arg has been identified as the primary kinase that phosphorylates cortactin on tyrosine 421 (Y421), which induces Na+/H+ exchanger-1 (NHE-1) and Nck1 recruitment to invadopodia (Oser et al., 2009; Mader et al., 2011; Magalhaes et al., 2011). NHE‑1 increases the local intracellular pH to release cofilin from its inhibitory interaction with cortactin (Magalhaes et al., 2011). This allows cofilin severing to generate free actin barbed ends, which elongate to form daughter filaments, on which Nck1 is believed to induce N‑WASp-Arp2/3–dependent actin polymerization at invadopodia (Oser et al., 2009). Although cortactin tyrosine phosphorylation and cofilin severing activity are not required for initial invadopodium precursor formation, they are critical for induction of actin polymerization and matrix degradation, which are collectively referred to as invadopodial maturation (Yamaguchi et al., 2005; Artym et al., 2006; Oser et al., 2009). Thus Arg-mediated cortactin phosphorylation is a key switch that initiates invadopodial maturation.
Although Arg has been shown to play a prominent role in regulating invadopodial function (Mader et al., 2011), the upstream regulators of Arg in invadopodia are poorly understood. Arg kinase activity is enhanced by cell adhesion in fibroblasts, and Arg was shown to bind to the β1 integrin cytoplasmic tail in vitro and in neurons (Hernandez et al., 2004; Lapetina et al., 2009; Warren et al., 2012). In the present study, we investigate how β1 integrin regulates invadopodial formation, maturation, and dynamics and evaluate whether β1 integrin interacts with Arg and affects its kinase activity at invadopodia.
RESULTS
β1 integrin is required for invadopodial maturation, stability and matrix degradation but not precursor formation
Metastatic human MDA-MB-231 breast carcinoma cells were selected to study the role of β1 integrin in regulating invadopodia because they produce numerous invadopodia, express high levels of β1 integrin relative to β3 and β5 integrins, and are highly invasive in vivo (Artym et al., 2006; Patsialou et al., 2009; Mierke et al., 2011). To assess whether β1 integrin regulates invadopodium formation, we treated MDA‑MB‑231 cells with control or β1 integrin siRNA. β1 integrin knockdown by two different small interfering RNAs (siRNAs) was highly efficient, resulting in >95% reduction in β1 integrin expression (Figure 1A and Supplemental Figure S2A). MDA-MB-231 cell spreading on gelatin requires both β1 integrin– and β3 integrin–mediated adhesion, as cell spreading is unaffected by siRNA knockdown of either protein alone but is impaired in β1/β3 integrin double-knockdown cells (Figure 1B and Supplemental Figure S1E). We took advantage of the fact that overall cell adhesion and focal adhesion formation on Alexa 405–labeled gelatin were modestly or not affected in β1 integrin–knockdown cells to evaluate its specific role in regulating the invadopodial compartment (Supplemental Figure S1, A–E).
FIGURE 1:

β1 integrin is required for invadopodial maturation, stability, and matrix degradation but not precursor formation. (A) Western blot analysis of MDA-MB-231 cells transfected with control or β1 integrin siRNA (SMARTpool) for 96 h. Blots were stained for β1 integrin and β-actin (loading control). (B, C) Steady-state invadopodial matrix degradation assay: MDA-MB-231 cells were plated on Alexa 405–labeled gelatin for 4 h. (B) Representative images of β1 integrin (C27), cortactin (sc-30771), and Tks5 staining. Red arrowheads denote mature invadopodia; blue arrowheads denote invadopodium precursors. Inset, magnified image of invadopodia in the box. Bar, 10 μm. (C) Quantification of mature invadopodia formed by control or β1 integrin siRNA (SMARTpool)–treated cells in the steady-state invadopodial matrix degradation assay. Mature invadopodia were scored as cortactin-Tks5–rich structures that colocalize with a degradation hole in Alexa 405–labeled gelatin. n > 100 cells; three independent experiments. *p < 0.0025 compared with control siRNA. (D) Invadopodium precursor formation assay: quantification of the number of cortactin- and Tks5-rich invadopodium precursors formed in MDA-MB-231 cells stimulated with EGF for 0 (untreated), 1, 3, or 5 min. Precursors were scored as punctate cortactin-Tks5–rich structures that do not colocalize with a degradation hole in Alexa 405–labeled gelatin. n > 45 cells; three independent experiments. *p < 0.017 compared with control siRNA 0 min; **p < 0.007 compared with β1 integrin siRNA 0 min. (E, F) TagRFP-cortactin– and GFP-Tks5–expressing control and β1 integrin–knockdown cells were plated on Alexa 405–labeled gelatin and imaged by time-lapse microscopy for 3 h. (E) Representative images of a TagRFP-cortactin– and GFP-Tks5–rich mature invadopodium formed by a control cell and a short-lived, invadopodium precursor formed by β1 integrin–depleted cells (see Supplemental Movies S1 and S2). Box, 3.85 μm. (F) Quantification of invadopodium lifetimes in control and β1 integrin siRNA (SMARTpool)–treated cells generated from time-lapse movies. n > 250 invadopodia; n > 22 cells; three independent experiments. *p < 0.0002 compared with control siRNA. (G) Quantification of invadopodial degradation area/field in the steady-state invadopodial matrix degradation assay normalized to the number of cells/field. *p < 0.003 compared with control siRNA.
MDA-MB-231 cells were plated on thin, Alexa 405–labeled gelatin for 4 h to visualize invadopodial matrix degradation (Mader et al., 2011) and immunostained for β1 integrin and the invadopodium markers cortactin and Tks5 (Seals et al., 2005; Chan et al., 2009; Oser et al., 2009). We found that β1 integrin localizes to the core of mature, actively degrading invadopodia in control cells, and silencing β1 integrin, but not β3 integrin, results in significant reduction in the number of mature invadopodia (Figure 1, B and C, and Supplemental Figures S1, G and H, and S2B; p < 0.0025; p < 0.01). Accordingly, there is a fourfold decrease in the mean degradation area/cell in β1 integrin–knockdown cells, indicating that these cells are less degradative during the 4-h plating period overall (Figure 1G). Knocking down β1 integrin in MTLn3 cells—another highly metastatic mammary adenocarcinoma cell line—also results in a decrease in the number of mature invadopodia, suggesting that β1 integrin may play a general role in regulating invadopodial maturation in metastatic breast cancer cells (Supplemental Figure S2, C and D).
Invadopodia initially form as nonproteolytic precursor structures, which polymerize actin and recruit MMPs to develop into fully functional, mature invadopodia (Artym et al., 2006; Sakurai-Yageta et al., 2008; Oser et al., 2009). To determine whether β1 integrin affects the early stages of invadopodium precursor formation, control and β1 integrin–depleted MDA‑MB-231 cells were serum starved overnight and stimulated with EGF to synchronously induce precursor formation (Oser et al., 2010). Control and β1 integrin–knockdown cells form nearly identical numbers of cortactin/Tks5–rich invadopodium precursors during the first few minutes after EGF stimulation (Figure 1D), suggesting that the early stages of EGF-induced invadopodium precursor formation can occur in a β1 integrin–independent manner. Consistent with a role in precursor maturation, β1 integrin is enriched by 1.7- and 1.6-fold after 3 and 5 min of EGF stimulation, respectively, and is enriched 1.8-fold in mature invadopodia compared with precursors at steady state (Supplemental Figure S3).
Synergistic cofilin/Arp2/3–dependent actin polymerization is required to stabilize invadopodia, allowing them to mature and degrade the ECM (Yamaguchi et al., 2005). Because integrins are adhesion receptors, we hypothesized that β1 integrin might also stabilize invadopodia by anchoring them to the ECM for efficient, focused matrix degradation. To test this, we plated control and β1 integrin–depleted cells expressing Tag red fluorescent protein (RFP)–cortactin and green fluorescent protein (GFP)-Tks5 on Alexa 405–labeled gelatin and used live-cell time-lapse microscopy to determine invadopodium lifetime. Invadopodium precursors were identified as punctate, nondegrading TagRFP-cortactin/GFP-Tks5–rich structures, whereas mature invadopodia were defined as structures that were actively degrading the ECM. Control cells form many long-lived invadopodia that often persist in a single location for >1 h; β1 integrin–depleted cells, on the other hand, form unstable invadopodia that often cannot anchor to the matrix and quickly disassemble (Supplemental Movies S1 and S2). When we quantified invadopodium lifetimes, we found that invadopodium precursor and mature invadopodium lifetimes were reduced by ∼50 and ∼40%, respectively, in cells treated with β1 integrin siRNA compared with control (Figure 1, E and F). Taken together, these data suggest that β1 integrin is dispensable for initial invadopodium precursor formation, but its function is critical for the subsequent stabilization and maturation of these precursors into fully functional, degradative invadopodia.
β1 integrin is activated in invadopodium precursors, and stimulation of β1 integrin–mediated adhesion accelerates invadopodial maturation in EGF-stimulated cells
Integrins are believed to exist in three major conformation states in cells: inactive (bent), activated/“primed” (extended but unligated) and adherent (extended and ligated to the ECM; Frelinger et al., 1988; Xiong et al., 2001; Takagi et al., 2002; Mould and Humphries, 2004; Legate et al., 2005; Nishida et al., 2006; Askari et al., 2010). To determine the activation status of β1 integrin during invadopodium precursor maturation, we used the EGF stimulation assay. Cells were treated with EGF and immunostained with antibodies against cortactin and Tks5 to identify invadopodia as well as activated β1 integrin (conformation-sensitive 9EG7 antibody) and total β1 integrin (P5D2 antibody; Bazzoni et al., 1995). The ratio of activated to total β1 integrin mean fluorescence intensity (MFI) at invadopodium precursor cores was used to determine the activation status of β1 integrin. We found that β1 integrin activation increases by 28 and 50% in invadopodium precursors after 3 and 5 min of EGF stimulation, respectively (Figure 2, A and B), indicating that β1 integrin adopts an adhesion-competent conformation during invadopodial maturation.
FIGURE 2:

β1 integrin is activated in invadopodium precursors, and stimulation of β1 integrin–mediated adhesion accelerates invadopodial maturation in EGF-stimulated cells. (A, B) MDA-MB-231 cells were stimulated with EGF, fixed, and stained with antibodies against total β1 integrin (P5D2), activated β1 integrin (9EG7), cortactin, and Tks5. (A) Representative images of activated β1 integrin staining at 0 min (untreated) and 3 and 5 min after EGF stimulation. The four leftmost panels show insets of the box on the far right. Arrowheads denote invadopodium precursors containing cortactin and Tks5. Bar, 10 μm. (B) Quantification of the ratio of activated β1 integrin:total β1 integrin MFI at the core of invadopodium precursors. n > 39 invadopodium precursors; n > 122 cells; three independent experiments. *p < 0.05 compared with 0 min. (C, D) Invadopodium maturation assay. MDA-MB-231 cells were plated on Alexa 405–labeled gelatin, pretreated with IgG, K20 β1 integrin antibody (nonactivating), TS2/16 β1 integrin antibody (activating), or mAb13 β1 integrin antibody (blocking) and stimulated with EGF for 0, 3, 15, or 30 min. (C) Representative merged images of cortactin- and Tks5-rich invadopodia formed by cells pretreated with IgG or TS2/16 and then stimulated with EGF for 0 or 15 min. Inset, magnified image of invadopodia in the box. Bar, 10 μm. (D) Quantification of cortactin- and Tks5-rich mature invadopodia at each time point. n > 40 cells; three independent experiments. *p < 0.047.
To further characterize the role of β1 integrin activation in regulating invadopodial function, we pretreated adherent cells with mouse immunoglobulin G (IgG) isotype control, a nonactivating β1 integrin antibody (K20), a function-stimulating β1 integrin antibody (TS2/16), or a function-blocking β1 integrin antibody (mAb13) and then stimulated them with EGF to induce invadopodium precursor formation (Supplemental Figure S7C; Mould et al., 1996; Byron et al., 2009). When cells are treated with control IgG or the K20 antibody, which clusters but does not activate β1 integrin (Miyamoto et al., 1995; Byron et al., 2009), invadopodium precursors mature and degrade Alexa 405–labeled gelatin 30 min after EGF stimulation (Figure 2, C and D). We found that cells pretreated with the TS2/16 antibody, which stimulates β1 integrin-mediated adhesion likely by favoring its extended conformation, form invadopodium precursors that mature within 15 min, suggesting that forced β1 integrin activation/adhesion accelerates invadopodium precursor maturation (Figure 2, C and D; Bharadwaj et al., 2005; Byron et al., 2009). Similar results were obtained in cells pretreated with Mn2+ to activate integrins, including β1 integrin (Supplemental Figure S4B). Treatment with the TS2/16 antibody does not, however, affect the formation of nonproteolytic precursors at any time point (Supplemental Figure S4A). Conversely, treatment with the mAb13 blocking antibody suppresses invadopodial maturation at both 15 and 30 min after EGF stimulation (Figure 2D). Collectively these data demonstrate that β1 integrin is activated in invadopodium precursors, and β1 integrin–mediated adhesion enhances the rate of invadopodial ECM proteolysis.
β1 integrin interacts with Arg at invadopodia
Cortactin tyrosine phosphorylation is a critical step in invadopodial maturation, initiating cofilin-dependent barbed-end formation and Arp2/3-dependent actin polymerization (Oser et al., 2009; Magalhaes et al., 2011). Mader et al. (2011) showed that Arg phosphorylates cortactin on tyrosine 421 in invadopodium precursors; however, the mechanism of Arg activation at invadopodia is not fully understood. Because β1 integrin binds Arg in vitro (Warren et al., 2012), we reasoned that Arg binding to β1 integrin might be necessary for its activation in invadopodium precursors to promote maturation.
To address whether β1 integrin from MDA-MB-231 cells is competent to bind Arg, we incubated lysates with beads coated with full-length Arg (Calderwood et al., 2003; Lapetina et al., 2009). β1 integrin is specifically pulled down by the Arg beads but not by the negative control bovine serum albumin (BSA)–coated beads (Figure 3A). We next asked whether β1 integrin interacted with Arg at invadopodia.
FIGURE 3:

β1 integrin interacts with Arg in invadopodia. (A) Representative Western blot from the Arg bead pull-down assay. MDA-MB-231 cell lysates were incubated with BSA (negative control) or full-length Arg-coated beads, and the resulting pull-downs were run on SDS–PAGE and stained for β1 integrin. Two independent experiments. (B, C) β1 integrin–Arg acceptor photobleaching FRET. (B) Representative images of FRET efficiency between β1 integrin and Arg at Tks5-rich mature invadopodia in MDA-MB-231 cells at steady state (s.s.) or after integrin activation with 1 mM MnCl2 for 30 min (Mn2+). Dashed white circles denote mature invadopodia. Inset, magnified view of β1 integrin and Arg colocalized at invadopodia and the associated FRET efficiency. LUT bar indicates linear scale of FRET efficiency from 0 to 16%. Bar, 10 μm. (C) Quantification of β1 integrin–Arg and β1 integrin–cortactin FRET efficiencies at invadopodia. n > 61 invadopodia; n > 23 cells; three independent experiments. *p < 0.003. (D) Principle of PLA. Cells are fixed, permeabilized, and stained with primary antibodies (proteins of interest depicted as red and blue spheres). Secondary antibodies conjugated to complementary oligonucleotides are added. The oligonucleotides are hybridized and ligated, and the circular DNA is replicated by rolling circle amplification, incorporating fluorescently labeled nucleotides that can be detected by microscopy. Adapted from Olink. (E) Representative maximum-intensity Z‑projection images of MDA-MB-231 cells showing colocalization of β1 integrin-–Arg PLA events (green) with mature, Tks5-rich invadopodia (red; arrowheads) and negative control β1 integrin–IgG PLA events, which almost never colocalize with mature invadopodia. (F) Quantification of PLA signal colocalization with mature invadopodia. n > 131 invadopodia; n > 62 cells; three independent experiments. *p < 9.34E-5.
To evaluate this, we plated MDA-MB-231 cells on Alexa 405–labeled gelatin and conducted acceptor photobleaching fluorescence resonance energy transfer (FRET) experiments between β1 integrin (acceptor) and Arg (donor). Regions containing Tks5-rich mature invadopodia were bleached, and β1 integrin–Arg FRET efficiency was calculated. Although there is a modest increase in FRET between β1 integrin and Arg at steady state, this interaction is significantly enhanced in cells treated with MnCl2 to activate integrins, including β1 integrin localized to invadopodia (Figure 3, B and C; p < 1.12E-5; Supplemental Figure S5A; Bazzoni et al., 1995). Under starvation conditions in which the levels of activated β1 integrin are low, however, relatively low β1 integrin–Arg FRET is observed in invadopodium precursors (Supplemental Figure S5C). Furthermore, the FRET between β1 integrin and Arg is ∼2-fold higher in the invadopodium core than the surrounding plasma membrane at steady state and ∼1.5-fold higher in the invadopodium cores of MnCl2-treated cells, suggesting that this interaction preferentially occurs in the invadopodial compartment (Supplemental Figure S5D). Last, to control for nonspecific interactions between antibodies within the invadopodial compartment, we calculated the FRET efficiency between β1 integrin and cortactin and found that it was minimal (∼2-fold less than steady state and ∼4-fold less than MnCl2-treated cells; Figure 3C and Supplemental Figure S5B).
To confirm the FRET results by an independent method, we used the in situ proximity ligation assay (PLA), a technique that uses DNA aptamer–conjugated antibodies to detect very close interactions between two proteins with high spatial resolution (Figure 3D; Fredriksson et al., 2002). PLA was performed with β1 integrin and Arg or IgG antibodies, the latter of which served as the negative control. Colocalization of PLA signals with Tks5-rich mature invadopodia was used to quantify the interaction. Results indicate that β1 integrin–Arg pairing in mature invadopodia yields a sixfold increase in PLA signal compared with the level observed using β1 integrin and a nonspecific IgG (Figure 3, E and F). Thus FRET and PLA experiments indicate that β1 integrin interacts with Arg in invadopodia, Arg preferentially interacts with activated β1 integrin, and β1 integrin might play a role in Arg activation.
β1 integrin is required for Arg-mediated cortactin Y421 phosphorylation at invadopodium precursors
Next we investigated whether β1 integrin is required for Arg recruitment to invadopodia. Of interest, despite interacting with β1 integrin, Arg recruitment is not affected in β1 integrin–knockdown cells (Supplemental Figure S6A), raising the possibility that the interaction between Arg and β1 integrin might be responsible for activating invadopodial Arg. To test this, we evaluated Arg kinase activity in invadopodium precursors using the EGF stimulation assay to measure cortactin phosphorylation status. Because cortactin Y421 phosphorylation at invadopodia is dependent upon Arg (Mader et al., 2011), we used a phospho-specific Y421 cortactin antibody to evaluate Arg kinase activity at invadopodium precursors in cells treated with control or β1 integrin siRNA. Whereas EGF stimulates a 60% increase in cortactin Y421 phosphorylation in control cells, no increase in cortactin phosphorylation was observed in cells treated with β1 integrin siRNA, with the level of cortactin pY421 remaining at the background observed in Arg-knockdown (KD) cells (raw 3-min ratio of pY421/total cortactin–β1 KD, 0.33, vs. Arg KD, 0.34; Figure 4, A and B). This suggests that EGFR-Src signaling alone is not sufficient to induce Arg-mediated cortactin phosphorylation, and β1 integrin is essential for Arg activation at invadopodium precursors.
FIGURE 4:

β1 integrin is required for Arg-mediated cortactin Y421 phosphorylation at invadopodium precursors in response to EGF stimulation. (A, B) Cortactin Y421 phosphorylation at precursors in the invadopodium precursor formation assay in MDA-MB-231 cells. (A) Representative images of control and β1 integrin siRNA (SMARTpool)–treated cells stimulated with EGF for 0 (untreated) or 3 min and stained with antibodies against cortactin, pY421 cortactin, and phalloidin (F-actin). Insets, Y421 phosphorylation status at cortactin-F-actin–rich invadopodium precursors. Bar, 10 μm. (B) Quantification of pY421 cortactin:total cortactin MFI at invadopodium precursors in cells treated with control or β1 integrin siRNA (left; n > 125 invadopodium precursors; n > 85 cells; three independent experiments) and control and Arg knockdown cells pretreated with IgG, K20, TS2/16, or mAb13 for 10 min before EGF stimulation (right; n > 80 invadopodia; n > 65 cells; three independent experiments). *p < 0.004; **p < 0.026 mAb13 compared with IgG, K20, and TS2/16 and Arg siRNA compared with control siRNA 3 min, IgG, K20, and TS2/16, respectively.
Given that β1 integrin activation enhances its interaction with Arg, we tested the hypothesis that promoting β1 integrin–mediated adhesion with function-stimulating antibodies could enhance Arg-dependent cortactin Y421 phosphorylation at invadopodium precursors. To do this, we repeated the cortactin phosphorylation assay in cells pretreated with the IgG, K20, TS2/16, or mAb13 antibodies. Whereas treatment of control cells with IgG or the K20 β1 integrin antibody induces cortactin phosphorylation similar to EGF alone (∼45% increase), β1 integrin activation with the TS2/16 antibody resulted in an 85% increase in cortactin phosphorylation (Figure 4B). Conversely, inhibiting β1 integrin activation by pretreatment with the mAb13 antibody completely blocked this phosphorylation event (Figure 4B). The additive effect of β1 integrin activation on cortactin phosphorylation was found to depend on Arg kinase activity, since cortactin phosphorylation remained at the basal level in Arg-knockdown cells after stimulation (Figure 4B and Supplemental Figure S6C). Taken together, these experiments identify β1 integrin as an upstream regulator of Arg and demonstrate that β1 integrin–mediated adhesion is an important trigger that promotes Arg kinase activity in invadopodium precursors.
β1 integrin is required for cofilin-mediated barbed-end formation and actin polymerization at invadopodium precursors
The finding that β1 integrin promotes cortactin Y421 phosphorylation led us to investigate whether β1 integrin is also important for actin polymerization at invadopodia, since cortactin phosphorylation is the primary regulator of cofilin activity in precursors (Oser et al., 2009; Magalhaes et al., 2011). In MTLn3 cells, cofilin severs F-actin in invadopodium precursors to produce free barbed ends that elongate and serve as sites for Arp2/3-dependent actin nucleation and polymerization (Yamaguchi et al., 2005; Oser et al., 2009). In MDA-MB-231 cells, EGF stimulation results in a peak in barbed-end intensity at 3 min (Oser et al., 2010). To determine whether barbed-end formation in MDA-MB-231 cells also requires cofilin severing activity, cells were treated with either control or cofilin siRNA for 48 h, and the barbed-end assay was performed as described previously (Figure 5E; Chan et al., 1998; Oser et al., 2009). As in MTLn3 cells, cofilin knockdown in MDA-MB-231 cells abrogated EGF-dependent barbed-end formation at invadopodium precursors (Figure 5D), indicating that barbed-end formation in MDA-MB-231 cells requires cofilin.
FIGURE 5:

β1 integrin is required for cofilin-mediated barbed-end formation and actin polymerization at invadopodium precursors. (A, B) Cofilin–β-actin FRET at invadopodium precursors in MDA-MB-231 cells. (A) Representative images of cofilin–β-actin FRET efficiency at precursors in cells treated with control and β1 integrin siRNA (SMARTpool) and stimulated with EGF for 3 min. Dashed white circles denote invadopodium precursors. Insets, cofilin-actin–containing invadopodium precursors and the associated FRET efficiency. LUT bar indicates linear scale of FRET efficiency from 0 to 16%. Bar, 10 μm. (B) Quantification of cofilin–β-actin FRET efficiency at precursors 0 (untreated) or 3 min after EGF stimulation (control, 0 min, 4.34%; β1, 0 min, 3.85%). n > 60 invadopodium precursors; n > 35 cells; three independent experiments. *p < 0.018. (C, D) Barbed-end assay. (C) Representative images of cells treated with control or β1 integrin siRNA (SMARTpool), stimulated with EGF for 0 (untreated) or 3 min, and stained for anti-biotin (barbed ends), cortactin, and Arp2. Inset, barbed-end intensity at cortactin-Arp2–containing invadopodium precursors. Bar, 10 μm. (D) Quantification of barbed-end MFI at invadopodium precursors. n > 190 invadopodium precursors; n > 126 cells; three independent experiments. *p < 1.22E‑11. (E) Western blot analysis of MDA-MB-231 cells transfected with control or cofilin siRNA for 48 h. Blots were stained for cofilin and β-actin (loading control).
To determine whether β1 integrin regulates cofilin activity and actin polymerization at invadopodium precursors, we used cofilin–β-actin acceptor photobleaching FRET and the barbed-end assay (Chan et al., 1998; Oser et al., 2009). Because cofilin is an F-actin–binding and –severing protein, cofilin–β-actin FRET correlates with increased cofilin activity. In control cells, EGF enhances the interaction between cofilin and actin at invadopodium precursors, resulting in ∼40% increase in FRET efficiency after 3 min (Figure 5, A and B). Cofilin binding to actin is significantly reduced in β1 integrin–depleted cells, in which the FRET efficiency at invadopodium precursors decreases by ∼20% after EGF stimulation (Figure 5, A and B; p < 0.018).
Although these data suggests that cofilin activity may be suppressed in β1 integrin–knockdown cells, the generation of free actin barbed ends is a more direct measurement of cofilin activity (Wang et al., 2007). Therefore we visualized the incorporation of biotinylated G-actin monomers at the barbed ends of preexisting actin filaments in invadopodium precursors to measure relative barbed-end number (Chan et al., 1998; Oser et al., 2009). Consistent with the FRET experiments, EGF stimulates a significant increase in barbed-end formation in control cells after 3 min; barbed-end intensity in invadopodium precursors in β1 integrin–knockdown cells, however, is significantly reduced at this time point (Figure 5, C and D; p < 1.22E-11). Taken together, these data demonstrate that β1 integrin is an important regulator of cofilin severing activity, free actin barbed-end formation, and actin polymerization at invadopodium precursors.
β1 integrin is essential for invadopodium formation in physiologically relevant three-dimensional matrix
To assess the role of β1 integrin in regulating invadopodia in a more physiological three-dimensional (3D) context, MDA-MB-231 cells were treated with control or β1 integrin siRNA, transfected with TagRFP-cortactin, and cultured in 3D extracellular matrix consisting of type I collagen, dequenched (DQ) type I collagen, and Matrigel for 24–36 h (Nystrom et al., 2005). In a 3D environment, invadopodia form as thin, 3- to 4-μm protrusions that are typically enriched in cortactin and MT1-MMP and >7 μm in length and degrade collagen along the length of and at the tip of the protrusion (Supplemental Figure S7A; Li et al., 2010; Magalhaes et al., 2011). Whereas control cells formed long, branched protrusions associated with local matrix degradation, β1 integrin–knockdown cells and cells treated with the β1 integrin function–blocking antibody (mAb13) formed sevenfold fewer degradative invadopodial protrusions (Figure 6, A and B, and Supplemental Movies S3 and S4). Furthermore, invadopodium length was decreased by >40%, and the amount of degradation associated with each protrusion was significantly decreased in β1 integrin-depleted cells in 3D matrix (Figure 6, C and D, and Supplemental Figure S7B; p < 3.15E-9). Thus the data implicating β1 integrin in regulating invadopodial actin polymerization and maturation in metastatic breast cancer cells in two dimensions is consistent with a role for β1 integrin in a more physiologically relevant 3D matrix.
FIGURE 6:

β1 integrin promotes invadopodial matrix degradation in 3D extracellular matrix. (A–D) 3D extracellular matrix invadopodium assay. (A) Representative maximum-intensity Z‑projection multiphoton images of MDA-MB-231 cells transiently transfected with control or β1 integrin siRNA and TagRFP-cortactin. Cells were embedded in a 3D ECM gel consisting of type I collagen, DQ-type I collagen, and Matrigel for 36 h. (B) Quantification of invadopodium number in control, β1 integrin–depleted, and mab13-treated cells in 3D matrix. n > 115 cells; three independent experiments. *p < 1.27E-15, compared with control siRNA. (C) Quantification of invadopodium length in 3D matrix. n > 115 cells; three independent experiments. *p < 0.013, compared with control siRNA. (D) Quantification of mean DQ-collagen (degradation) MFI per protrusion > 7 μm. n > 115 cells; three independent experiments. *p < 3.15E-9, compared with control siRNA.
DISCUSSION
Invadopodia are believed to facilitate multiple stages of tumor cell metastasis, including local invasion, migration through the stroma, and intravasation (Artym et al., 2006; Huttenlocher and Horwitz, 2011; Linder et al., 2011; Magalhaes et al., 2011; Bravo-Cordero et al., 2012). In this study, we investigated the roles of β1 and β3 integrins in regulating invadopodial function in metastatic human breast cancer cells. On the basis of the data presented here, we propose the following model in which β1 integrin is not required for the initial formation of invadopodium precursors but drives invadopodial maturation, whereas β3 integrin is important for overall adhesion but is not required for invadopodial function (Figure 7). β1 integrin interacts with Arg at invadopodia and is required for its kinase activity in MDA-MB-231 cells. Depletion of β1 integrin by siRNA completely blocks Arg-dependent cortactin Y421 phosphorylation and cofilin activity, leading to a decrease in the number and stability of mature, actively degrading invadopodia. Thus we provide evidence that β1 integrin is a critical regulator of invadopodial maturation by stimulating the Arg–cortactin–cofilin pathway.
FIGURE 7:
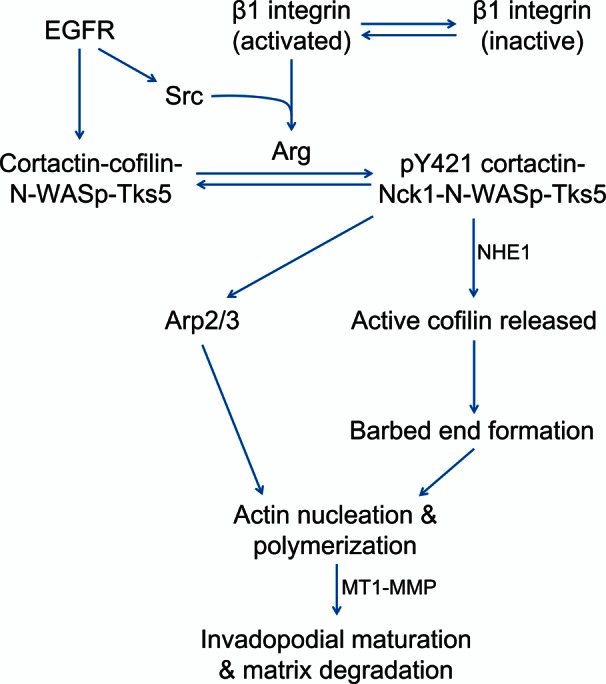
Model of β1 integrin–dependent regulation of invadopodia. β1 integrin is activated in invadopodium precursors, leading to increased local β1 integrin adhesion in the protrusion/core, which is a key switch that drives Arg activation, cortactin phosphorylation, and MMP recruitment during invadopodial maturation. It is proposed that β1 integrin-EGFR cross-talk activates Arg by a three-step mechanism: 1) Arg binding to the β1 integrin cytoplasmic tail is believed to disrupt its autoinhibitory conformation, unmasking the Y272 autophosphorylation site and Y439 on the activation loop. 2) Integrin-mediated clustering of Arg likely facilitates autophosphorylation on Y272. 3) EGFR activation induces Src-dependent Arg phosphorylation on Y439, resulting in full Arg kinase activation. Arg-dependent cortactin phosphorylation results in the recruitment of NHE-1 to increase the local intracellular pH, resulting in disruption of the inhibitory interaction between cortactin and cofilin and recruitment of Nck1 for N‑WASp activation to induce Arp2/3-dependent actin polymerization. In this way, β1 integrin acts as a critical upstream regulator of Arg kinase activity, actin polymerization, and subsequent protease recruitment to promote efficient invadopodial matrix degradation.
β1 integrin activation and adhesion regulate invadopodial function
The role of integrin activation and adhesion in regulating invadopodial function is incompletely understood (Buccione et al., 2009; Linder et al., 2011; Murphy and Courtneidge, 2011). We demonstrate that β1 integrin is activated in invadopodial protrusions (core), favoring adhesion to the ECM. Stimulation of β1 integrin–mediated adhesion increases the rate of invadopodial matrix proteolysis, enhances the interaction between β1 integrin and Arg, and induces Arg kinase activity. Conversely, inhibiting β1 integrin adhesion completely blocks Arg activation, as well as invadopodial maturation. Taken together, these experiments identify β1 integrin activation and adhesion to the ECM as a key upstream switch in invadopodial maturation that drives Arg activation, cortactin phosphorylation, and recruitment of MT1-MMP to activate MMP-2 and MMP-9 for matrix degradation (Deryugina et al., 2001).
Recently a number of groups have shown that invadopodia are surrounded by a ring of proteins, including Hic-5, paxillin, and β1 integrin, as well as RhoC GTPase activity (Bravo-Cordero et al., 2011; Branch et al., 2012; Pignatelli et al., 2012). These proteins play a critical role in regulating the geometry of the invadopodial protrusion, as well as the efficiency at which it degrades the underlying extracellular matrix. The differences in β1 integrin localization at invadopodia observed in MCF10A-CA1D breast epithelial cells (ring; Branch et al., 2012) compared with the localization in highly metastatic breast cancer and melanoma cells reported here and elsewhere (core) raises the intriguing possibility that there is a switch in integrin localization as tumor cells progress through the metastatic cascade (Mueller et al., 1999). It will be interesting to explore which molecules regulate the transition from β1 integrin distribution in a ring to the invadopodium core and how differential integrin localization affects the metastatic potential of tumor cells.
β1 integrin–EGFR cross-talk is required to activate Arg at invadopodia
Cross-talk between integrins and growth factor receptors is a well-established phenomenon (Ivaska and Heino, 2011). Classically, integrin–growth factor receptor cross-talk has been associated with proliferation, but more recently these cross-talk mechanisms have been implicated in tumor cell migration and invasion. Norman and coworkers showed that β1 integrin and EGFR interact in the endocytic compartment to stimulate Akt phosphorylation and invasion in ovarian carcinoma cells (Caswell et al., 2008). In the present study, we show that β1 integrin–EGFR cross-talk in invadopodia regulates Arg kinase activity and actin polymerization, revealing yet another context in which the two receptors cooperate to promote tumor cell invasion.
EGFR signaling through Src was shown to phosphorylate Arg on its activation loop (Mader et al., 2011); here we demonstrate that β1 integrin interacts with Arg in invadopodia. Because both of these events are required for Arg kinase activity at invadopodium precursors, we propose the following three-step model of Arg activation in invadopodia: Arg is likely recruited to invadopodia via its interactions with actin and/or cortactin (Wang et al., 2001; Lapetina et al., 2009). 1) Arg binding to β1 integrin is believed to disrupt the Arg autoinhibitory (folded) conformation, resulting in extension of the molecule and unmasking of the Y272 autophosphorylation site and Y439 on the activation loop (Tanis et al., 2003). 2) Integrin-mediated clustering of Arg would then facilitate autophosphorylation on Y272, whereas 3) EGFR activation induces Src-dependent Arg phosphorylation on Y439, resulting in full Arg kinase activation (Mader et al., 2011). This model is supported by three main lines of evidence: 1) β1 integrin interacts with Arg at invadopodia, and this binding event is required to activate Arg in vitro (Simpson, Bradley, and Koleske, unpublished data), 2) Arg kinase activity at invadopodia is abrogated in β1 integrin–depleted cells, and 3) in the absence of EGF/serum, β1 integrin activation alone is not sufficient to induce Arg-dependent cortactin Y421 phosphorylation (Supplemental Figure S6B). Collectively these findings indicate that β1 integrin and EGFR signaling converge at the level of Arg (Figure 7), as both signals are required for efficient Arg activation at invadopodia.
β1 integrin regulates actin dynamics at invadopodia
β1 integrin is required for Arg-mediated cortactin phosphorylation, which, in turn, promotes cofilin severing activity and actin polymerization at invadopodia. In melanoma cells, β1 integrin stimulation with laminin peptides induces p190RhoGAP phosphorylation and activation at invadopodia (Nakahara et al., 1998); however, the kinase responsible for this phosphorylation event has not been identified. Of interest, integrins have been shown to activate Arg-dependent p190RhoGAP phosphorylation in fibroblasts and neurons (Hernandez et al., 2004; Bradley et al., 2006; Warren et al., 2012). Because p190RhoGAP localizes to the invadopodium core and inhibits RhoC to allow cofilin to become activated (Bravo-Cordero et al., 2011), it is tempting to speculate that β1 integrin–dependent Arg activation promotes cofilin-mediated barbed-end formation both by phosphorylating cortactin to relieve cortactin-dependent cofilin sequestration and by deactivating RhoC in invadopodia to prevent inhibitory cofilin phosphorylation. In this way, β1 integrin signaling through Arg may be the predominant mechanism that spatiotemporally regulates actin polymerization at invadopodia.
Here we show that β1 integrin is a major regulator of invadopodial maturation in both two- and three-dimensional extracellular matrices. β1 integrin promotes metastatic dissemination in a number of tumor models, including breast and ovarian cancer (Huck et al., 2010; Mitra et al., 2011); however, the precise mechanism by which β1 integrin enhances the metastatic potential of tumor cells is not known. Our data indicate that β1 integrin likely promotes breast cancer metastasis via the β1 integrin–Arg pathway described here. Because invadopodia are believed to facilitate transendothelial migration and invadopodial protrusions on tumor cells have been recently identified adjacent to blood vessels in vivo (Gligorijevic et al., 2012), it will be interesting to explore the possibility that β1 integrin enhances breast cancer cell metastasis by promoting invadopodial basement membrane degradation and intravasation in vivo.
MATERIALS AND METHODS
Cell culture
MDA-MB-231 cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics. For EGF stimulation experiments, MDA-MB-231 cells were serum starved in 0.5% FBS and 0.8% BSA in DMEM for 12–16 h. Cells were then starved in 0.345% BSA in L15 media for 10 min immediately before stimulation with 2.5 nM EGF. Live-cell imaging experiments were conducted at 37°C using L15 media and 10% FBS. MTLn3 cells were grown in αMEM supplemented with 5% FBS and antibiotics.
Antibodies and reagents
The β1 integrin (C27) antibody was obtained from Vitatex (Stony Brook, NY). Cortactin (sc-30771), Tks5 (sc-30122), Arp2 (sc-H-84), and the K20 (sc-18887) and P5D2 (sc-13590) β1 integrin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The cortactin (ab33333) antibody was from Abcam (Cambridge, MA). The activated β1 integrin antibody (9EG7; 550531) and the function-blocking β1 integrin antibody (mAb13; 552828) were from BD PharMingen Biosciences (San Diego, CA). The function-stimulating β1 integrin antibody (TS2/16; MA2910) was from Thermo Scientific (Waltham, MA). The β-actin (AC-15) and phospho-Y421 cortactin antibodies were from Sigma-Aldrich (St. Louis, MO). Fluorescein isothiocyanate (FITC)–anti-biotin antibody was from Jackson ImmunoResearch Laboratories (West Grove, PA). Mouse IgG1 (MAB002) was from R&D Systems (Minneapolis, MN). Anti-cofilin (AE774) was custom made as described previously (Yamaguchi et al., 2005). The Arg (AR19) antibody was a generous gift from Peter Davies (Albert Einstein School of Medicine, New York, NY). All Alexa Fluor secondary antibodies were from Molecular Probes (Life Technologies, Carlsbad, CA). For Western blots, the following antibodies were used: β1 integrin (610467; BD PharMingen Biosciences), Arg (AR19), cofilin (AE774), and β-actin (AC-15).
DNA constructs, RNA interference, and transfection
For live-cell imaging, 105 MDA-MB-231 cells were plated on a six-well plate and then transfected with 0.5 μg of TagRFP-cortactin (Oser et al., 2009) and GFP-Tks5 (kindly provided by Sara Courtneidge, Burnham Institute for Medical Research, La Jolla, CA; Stylli et al., 2009) using Lipofectamine LTX and PLUS reagent 24 h before imaging per manufacturer's instructions. MT1-MMP-GFP was described previously (Galvez et al., 2002). The control empty vector and stable β1 integrin shRNA cell lines were generated using pGIPZ shRNA lentiviral vectors (Open Biosystems, Huntsville, AL). ON-TARGETplus SMARTpool β1 integrin (human, L-004506-00–MDA-MB-231 cells; rat, L-089600-01–MTLn3 cells), β3 integrin (L-004124-00-0010), Arg (L-003101-00), and cofilin (L-012707-00) siRNAs were obtained from Dharmacon (Lafayette, CO). The siGENOME β1 integrin siRNA (D-004506-03) was also from Dharmacon. Nonsilencing control siRNA was obtained from Qiagen (Valencia, CA). For siRNA transfection, 106 MDA‑MB-231 cells were transfected with 2 μM siRNA using the Amaxa V nucleofection system (Lonza, Basel, Switzerland) 48–96 h before each experiment. MTLn3 cells were transfected with 100 nM β1 integrin siRNA in Oligofectamine for 48 h as described previously (Kempiak et al., 2005).
Invadopodial matrix degradation assay, immunofluorescence, and fixation protocols
The invadopodial matrix degradation assay was conducted as described previously (Mader et al., 2011; Sharma et al., 2013). Briefly, gelatin was conjugated with an Alexa 405 dye (Molecular Probes, Eugene, OR). MatTek dishes (MatTek Corporation, Ashland, MA) were treated with 1 N HCl and coated with 50 μg/ml poly-l-lysine. A 0.2% gelatin solution was prepared in phosphate-buffered saline (PBS), and a 1:40 mixture of Alexa 405–labeled gelatin:unlabeled gelatin was warmed to 37°C before addition to the poly-l-lysine. Gelatin was cross-linked with 0.01% glutaraldehyde, which was then quenched with 5 mg/ml sodium borohydride. MDA-MB-231, 2 × 105 cells, was plated on Alexa 405–labeled gelatin for 4 h, and 7.5 × 104 MTLn3 cells were plated overnight before fixation. Unless otherwise indicated, cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100, and blocked with 1% FBS and 1% BSA in PBS. All primary and secondary antibodies were diluted in blocking buffer. For Arg staining, cells were fixed in 2% PFA. For cofilin staining, cells were fixed and stained using the protocol described previously (Eddy et al., 2000).
Invadopodium precursors were identified as cortactin-Tks5–rich punctate structures that do not colocalize with degradation holes, whereas mature invadopodia are similar structures that do colocalize with degradation holes. Degradation area was calculated as the total area covered by degradation holes/field in thresholded images using the Analyze Particles tool in ImageJ (National Institutes of Health, Bethesda, MD) and normalized to the number of cells in each field to give degradation area/cell. All fixed cell images (except FRET and 3D) were acquired on a DeltaVision Core Microscope (Applied Precision, Issaquah, WA) using a CoolSnap HQ2 camera (Photometrics, Tucson, AZ), 60×/numerical aperture (NA) 1.4 oil objective, standard four-channel filter set, and softWoRx software. Fixed cells were imaged in PBS at room temperature.
Invadopodia lifetime analysis
Cells were transfected with control or β1 integrin siRNA 96 h before imaging, cotransfected with TagRFP-cortactin and GFP-Tks5 for 24 h, and plated on Alexa 405–labeled gelatin for 2 h before imaging on the DeltaVision Core Microscope in a 37°C heated chamber. Images were acquired every 2 min for the RFP and GFP channels and every 10 min for the Alexa 405 channel for 3 h in 15 random fields. Because invadopodia in MDA-MB-231 cells are stationary structures (in contrast to MTLn3 cells; Yamaguchi et al., 2005), stationary cortactin-Tks5–rich dots (1–2 μm) were identified as nondegrading invadopodium precursors and mature, degrading invadopodia as described earlier. Lifetimes were calculated as the time from initial invadopodia assembly (appearance slice) to the disappearance of the structure using a custom invadopodia tracker plug-in (Sharma et al., 2013). For Figure 1E, still images of representative invadopodia were generated by cropping areas surrounding invadopodia in background-subtracted images.
Invadopodium maturation assay
Cells were plated on Alexa 405–labeled gelatin in the presence of the broad-spectrum MMP inhibitor GM6001 (25 μM; Enzo Life Sciences, Plymouth Meeting, PA) to prevent degradation of the gelatin before the assay. Cells were then starved for 12–16 h as described and pretreated with 2.5 μg/ml mouse IgG, nonactivating K20 β1 integrin antibody, function-stimulating TS2/16 β1 integrin antibody, or function-blocking mAb13 β1 integrin antibody in DMEM starvation media for 30 min before EGF stimulation. Cells were stimulated with 2.5 nM EGF (+ antibody) for 0, 3, 15, or 30 min, fixed, and stained for cortactin and Tks5. Mature invadopodia were identified as in the invadopodial matrix degradation assay.
Quantitative immunofluorescence
For quantitative immunofluorescence, cells from each sample were imaged using the same neutral density (ND) filter and exposure time. Invadopodium precursors were identified using two markers, and the mean gray value (MGV) within invadopodia was measured in ImageJ by drawing regions of interest around the invadopodium in background (BG)-subtracted images. In EGF stimulation experiments, MGV at invadopodium precursors after EGF stimulation was normalized to 0-min (untreated) MGV for that condition (Mader et al., 2011). For quantification of β1 integrin enrichment at invadopodia, MGVs at the invadopodia and the surrounding 1 μm were determined in background-subtracted images. β1 integrin enrichment at invadopodia was calculated using (β1 integrin MGVinvadopodia-BG/β1 integrin MGVsurrounding cytosol-BG) – 1.
Acceptor photobleaching FRET
MDA-MB-231 cells were plated on Alexa 405–labeled gelatin, fixed, and immunostained as described. The following antibody combinations were used for FRET experiments: 1) Arg AR19/Alexa Fluor 488–donkey anti-mouse (donor) and β1 integrin (C27)/Alexa Fluor 555–goat anti-rat (acceptor), 2) cortactin/Alexa Fluor 488–donkey anti-mouse (donor) and β1 integrin (C27)/Alexa Fluor 555–goat anti-rat (acceptor), and 3) chicken cofilin 774/Alexa Fluor 488–goat anti-chicken (donor) and β‑actin AC-15/Alexa Fluor 555–goat anti-mouse (acceptor). For the β1 integrin–Arg and –cortactin experiments, colocalization of Tks5 and cortactin with degradation holes in Alexa 405–labeled gelatin were used to identify invadopodia, respectively. For β1 integrin activation experiments, cells were stimulated with 1 mM MnCl2 for 30 min before fixation to avoid antibody species conflicts with the TS2/16 antibody. Because our Zeiss laser scanning microscope (LSM5 LIVE DuoScan; Zeiss, Jena, Germany) does not have a far-red laser, Tks5- and cortactin-rich invadopodia were imaged on the DeltaVision Core Microscope as described, and the same cells were then located using gridded coverslips (MatTek) and imaged on the Zeiss microscope. For all FRET experiments, cells were imaged in PBS at room temperature on the Zeiss laser scanning microscope (LSM5 LIVE DuoScan) using a 60×/NA 1.4 oil objective, the LSM 5 Live DuoScan software, and a cooled charge-coupled device camera. A region of interest surrounding invadopodia was bleached using 100% 561-nm laser power (acceptor channel), and images were acquired in both the 488- and 561-nm channels before and after bleaching. FRET efficiency was calculated as the E = 1 – (donor pre/donor post) in background-subtracted images and was corrected for fluctuations in laser power and donor bleaching in ImageJ. As a control for the β1 integrin–Arg FRET experiment, cells were stained only with the AR19 Arg antibody/Alexa 488 and Tks5/Alexa 647. Regions surrounding Tks5-rich invadopodia were then bleached using the 561-nm laser, and the FRET efficiency was calculated as described. This resulted in a minimal increase in FRET efficiency in the Alexa 488 channel (mean, 0.9%). The acceptor photobleaching FRET controls for the cofilin–β-actin secondary antibody pairs were described previously (Oser et al., 2009).
Barbed-end assay
The barbed-end assay was performed as described previously (Chan et al., 1998; Oser et al., 2009). Briefly, cells were treated with control, β1 integrin, or cofilin siRNA and serum starved for 12–16 h as described. Cells were stimulated with 2.5 nM EGF and permeabilized with 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.5), 138 mM KCl, 4 mM MgCl2, 3 mM ethylene glycol tetraacetic acid, 0.2 mg/ml saponin, 1 mM ATP, and 1% BSA. Cells were incubated with 0.4 μM biotin–actin (Cytoskeleton, Denver, CO) for 1 min at 37°C and then fixed with 4% PFA, blocked with 1% FBS/1% BSA/3μM phalloidin in PBS, and immunostained with FITC–anti-biotin, as well as cortactin and Arp2 to identify invadopodium precursors. Cells were imaged on the DeltaVision Core Microscope, and the barbed-end intensity at each invadopodium was determined by measuring the FITC–biotin MGV in background-subtracted images. Barbed-end intensities were normalized to the 0-min MGV for each experiment (Mader et al., 2011).
Three-dimensional invadopodium matrix degradation assay
MDA-MB-231 cells were transfected with control or β1 integrin siRNA (SMARTpool) for 96 h and then transiently transfected with TagRFP-cortactin as described. We resuspended 3.5 × 104 cells in a 3D matrix containing type I collagen (BD PharMingen Biosciences) and Matrigel (Invitrogen) at final concentrations of 4.6 and 2.2 mg/ml, respectively, on a MatTek dish for 1 h at 37°C before adding DMEM/10% FBS as described previously (Nystrom et al., 2005; Magalhaes et al., 2011). To visualize invadopodial matrix degradation, DQ-type I collagen was added in a 1:10 ratio with unlabeled type I collagen (Invitrogen, Carlsbad, CA). Cells were allowed to invade for 24–36 h before fixation in 4% PFA for 7 min at 4°C, and a 25- to 50-μm z-stack (0.5-μm step) was acquired using a 25×/1.05 NA water immersion objective on an inverted Olympus IX70 microscope (Olympus America, Melville, NY; Wyckoff et al., 2000). Cells were imaged in PBS at room temperature. Invadopodia were quantified as 2- to 3-μm-wide protrusions that were ≥7 μm in length and colocalize with degradation (enhanced DQ-type I collagen signal; Magalhaes et al., 2011). Invadopodium length was calculated in ImageJ. Degradation area was calculated as the MFI in the DQ-type I collagen channel along the length of all protrusions ≥7 μm in length. The 3D reconstructions were generated from z-stacks using Imaris (Bitplane, Zurich, Switzerland).
Proximity ligation assay
MDA-MB-231 cells were plated for the invadopodial matrix degradation assay, fixed, permeabilized, and stained with primary antibodies (C27, β1 integrin; AR19, Arg; IgG) as described. The assay was conducted according to manufacturer's instructions for the Duolink II Probe Maker PLUS, PLA probe anti-mouse MINUS, and detection reagent orange (Olink Bioscience, Uppsala, Sweden).
Western blotting
Cells were transfected with control, nontargeting siRNA or cofilin, Arg, or β1 integrin siRNA for the specified times. Cells were then lysed using SDS–PAGE sample buffer, sonicated, and boiled at 95°C. A polyacrylamide gel was prepared, and lysates were run at 120 V for 1.5 h. Protein was then transferred to nitrocellulose paper, blocked using the Odyssey solution (LI-COR Biosciences, Lincoln, NE), and immunostained. Odyssey anti-mouse 680, anti-rabbit 800, and anti-chicken 800 secondary antibodies were used. All primary and secondary antibodies were diluted in Odyssey blocking solution. Fluorescent secondary antibodies were detected using the Odyssey scanner. Quantitation of protein expression was performed by measuring the MGV of each band in background-subtracted images in ImageJ.
Bead pull-down assay
Full-length histidine-tagged Arg was purified from insect cells using the Bac-to-Bac baculovirus expression system (Invitrogen) and coupled to AminoLink beads (Pierce, Rockford, IL) as described previously (Lapetina et al., 2009; Warren et al., 2012). Briefly, purified recombinant His-Arg was buffer exchanged into 3.65× PBS (36.5 mM phosphate, 500 mM NaCl, 9.5 mM KCl) and linked to AminoLink beads at 4°C rotating overnight to a final concentration of 14 μg Arg/μl beads. Sodium cyanoborohydride was added to a final concentration of 50 mM to catalyze the coupling reaction. After linking, remaining active sites on the beads were blocked with 100 mg/ml BSA in 1 M Tris-HCl, pH 7.4. Beads were also linked with the same BSA solution in the absence of Arg as a negative control. Beads were washed and resuspended in a buffer containing 25 mM HEPES, pH 7.25, 100 mM NaCl, 5% glycerol, and 0.01% Triton X-100 before use. For pull-down assays, one million MDA-MB-231 cells were lysed in a pull-down buffer (10 mM 1,4-piperazinediethanesulfonic acid, 50 mM NaCl, 150 mM sucrose, 1 mM Na3VO4, 50 mM NaF, 40 mM Na4P2O7, 0.05% Triton X-100, pH 6.8; Lad et al., 2007). Cells were then incubated with either BSA or Arg beads for 2 h, washed, and run on SDS–PAGE. Blots were then probed for β1 integrin.
Statistical analysis
Statistical analysis was conducted using an unpaired, two-tailed Student's t test. Statistical significance was defined as p < 0.05. Error bars represent SEM. All graphs are displayed as mean ± SEM.
Supplementary Material
Acknowledgments
We thank Sara Courtneidge and Peter Davies for kindly providing the GFP-Tks5 construct and AR19 antibody, respectively. We also thank Minna Roh-Johnson and Esther Arwert for help with the 3D culture and multiphoton imaging, as well as Antonia Patsialou and the Einstein shRNA core facility for guidance in generating the stable β1 integrin shRNA cell line. We thank Allison Harney and the Cox, Segall, Hodgson, and Gertler labs for helpful review of the manuscript and thoughtful discussion. We also thank Vera DesMarais and Danny van der Helm for help in evaluating β1 integrin antibodies in various breast cancer cell lines. We thank the Analytical Imaging Facility and Gruss Lipper Biophotonics Center, Albert Einstein College of Medicine, especially Vera DesMarais and Peng Guo, for technical support. This work was funded by National Institutes of Health Grant CA150344 (J.C.), Komen postdoctoral fellowship KG111405 (V.P.S.), and National Institutes of Health Grants CA133346 and GM100041 (A.J.K.).
Abbreviations used:
- ECM
extracellular matrix
- EGF
epidermal growth factor
- GFP
green fluorescent protein
- MMP
matrix metalloprotease
- RFP
red fluorescent protein
- siRNA
small interfering RNA
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0908) on April 3, 2013.
REFERENCES
- Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–3043. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- Askari JA, Tynan CJ, Webb SED, Martin-Fernandez ML, Ballestrem C, Humphries MJ. Focal adhesions are sites of integrin extension. J Cell Biol. 2010;188:891–903. doi: 10.1083/jcb.200907174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G, Shih DT, Buck CA, Hemler ME. Monoclonal-antibody 9EG7 defines a novel beta(1) integrin epitope induced by soluble ligand and manganese, but inhibited by calcium. J Biol Chem. 1995;270:25570–25577. doi: 10.1074/jbc.270.43.25570. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S, Thanawala R, Bon G, Falcioni R, Prasad GL. Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene. 2005;24:8291–8303. doi: 10.1038/sj.onc.1208993. [DOI] [PubMed] [Google Scholar]
- Bradley WD, Hernandez SE, Settleman J, Koleske AJ. Integrin signaling through Arg activates p190RhoGAP by promoting its binding to p120RasGAP and recruitment to the membrane. Mol Biol Cell. 2006;17:4827–4836. doi: 10.1091/mbc.E06-02-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch KM, Hoshino D, Weaver AM. Adhesion rings surround invadopodia and promote maturation. Biol Open. 2012;1:1–12. doi: 10.1242/bio.20121867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol. 2012;24:277–283. doi: 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Oser M, Chen X, Eddy R, Hodgson L, Condeelis J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr Biol. 2011;21:635–644. doi: 10.1016/j.cub.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccione R, Caldieri G, Ayala I. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 2009;28:137–149. doi: 10.1007/s10555-008-9176-1. [DOI] [PubMed] [Google Scholar]
- Byron A, Humphries JD, Askari JA, Craig SE, Mould AP, Humphries MJ. Anti-integrin monoclonal antibodies. J Cell Sci. 2009;122:4009–4011. doi: 10.1242/jcs.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood DA, Fujioka Y, de Pereda JM, Garcia-Alvarez B, Nakamoto T, Margolis B, McGlade CJ, Liddington RC, Ginsberg MH. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: A structural prototype for diversity in integrin signaling. Proc Natl Acad Sci USA. 2003;100:2272–2277. doi: 10.1073/pnas.262791999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell PT, Chan M, Lindsay AJ, McCaffrey MW, Boettiger D, Norman JC. Rab-coupling protein coordinates recycling of alpha 5 beta 1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J Cell Biol. 2008;183:143–155. doi: 10.1083/jcb.200804140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AY, Raft S, Bailly M, Wyckoff JB, Segall JE, Condeelis JS. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J Cell Sci. 1998;111:199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- Chan KT, Cortesio CL, Huttenlocher A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J Cell Biol. 2009;185:357–370. doi: 10.1083/jcb.200809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, Ratnikov B, Monosov E, Postnova TI, DiScipio R, Smith JW, Strongin AY. MT1-MMP initiates activation of pro-MMP-2 and integrin alpha v beta 3 promotes maturation of MMP-2 in breast carcinoma cells. Exp Cell Res. 2001;263:209–223. doi: 10.1006/excr.2000.5118. [DOI] [PubMed] [Google Scholar]
- Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372–386. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy RJ, Pierini LM, Matsumura F, Maxfield FR. Ca2+-dependent myosin II activation is required for uropod retraction during neutrophil migration. J Cell Sci. 2000;113:1287–1298. doi: 10.1242/jcs.113.7.1287. [DOI] [PubMed] [Google Scholar]
- Felding-Habermann B, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci USA. 2001;98:1853–1858. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- Frelinger AL, Lam SCT, Plow EF, Smith MA, Loftus JC, Ginsberg MH. Occupancy of an adhesive glycoprotein receptor modulates expression of an antigenic site involved in cell-adhesion. J Biol Chem. 1988;263:12397–12402. [PubMed] [Google Scholar]
- Galvez BG, Matias-Roman S, Yanez-Mo M, Sanchez-Madrid F, Arroyo AG. ECM regulates MT1-MMP localization with beta 1 or alpha v beta 3 integrins at distinct cell compartments modulating its internalization and activity on human endothelial cells. J Cell Biol. 2002;159:509–521. doi: 10.1083/jcb.200205026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125:724–734. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiak JJ, Cao HST, Burton DW, Kaushal S, Vargas F, Clopton P, Snyder CS, Deftos LJ, Hoffman RM, Bouvet M. Knockdown of the beta(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int J Cancer. 2011;129:2905–2915. doi: 10.1002/ijc.25942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- Hernandez SE, Krishnaswami M, Miller AL, Koleske AJ. How do Abl family kinases regulate cell shape and movement. Trends Cell Biol. 2004;14:36–44. doi: 10.1016/j.tcb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Huck L, Pontier SM, Zuo DM, Muller WJ. β1-integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc Natl Acad Sci USA. 2010;107:15559–15564. doi: 10.1073/pnas.1003034107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Lakonishok M, Kinder M, Wu S, Truong T, Knudsen KA, Horwitz AF. Integrin and cadherin synergy regulates contact inhibition of migration and motile activity. J Cell Biol. 1998;141:515–526. doi: 10.1083/jcb.141.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaska J, Heino J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol. 2011;27:291–320. doi: 10.1146/annurev-cellbio-092910-154017. [DOI] [PubMed] [Google Scholar]
- Kempiak SJ, Yamaguchi H, Sarmiento C, Sidani M, Ghosh M, Eddy RJ, DesMarais V, Way M, Condeelis J, Segall JE. A neural Wiskott-Aldrich syndrome protein-mediated pathway for localized activation of actin polymerization that is regulated by cortactin. J Biol Chem. 2005;280:5836–5842. doi: 10.1074/jbc.M410713200. [DOI] [PubMed] [Google Scholar]
- Lad Y, Harburger DS, Calderwood DA. Integrin cytoskeletal interactions. Methods Enzymol. 2007;426:69–84. doi: 10.1016/S0076-6879(07)26004-5. [DOI] [PubMed] [Google Scholar]
- Lahlou H, Muller WJ. Beta1-integrins signaling and mammary tumor progression in transgenic mouse models: implications for human breast cancer. Breast Cancer Res. 2011;13:229. doi: 10.1186/bcr2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapetina S, Mader CC, Machida K, Mayer BJ, Koleske AJ. Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J Cell Biol. 2009;185:503–519. doi: 10.1083/jcb.200809085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate KR, Montañez E, Kudlacek O, Füssler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2005;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- Li A, Dawson JC, Forero-Vargas M, Spence HJ, Yu XZ, Konig I, Anderson K, Machesky LM. The actin-bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr Biol. 2010;20:339–345. doi: 10.1016/j.cub.2009.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Wiesner C, Himmel M. Degrading devices: invadosomes in proteolytic cell invasion. Annu Rev Cell Dev Biol. 2011;27:185–211. doi: 10.1146/annurev-cellbio-092910-154216. [DOI] [PubMed] [Google Scholar]
- Mader CC, Oser M, Magalhaes MAO, Bravo-Cordero JJ, Condeelis J, Koleske AJ, Gil-Henn H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011;71:1730–1741. doi: 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes MAO, Larson DR, Mader CC, Bravo-Cordero JJ, Gil-Henn H, Oser M, Chen X, Koleske AJ, Condeelis J. Cortactin phosphorylation regulates cell invasion through a pH-dependent pathway. J Cell Biol. 2011;195:903–920. doi: 10.1083/jcb.201103045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschler S, Wirl G, Spring H, Bredow DV, Sordat I, Beug H, Reichmann E. Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene. 2005;24:2032–2041. doi: 10.1038/sj.onc.1208423. [DOI] [PubMed] [Google Scholar]
- Mierke CT, Frey B, Fellner M, Herrmann M, Fabry B. Integrin α5β1 facilitates cancer cell invasion through enhanced contractile forces. J Cell Sci. 2011;124:369–383. doi: 10.1242/jcs.071985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra AK, Sawada K, Tiwari P, Mui K, Gwin K. A novel, ligand independent activation of c-Met by alpha 5 beta 1-integrin regulates ovarian cancer invasion and metastasis. Oncogene. 2011;28:181–181. doi: 10.1038/onc.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Akiyama SK, Yamada KM. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Mould AP, Akiyama SK, Humphries MJ. The inhibitory anti-beta1 integrin monoclonal antibody 13 recognizes an epitope that is attenuated by ligand occupancy—evidence for allosteric inhibition of integrin function. J Biol Chem. 1996;271:20365–20374. doi: 10.1074/jbc.271.34.20365. [DOI] [PubMed] [Google Scholar]
- Mould AP, Humphries MJ. Regulation of integrin function through conformational complexity: not simply a knee-jerk reaction. Curr Opin Cell Biol. 2004;16:544–551. doi: 10.1016/j.ceb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Mueller SC, Ghersi G, Akiyama SK, Sang QXA, Howard L, Pineiro-Sanchez M, Nakahara H, Yeh Y, Chen WT. A novel protease-docking function of integrin at invadopodia. J Biol Chem. 1999;274:24947–24952. doi: 10.1074/jbc.274.35.24947. [DOI] [PubMed] [Google Scholar]
- Murphy DA, Courtneidge SA. The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara H, Mueller SC, Nomizu M, Yamada Y, Yeh YY, Chen WT. Activation of beta 1 integrin signaling stimulates tyrosine phosphorylation of p190(RhoGAP) and membrane-protrusive activities at invadopodia. J Biol Chem. 1998;273:9–12. doi: 10.1074/jbc.273.1.9. [DOI] [PubMed] [Google Scholar]
- Nishida N, Xie C, Shimaoka M, Cheng YF, Walz T, Springer TA. Activation of leukocyte beta(2) integrins by conversion from bent to extended conformations. Immunity. 2006;25:583–594. doi: 10.1016/j.immuni.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Nystrom ML, Thomas GL, Stone M, Mackenzie IC, Hart IR, Marshall JF. Development of a quantitative method to analyse tumour cell invasion in organotypic culture. J Pathol. 2005;205:468–475. doi: 10.1002/path.1716. [DOI] [PubMed] [Google Scholar]
- Oser M, Mader CC, Gil-Henn H, Magalhaes M, Bravo-Cordero JJ, Koleske AJ, Condeelis J. Specific tyrosine phosphorylation sites on cortactin regulate Nck1-dependent actin polymerization in invadopodia. J Cell Sci. 2010;123:3662–3673. doi: 10.1242/jcs.068163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser M, Yamaguchi H, Mader CC, Bravo-Cordero JJ, Arias M, Chen X, DesMarais V, van Rheenen J, Koleske AJ, Condeelis J. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol. 2009;186:571–587. doi: 10.1083/jcb.200812176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CC, Zhang H, Paravicini M, Gray JW, Baehner F, Park CJ, Bissell MJ. Beta(1) integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006;66:1526–1535. doi: 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69:9498–9506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli J, Tumbarello DA, Schmidt RP, Turner CE. Hic-5 promotes invadopodia formation and invasion during TGF-β-induced epithelial-mesenchymal transition. J Cell Biol. 2012;197:421–437. doi: 10.1083/jcb.201108143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai-Yageta M, Recchi C, Le Dez G, Sibarita JB, Daviet L, Camonis J, D'Souza-Schorey C, Chavrier P. The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J Cell Biol. 2008;181:985–998. doi: 10.1083/jcb.200709076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sameni M, Dosescu J, Yamada KM, Sloane BF, Cavallo-Medved D. Functional live-cell imaging demonstrates that beta(1)-integrin promotes type IV collagen degradation by breast and prostate cancer cells. Mol Imaging. 2008;7:199–213. [PMC free article] [PubMed] [Google Scholar]
- Seals D, Azucenajr E, Pass I, Tesfay L, Gordon R, Woodrow M, Resau J, Courtneidge S. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–165. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Sharma VP, Entenberg D, Condeelis J. High-resolution live-cell imaging and time-lapse microscopy of invadopodium dynamics and tracking analysis. Methods Mol Biol. 2013 doi: 10.1007/978-1-62703-538-5_21. in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylli SS, Stacey TTI, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J Cell Sci. 2009;122:2727–2740. doi: 10.1242/jcs.046680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215. doi: 10.1186/gb-2007-8-5-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- Tanis KQ, Veach D, Duewel HS, Bornmann WG, Koleske AJ. Two distinct phosphorylation pathways have additive effects on Abl family kinase activation. Mol Cell Biol. 2003;23:3884–3896. doi: 10.1128/MCB.23.11.3884-3896.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha M, Declerck YA, Markland FS. Contortrostatin, a snake venom disintegrin, inhibits beta 1 integrin-mediated human metastatic melanoma cell adhesion and blocks experimental metastasis. Cancer Res. 1994;54:4993–4998. [PubMed] [Google Scholar]
- Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer. 2007;7:429–440. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WG, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, Singer RH, Segall JE, Condeelis JS. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- Wang YX, Miller AL, Mooseker MS, Koleske AJ. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc Natl Acad Sci USA. 2001;98:14865–14870. doi: 10.1073/pnas.251249298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren MS, Bradley WD, Gourley SL, Lin YC, Simpson MA, Reichardt LF, Greer CA, Taylor JR, Koleske AJ. Integrin β1 signals through Arg to regulate postnatal dendritic arborization, synapse density, and behavior. J Neurosci. 2012;32:2824–2834. doi: 10.1523/JNEUROSCI.3942-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DE, Kurpios NA, Zuo DM, Hassell JA, Blaess S, Mueller U, Muller WJ. Targeted disruption of beta 1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6:159–170. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Wyckoff JB, Jones JG, Condeelis JS, Segall JE. A critical step in metastasis: In vivo analysis of intravasation at the primary tumor. Cancer Res. 2000;60:2504–2511. [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Diefenbach B, Zhang RG, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin alpha V beta 3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.