

p190RhoGAP-B mediates Rho activity and ductal morphogenesis in response to 3D collagen stiffness. p190B associates with p120-catenin at cell–cell contacts, where RhoA activity is decreased compared to cell–ECM adhesions. This suggests that Rho is in an inactive pool at cell–cell contacts and is recruited to cell–ECM contacts in stiff matrices.
Abstract
Breast epithelial cells cultured in three-dimensional (3D) collagen gels undergo ductal morphogenesis when the gel is compliant and they can achieve tensional homeostasis. We previously showed that this process requires down-regulation of Rho in compliant collagen gels, but the mechanism remains undefined. In this study, we find that p190RhoGAP-B, but not p190RhoGAP-A, mediates down-regulation of RhoA activity and ductal morphogenesis in T47D cells cultured in compliant 3D collagen gels. In addition, both RhoA and p190RhoGAP-B colocalize with p120-catenin at sites of cell–cell contact. The association between p190RhoGAP-B and p120-catenin is regulated by matrix compliance such that it increases in compliant vs. rigid collagen gels. Furthermore, knockdown of p120-catenin disrupts ductal morphogenesis, disregulates RhoA activity, and results in loss of p190B at cell–cell contacts. Consistent with these findings, using a RhoA-specific FRET biosensor (RhoA-FLARE.sc), we determined spatial RhoA activity to be significantly decreased at cell–cell contacts versus cell–ECM adhesions, and, of importance, spatial RhoA activity is regulated by p190B. This finding suggests that RhoA exists as an inactive pool at cell–cell contacts and is recruited to cell–ECM contacts within stiff matrices. Overall, these results demonstrate that RhoA is down-regulated at cell–cell contacts through p190RhoGAP-B, which is localized to cell–cell contacts by association with p120-catenin that is regulated by tensional homeostasis.
INTRODUCTION
Increased mammographic tissue density is a significant risk factor for breast carcinoma (Boyd et al., 2007, 1998). These dense regions of mammary tissue primarily comprise type I collagen, and it is in the areas of increased collagen density that tumors first arise (Guo et al., 2001). The increase in collagen density has been associated with an increase in extracellular matrix (ECM) stiffness (Roeder et al., 2002; Gehler et al., 2009). Changes in the stiffness of the local collagen microenvironment alter cellular signaling pathways and dramatically influence cellular contractility and morphology (Guo et al., 2001; Wozniak et al., 2003; Paszek and Weaver, 2004; Discher et al., 2005). To respond to changes in extracellular stiffness, cells pull against the matrix using Rho/ROCK-mediated contractility (Wozniak et al., 2003). When the matrix is sufficiently compliant, the cells contract and remodel the matrix and achieve a state termed tensional homeostasis, which for epithelial cells is necessary for subsequent ductal morphogenesis (Wozniak et al., 2003; Gehler et al., 2009). In contrast, when cells encounter a stiff matrix, Rho-mediated contractility is countered by the stiffness of the ECM, and the cells continue to generate tension between the ECM and the actin cytoskeleton. This results in increased matrix adhesions (Choquet et al., 1997), activation of focal adhesion proteins, and fundamentally different signaling events (Wozniak et al., 2003; Paszek et al., 2005; Giannone and Sheetz, 2006; Provenzano et al., 2008b). However, the upstream pathways regulating Rho-mediated intracellular force generation and morphogenesis have not been completely defined.
The small GTPase Rho is emerging as a central mechanism by which cells sense and respond to the stiffness of the ECM (Paszek et al., 2005; Provenzano et al., 2009). Several labs found that Rho is responsive to changes in mechanical perturbations, such as fluid shear stress, as well as changes in mechanical properties of the ECM (Pavalko et al., 1998; Numaguchi et al., 1999; Sahai and Marshall, 2003; Wozniak et al., 2003; Yoshizaki et al., 2003; Paszek et al., 2005). Functionally, Rho is a key regulator of cytoskeletal dynamics, which influences a wide range of cellular processes, including cell migration and metastasis (Heasman and Ridley, 2008; Tang et al., 2008). The use of fluorescent reporters to visualize the precise spatial and temporal activity of Rho and the GTPase family members has greatly broadened our understanding of Rho signaling during cell migration, protrusion, and cancer progression primarily in two dimensions (Chamberlain et al., 2000; Malliri and Collard, 2003; Yoshizaki et al., 2003; Nalbant et al., 2004; Kurokawa and Matsuda, 2005; Machacek et al., 2009; Kardash et al., 2010; Pertz, 2010; Omelchenko and Hall, 2012). However, the spatial regulation of RhoA activity and the upstream mechanisms involved in this regulation within three-dimensional (3D) matrices or in response to ECM stiffness are largely unknown.
We previously demonstrated that proper down-regulation of Rho activity is required for ductal morphogenesis of breast epithelial cells within a compliant collagen gel (Wozniak et al., 2003). Particularly striking was the finding that cells have high baseline Rho activity even when held in suspension in the absence of an ECM but down-regulate Rho activity when cultured in a compliant, but not a stiff, ECM (Wozniak et al., 2003). These data suggested that the default state is high Rho activity and that a compliant matrix plays an active role in the down-regulation of Rho activity by a Rho GTPase-activating protein (GAP). In the mouse mammary gland, the Rho-specific GAP, p190RhoGAP-B (p190B), is localized specifically at the terminal end buds of the developing mouse mammary gland (Chakravarty et al., 2000), and mammary-specific knockdown of p190B disrupts ductal morphogenesis of the gland (Chakravarty et al., 2003). In this study we found that the specific knockdown of p190B, but not p190RhoGAP-A (p190A), is required for proper regulation of RhoA activity in response to changes in 3D collagen matrix stiffness. Both RhoA and p190B accumulate at sites of cell–cell contacts, and this is facilitated by the junctional protein, p120-catenin. The expression of a Förster resonance energy transfer (FRET; Lakowicz, 1999)–based biosensor for RhoA activity allowed us to determine that RhoA activity is down-regulated at sites of cell–cell contact compared with sites of cell–matrix interactions. Taken together, these results demonstrate that a p190B–p120-catenin complex down-regulates RhoA at sites of cell–cell contact in compliant 3D collagen gels, a process that is necessary for ductal morphogenesis.
RESULTS
p190RhoGAP-B is required for proper RhoA regulation and ductal morphogenesis in 3D collagen gels
Breast epithelial cells cultured in a compliant 3D collagen gel down-regulate the activity of the small GTPase Rho (Wozniak et al., 2003). Of the known Rho regulatory proteins, both A and B family members of the Rho-GTP activating protein p190RhoGAP are expressed in human tissues (Settleman et al., 1992b; Burbelo et al., 1995b; Chakravarty et al., 2000). To determine which of these family members may be involved in regulation of RhoA activity in response to changes in collagen compliance, we transfected human T47D breast cells with short hairpin RNA (shRNA) constructs specific for either p190A or p190B. Of the shRNAs tested, four specifically knocked down expression of p190B (by ∼30–95%) but did not alter the levels of p190A (Figure 1A and Supplemental Figure S1). In addition, two shRNAs were identified that reduced p190A levels (by ∼50%) but did not change the levels of p190B (Figure 1A and Supplemental Figure S1).
FIGURE 1:

p190RhoGAP-B, but not p190RhoGAP-A, mediates Rho regulation and morphogenesis in compliant collagen gels. (A) Of the shRNAs producing knockdown (two for p190A and four for p190B, characterized in Supplemental Figures S1 and S2), we chose p190A-2 and p190B-1 for all subsequent experiments, based on stability of knockdown and the compatibility of these particular vectors for FRET experiments. p190RhoGAP-A– or B–specific knockdown in T47D cells was confirmed by real-time PCR. Primers specific for either p190A or p190B were used to determine the levels of cDNA in T47D cells stably transfected with control vectors, pLK0.1 or pSM2c, or shRNA constructs, p190A-2 and p190B-1. Isoform-specific knockdown was also determined by Western blot analysis using p190B- and p190A-specific antibodies. Vinculin was used as a loading control. (B) T47D cells expressing p190A or p190B shRNA or the appropriate vector control plasmids (pLK0.1 and pSM2c, respectively) were cultured in attached (rigid) or floating (compliant) 1.3 mg/ml collagen gels for 10 d. Phase contrast images show disruption of normal tubulogenesis in floating gels only when p190RhoGAP-B is knocked down (scale bar, 100 μm). (C) RhoA activity was measured by G-LISA in T47D cells cultured in compliant vs. rigid collagen gels. Active RhoA levels were normalized to total RhoA in each sample (determined by Western blot with RhoA-specific antibodies). In untransfected T47D, vector control (pSM2c and pLK0.1) cells and p190A shRNA–expressing cells, RhoA activity is significantly decreased in compliant vs. rigid gels (*rigid vs. compliant: untransfected, p = 0.0001; pSM2c, p = 0.0226; pLK0.1, p = 0.0453; p190A shRNA, p = 0.0254. n = 5). RhoA activity is no longer regulated by matrix compliance and is elevated in both rigid and compliant gels when p190B is knocked down (#p190B vs. pSM2c vector controls: rigid, p = 0.0450; compliant, p = 0.0110. n = 5).
T47D cells expressing p190A- or p190B-specific shRNA or control vectors were cultured in compliant (floating) versus rigid (attached) 1.3 mg/ml collagen gels. After 10 d in culture, the gels were imaged by phase contrast microscopy to assess ductal morphology. T47D cells expressing control vectors underwent ductal morphogenesis when cultured in 3D compliant collagen gels but not in rigid gels (Figure 1B and Supplemental Figure S2). Of interest, knockdown of p190A did not disrupt normal morphogenesis in compliant collagen gels (Figure 1B and Supplemental Figure S2). However, complete disruption of ductal morphogenesis in compliant gels was observed in p190B-knockdown cells, and the resulting phenotype was indistinguishable from cells cultured in rigid gels (Figure 1B and Supplemental Figure S2). This finding suggests that p190B, but not p190A, is required for ductal morphogenesis in a compliant collagen gel.
We previously demonstrated that ductal morphogenesis requires proper regulation of the Rho-ROCK pathway (Wozniak et al., 2003). To determine whether either p190A or p190B plays a role in regulation of RhoA activity in response to changes in collagen density, we measured RhoA activity in T47D cells expressing shRNA against p190A or p190B in compliant or rigid collagen gels. In control cells, RhoA activity was significantly decreased (∼20%) in cells cultured in compliant gels compared with rigid gels (Figure 1C). Knockdown of p190A did not alter the down-regulation of RhoA activity. However, when p190B was knocked down, the regulation of RhoA activity in response to changes in collagen matrix compliance was inhibited (Figure 1C and Supplemental Figure S1). In fact, RhoA activity increased in both compliant and rigid collagen gels in all four p190B knockdown cell lines (Supplemental Figure S1) compared with untransfected and vector control T47D cells (Figure 1C and Supplemental Figure S1). Subsequently p190B shRNA-1 was chosen for further experiments because this shRNA vector was conducive for FRET analysis of RhoA activity, as it did not coexpress a fluorescent marker. The loss of RhoA down-regulation in compliant conditions corresponds with the disruption of ductal morphogenesis in p190B-knockdown cells and demonstrates that p190B, but not p190A, is required for regulation of RhoA activity in 3D collagen matrices.
The association of RhoA, p190B, and p120-catenin increases in compliant gels
We next sought to determine the upstream mechanisms regulating p190B in response to changes in collagen compliance. To this end, we determined the localization of p190B by immunofluorescence in 3D collagen gels. T47D cells stably expressing green fluorescent protein (GFP)–tagged RhoA were cultured for 10 d in compliant and rigid collagen gels. Using antibodies specific for either p190A or p190B, we found that GFP-RhoA localizes with both p190A and p190B at sites of cell–cell contact. The colocalization of GFP-RhoA with p190A or p190B was similar in both compliant and rigid matrices (Figure 2A).
FIGURE 2:

p190RhoGAP-B association with RhoA increases in compliant collagen gels. (A) T47D cells stably expressing a GFP-RhoA construct were cultured for 10 d in either compliant or rigid 1.3 mg/ml collagen gels. The gels were fixed, and an antibody specific for p190A or B was used to determine localization. Images show that GFP-RhoA localizes with p190A and p190B at sites of cell–cell contact (scale bar, 100 μm). (B) T47D cells were cultured in compliant vs. rigid 1.3 mg/ml collagen gels, and RhoA was coimmunoprecipitated. Densitometric analysis of the Western blots was used to determine p190B and p120-catenin associated with RhoA in compliant and rigid conditions. p190B association with RhoA significantly increased in compliant vs. rigid collagen gels (p = 0.026, n = 6). p120-catenin association with RhoA significantly increased 1.9-fold in compliant vs. rigid collagen gels (*rigid vs. compliant p = 0.05, n = 6).
Others demonstrated that p190A regulates RhoA activity at sites of cell–cell contact and that p120-catenin plays a role in coordinating this regulation (Wildenberg et al., 2006). Consistent with reports of p190A and p120-catenin, we found that both p190A and p190B colocalized with p120-catenin in T47D cells cultured in compliant and rigid collagen gels (Figure 3A). The localization of either p190RhoGAP-A or -B and p120-catenin did not appear to dramatically change in response to collagen compliance by immunofluorescence (Figure 3A). However, coimmunoprecipitation of p190B with p120-catenin and RhoA increased in compliant versus rigid conditions (Figure 3B). The same result holds true when the complex of p190B:p120-catenin:RhoA is immunoprecipitated with an antibody against RhoA (Figure 2B). Of interest, in our hands both p190A:RhoA and p190A:p120-catenin association was very weak and not regulated by matrix compliance (data not shown). Taken together, these results suggest that p190B and p120-catenin may function together to down-regulate RhoA activity in response to matrix compliance, potentially at sites of cell–cell contacts.
FIGURE 3:

p120-catenin associates with p190 RhoGAP-B, and this association increases under compliant conditions. (A) Localization of p190B, p190A, and p120-catenin in compliant vs. rigid collagen gels by immunofluorescence using antibodies against p120-catenin, p190A, and p190B. p190B and p190A localize with p120-catenin at sites of cell–cell contact in compliant and rigid 3D collagen gels (scale bar, 100 μm). (B) Immunoblot analysis of RhoA and p120-catenin levels from p190B coimmunoprecipitations demonstrate that the association between p190B and p120-catenin is enhanced twofold in compliant vs. stiff collagen gels. (*rigid vs. compliant p = 0.0056, n = 5). The association of p190B and Rho trended toward an increase under compliant conditions; however, it is not significant (p = 0.073, n = 6). (C) GST pull-down to determine binding interactions of p190B and p120-catenin. Left, schematic of p120-catenin isoforms 3A, 4A, and 4AΔ560–628 (isoform 4A with a deletion of the RhoA-binding domain, amino acids 560–528) tagged with GST. Using these purified GST-p120-catenin proteins incubated with T47D lysates, we determined that p190B can bind to all of the p120-catenin constructs. Quantification of p190B bound to p120CTN-4A showed a 57% decrease compared with p190B bound to p120CTN-3A. The Rho binding domain deletion, p120CTN-4A-ΔRBD, also bound less p190B than did p120CTN-3A (62% less), but the association of p190B with p120CTN-4A or p120CTN-4A-ΔRBD was not different (N.S.). Thus the interaction between p120-catenin and p190B is not mediated by RhoA.
To test the hypothesis that p120-catenin binding to RhoA serves as a scaffold for p190B interaction, we used GST pull-down assays to determine whether these two regulatory proteins interact via RhoA. p120-catenin isoforms 3A, 4A, and a mutant of isoform 4A (4A Δ560–628) that deletes the RhoA-binding domain (schematic shown in Figure 3C) were expressed as glutathione S-transferase (GST)–tagged fusion proteins, purified, and incubated with T47D cell lysates to determine binding of p190B. Our results demonstrate that p190B bound robustly to p120-catenin isoform 3A, which is the most predominant isoform found in T47D cells. In addition, p190B bound isoform 4A and the Rho-binding domain deletion mutant 4A Δ560–628 to a lesser extent compared with 3A binding (43 and 38%, respectively; Figure 3C). The difference in binding between the 4A isoform and 4A Δ560–628 was not significant. Thus we can conclude that the interaction between p190B and p120-catenin is not dependent on RhoA binding to p120-catenin.
Further attempts to disrupt the interaction between p120-catenin and p190B using overexpression of p190B, expression of p190B deletion mutants, or rescue with shRNA resistant GFP-p190B in T47D cells resulted in caspase-mediated cell death (data not shown). These results are similar to reports from Ludwig and Parsons (2011) demonstrating that ectopic expression of p190RhoGAP induces apoptosis within 24 h of overexpression and suggest that the levels of p190RhoGAP are tightly regulated by the cell.
p120-catenin regulates RhoA activity and p190B localization in 3D collagen gels
To investigate whether p120-catenin may be part of the mechanism by which p190B regulates RhoA activation in response to matrix compliance, we transfected T47D cells with shRNA directed against human p120-catenin or pRS-vector alone as a control. Similar to published results in MDA-231 breast carcinoma cells (Yanagisawa and Anastasiadis, 2006), p120-catenin shRNA dramatically diminished p120-catenin protein levels compared with untransfected T47D cells and vector controls (Figure 4D).
FIGURE 4:

p120-catenin contributes to localizing p190RhoGAP-B to cell–cell contacts and to regulating activity of RhoA. (A) T47D cells expressing p120-catenin shRNA or the appropriate vector control plasmid were cultured in attached or floating 1.3 mg/ml collagen gels for 10 d. Phase contrast images show that shRNA against p120-catenin disrupts tubulogenesis in floating gels, as well as normal cell growth in attached collagen gels (scale bar, 100 μm). (B) In these cell lines, RhoA activity was down-regulated in T47D control cells and in p120-catenin shRNA cells cultured in compliant vs. rigid collagen gels (*compliant vs. rigid: untransfected, p = 0.0001; pRS, p = 0.0011; p120shRNA, p = 0.0464; n = 5). Of interest, p120-catenin is necessary for the proper level of RhoA activity in both compliant and rigid collagen gels, as RhoA activity is significantly elevated in p120-catenin shRNA–expressing cells compared with untransfected and vector control cells (*p120shRNA vs. untransfected, p = 0.0214; p120shRNA vs. pRS, p = 0.0141; n = 5). (C) Top, Immunofluorescence analysis of p120-catenin localization in control and p190B shRNA cells. Knockdown of p190B did not alter the localization of p120-catenin in compliant or rigid collagen gels. Bottom, analysis of p190B localization in control vs. p120-catenin shRNA cells completed after culture in compliant and rigid collagen gels. In contrast to p190B shRNA cells, knockdown of p120-catenin results in the visible loss of p190B at cell–cell contacts. (D) Western blot analysis confirmed that the total level of p190B was not altered in control, human-specific p120-catenin-shRNA or mouse p120-catenin-3A rescue cell lines. z was used as a loading control. Quantification of p190B immunofluorescence in regions of interest demonstrate a significant decrease in p190B intensity at sites of cell–cell contact in both rigid and compliant collagen gels when p120-catenin is knocked down (scale bar, 100 μm; *rigid and compliant pRS vs. p120-catenin shRNA, p = 0.004; n = 9 fields of cells in three experiments). The localization of p190B to cell–cell contacts was restored to 75% of control levels and significantly increased above knockdown levels upon rescue of p120-catenin with mouse p120CTN-3A (*rigid and compliant: p120-catenin shRNA vs. mouse-p120CTN-3A rescue, p = 0.042; n = 9 fields of cells in three experiments).
The increase in association of p190B with p120-catenin under compliant collagen gel conditions correlates with decreased RhoA activity. To determine whether p120-catenin plays a role in regulation of RhoA activity in response to matrix compliance, we analyzed RhoA activity in our cells expressing p120-catenin shRNA. of interest, similar to control cells, p120-catenin–knockdown cells exhibited the same down-regulation of RhoA activity in compliant gels as in rigid gels. However, the overall levels of Rho activity were significantly increased compared with untransfected and vector control cells (Figure 4C). These findings are similar to results demonstrating that depletion of p120-catenin enhances RhoA activation (Yanagisawa and Anastasiadis, 2006; Yanagisawa et al., 2008), suggesting that p120-catenin is needed for the proper regulation of RhoA activity in a compliant matrix.
Consistent with the effect on RhoA activation, p120-catenin–knockdown cells exhibited disrupted morphology under both compliant and rigid conditions. In rigid gels the untransfected cells and pRS vector control cells grew in large colonies; in contrast, cells expressing the p120-catenin shRNA construct grew as single cells or in two- to three-cell clusters (Figure 4D). Unlike control cells, which underwent ductal morphogenesis in compliant gels, p120-catenin–knockdown cells had disrupted ductal morphogenesis (Figure 4D). It is unclear whether the disruption of ductal morphogenesis is due to misregulation of RhoA activity or results from the role of p120-catenin in clustering cadherins to stabilize cell–cell contacts. In either case, our findings demonstrate that p120-catenin knockdown results in the disregulation of both RhoA activity and ductal morphogenesis in 3D collagen gels.
Next we analyzed the localization of p190B and p120-catenin in p120-catenin– or p190B-knockdown cells. Loss of p190B did not disrupt the localization of p120-catenin to cell–cell contacts (Figure 4C). However, knockdown of p120-catenin in T47D cells resulted in a dramatic (50% decrease in intensity) loss of p190B at sites of cell–cell contact compared with vector control cells (Figure 4, C and D). Using a mouse p120-catenin construct, we were able to rescue p120-catenin expression (Figure 4D) and determine localization of p190B. Although total p190B levels measured by Western blot were not changed in either p120-catenin–knockdown or p120-catenin–rescue cells (Figure 4D), p120-catenin–rescue cell lines restored the localization of p190B to sites of cell–cell adhesion to 75% of controls as visualized by immunofluorescence. This result suggests that p120-catenin may help to spatially localize p190B to cell–cell contacts in both compliant and rigid collagen matrices.
Spatial regulation of RhoA activity in 3D collagen gels
If the localization of p190B and p120-catenin is important to the regulation of RhoA activity, then we hypothesized that RhoA activity will be decreased at sites of cell–cell contact where both p190B and p120-catenin localize. To this end, we cultured T47D cells expressing a RhoA biosensor (RhoA-FLARE.sc) described by Pertz et al. (2006) in compliant or rigid collagen gels for 10 d to determine spatial regulation of RhoA activity. A confocal (White, 1987) image was taken in an optical plane midway through the cells, and RhoA activity was determined by ratiometric FRET (Tadross et al., 2009). The analysis included regions of cell–matrix interaction, which can be visualized by second-harmonic generation imaging (SHG; Campagnola and Loew, 2003) of collagen (Provenzano et al., 2008a), and regions of cell–cell contact visualized by RhoA-FLARE.sc (Figure 5A). After segmenting of the images into two separate regions of interest (ROIs; cell–matrix ROI, red; cell–cell ROI, blue), quantitative image analysis demonstrated that RhoA activity was significantly higher at sites of cell–matrix interaction than at sites of cell–cell contact (Figure 5, B and C). In addition, RhoA activity at cell–matrix adhesions in rigid gels was significantly higher than activity at the cell–matrix adhesions in compliant gels (Figure 5B). This result correlates with our biochemical measurements demonstrating that total RhoA activity is significantly higher in rigid gels than in compliant gels (Figure 1C) and demonstrates local activation of RhoA at the cell–matrix contact in response to matrix compliance.
FIGURE 5:

RhoA activity is spatially regulated at cell–cell vs. cell–matrix attachments through a mechanism involving p190B. (A) T47D cells stably expressing a RhoA biosensor (RhoA-FLARE.sc) were cultured in floating or attached 1.3 mg/ml collagen gels (scale bar, 100 μm). Multiphoton microscopy was used to visualize CFP (green), and collagen was visualized by second harmonic generation (red). (B) The same cells were imaged by confocal microscopy to collect direct CFP emission (shown in a and b) and fretted YFP emission. The images were segmented into regions of cell–matrix and cell–cell interactions. Spatial RhoA activity was determined by ratiometric FRET analysis. (C) RhoA activity in control cells was determined to be significantly enhanced at sites of cell–matrix interactions compared with sites of cell–cell interactions. In addition, RhoA activity at the cell–matrix contact was significantly higher under rigid than under compliant conditions. In rigid conditions the loss of p190B resulted in a significant increase in RhoA activity at the cell–matrix contact, similar to that of controls. However, under compliant conditions the spatial regulation of RhoA activity was lost in p190B-knockdown cells, such that there is no longer a significant increase in RhoA activity at cell–matrix compared with cell–cell contacts. Of importance, comparing RhoA activity at cell–matrix ROIs, we find that knockdown of p190B significantly elevates RhoA activity at the matrix compared with control cells (*cell–cell vs. cell–matrix ROIs: control rigid, p = 0.038; control compliant, p = 0.005; p190BshRNA rigid, p = 0.003; #vs. compliant cell–matrix ROI: control rigid, p = 0.026; p190BshRNA compliant, p = 0.039; n = 9, three fields of cells from each of three independent experiments).
Of interest, the pool of active RhoA at cell–cell contacts does not change in response to matrix stiffness (Figure 5C), and we previously demonstrated a decrease in association of p190B with p120-catenin and RhoA in rigid gels (Figures 2 and 3). The decreased association may result in a reduced pool of inactive RhoA at cell–cell contacts allowing the unbound RhoA to localize to the cell–matrix contact, where it can be activated (Figure 6). To determine whether p190B plays a role in the spatial regulation of RhoA activity, we expressed RhoA-FLARE.sc in p190B-knockdown cells. Similar to controls, RhoA activity in p190B-knockdown cells increased at sites of cell–matrix interaction compared with regions of cell–cell contact; however, the increase was only significant under rigid conditions (Figure 5C). Loss of p190B did not alter the level of RhoA activity at cell–cell contacts but resulted in a significant increase in RhoA activity at the cell–matrix contact in compliant conditions compared with controls (Figure 5C). These results further support the idea that p190B localizes to the cell–cell junctions to maintain a pool of inactive RhoA. In the absence of p190B, these results suggest that the pool of inactive RhoA is released from cell–cell adhesions, allowing RhoA to localize to the cell–matrix contact, where it can be activated. Thus this novel finding demonstrates spatial regulation of RhoA activity at cell–matrix versus cell–cell adhesions in response to matrix compliance through a mechanism involving p190B.
FIGURE 6:
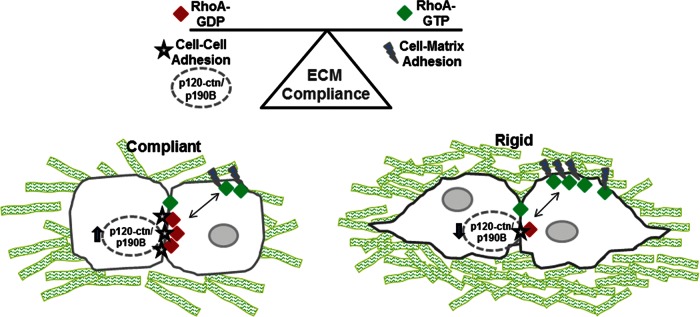
Model of RhoA regulation by cross-talk between cell–cell contacts and cell–matrix adhesions. Changes in collagen matrix compliance regulate spatial RhoA activity within mammary epithelial cells. The activity is regulated by a complex of p120-catenin and p190B localized at cell–cell contacts. In a compliant microenvironment this complex is up-regulated to hold a pool of RhoA inactive at the cell–cell contacts and at the same time may help to stabilize cell–cell contacts. Under rigid conditions, less p190B associates with p120-catenin, and more RhoA is free to localize to sites of cell–matrix adhesion, where RhoA is activated, presumably by a RhoGEF. This local activation of RhoA likely helps to counterbalance the stiffness of the extracellular environment. This model suggests that cross-talk between cell–cell contacts and cell–matrix adhesions is generated by the p190B–p120-catenin complex to spatially regulate RhoA activity in response to changes in tensional homeostasis.
DISCUSSION
Changes in stiffness of the local collagen microenvironment alter cellular signaling pathways, including RhoGTPases, and dramatically influence cellular contractility and morphology (Guo et al., 2001; Wozniak et al., 2003; Paszek and Weaver, 2004; Discher et al., 2005); however, very little is known about the upstream mechanisms regulating RhoA activity. This study demonstrates that RhoA activity is spatially down-regulated at sites of cell–cell contact compared with sites of cell–matrix interactions within a 3D collagen environment. We show that p190RhoGAP-B, but not p190RhoGAP-A, is required for the proper down-regulation of RhoA activity specifically at sites of cell–cell contact when mammary epithelial cells achieve tensional homeostasis within compliant collagen gels. In addition, p120-catenin facilitates the down-regulation of RhoA activity at cell–cell contacts, as both total RhoA and p190B accumulate at these contacts and p120-catenin is required for p190B localization to cell–cell contacts. Thus we present a novel pathway by which matrix compliance increases association of p190B with p120-catenin to locally down-regulate RhoA activity and promote ductal morphogenesis.
Although spatiotemporal measurements of the small GTPases have been analyzed in two dimensions, minimal data exist on spatial RhoA activity in 3D environments. Fluorescent biosensors for the Rho-family GTPases have been developed and used extensively by several labs to assess the role of the GTPases in 2D migration, cytokinesis, and cytoskeletal reorganization (Yoshizaki et al., 2003; Parsons et al., 2005; Su et al., 2009; Pertz, 2010). Timpson et al. (2011) analyzed spatial regulation of RhoA activity in 3D extracellular matrices during pancreatic cell invasion in vitro and in vivo. Their findings suggest that RhoA is activated at the poles of invading cells, whereas basal levels of RhoA activity are maintained in the cell body (Timpson et al., 2011). Here we demonstrate subcellular localization of RhoA activity in polarized mammary epithelial cells undergoing ductal morphogenesis (Figure 5). Our results are consistent with the idea that RhoA activity is spatially regulated in areas of membrane–ECM interactions compared with sites of cell–cell contacts, and this spatial regulation is responsive to changes in collagen matrix compliance.
Our ability to adapt FRET biosensors for 3D use not only allowed for analysis of spatial RhoA activity within 3D collagen gels, but it also helped us to define one of the mechanisms involved in the regulation of RhoA activity in response to changes in 3D matrix compliance. FRET measurements suggest that the large pool of RhoA localized to the cell–cell contacts is largely inactive; knockdown and coimmunoprecipitation results demonstrate that it is a complex of p120-catenin and p190B that inactivates the pool of RhoA at cell–cell contacts. Furthermore, this complex is regulated by the compliance of the ECM, suggesting that p120-catenin is necessary for the accumulation of p190B at sites of cell–cell contact, where RhoA is inactivated and sequestered in compliant conditions. Recently Omelchenko and Hall (2012) determined that another RhoGAP-containing protein, myosin-IXA, is required to down-regulate RhoA activity at cell–cell junctions, and this regulation is necessary for collective cell migration of bronchial epithelial cells. Coupled with our findings, this suggests specific spatiotemporal regulation of RhoA at cell–cell and cell–matrix contacts, which requires tight regulation by both GAPs and guanine nucleotide exchange factors (GEFs).
Evidence establishes an important role for p120-catenin in mammary development and tumor progression. Kurley et al. (2012) determined that mammary-specific knockdown of p120-catenin at puberty onset disrupts terminal end bud function and mammary gland development. Although they attributed the disruption of mammary morphogenesis to the loss of E-cadherin, failure of cell–cell adhesion, and loss of the collective migration process, their results are strikingly similar to those reported for mammary-specific knockdown of p190B (Chakravarty et al., 2003). In the latter studies, p190B RhoGAP localized to the terminal end buds during development, and loss of p190B disrupted mammary gland morphogenesis, a loss attributed to altered insulin-like growth factor 1 (IGF-1) signaling (Chakravarty et al., 2000, 2003). Here we demonstrate a link between p120-catenin and p190B RhoGAP in the spatiotemporal regulation of RhoA activity and downstream morphogenesis of breast epithelial cells in response to collagen compliance. Although similar to the model of Wildenberg et al. (2006), in which association of p190A and p120-catenin modulates Rho/Rac antagonism and adherens junction formation in NIH 3T3 cells in 2D culture conditions, our results are unique in that we find a role specifically for the p190B RhoGAP family member, and not p190A, in regulating RhoA activity in 3D microenvironments. It is possible that the differential association between p120-catenin and either p190A or p190B may be due, in part, to isoform-specific expression of p120-catenin. Several labs demonstrated switching of p120-catenin isoforms 1, 3, and 4 during tumor progression such that p120-catenin isoform-1 is up-regulated and involved in mesenchymal tumor cell invasion (Husmark et al., 1999; Tran et al., 1999; Eger et al., 2000; Ohkubo and Ozawa, 2004; Vandewalle et al., 2005; Yanagisawa et al., 2008). We note that T47D breast epithelial cells primarily express isoform-3, and p190B bound primarily to this isoform. In these experiments, we did not notice any change in the expression levels of the p120-catenin isoforms in response to changes in matrix compliance. However, it would be interesting to determine whether switching of p120-catenin isoforms would result in altered cellular localization of p190B or RhoA or alter downstream RhoA activity in other breast carcinoma cell lines.
The observation of specific function for the RhoGAP family member p190B in regulation of RhoA activity in response to tensional homeostasis due to 3D collagen compliance or stiffness is novel. The p190RhoGAP subfamily is composed of two closely related proteins, p190A and p190B, which are >50% homologous in amino acid sequence (Burbelo et al., 1995a). Functionally both isoforms have GAP activity for Rac, Cd42, and Rho, with specificity for Rho in vivo, and show ubiquitous tissue expression (Settleman et al., 1992a; Ridley et al., 1993; Burbelo et al., 1995a, 1998). Despite the similarities, tyrosine phosphorylation of p190A regulates its interaction with accessory proteins, such as p120RasGAP or TFII (McGlade et al., 1993; Roof et al., 1998, 2000; Jiang et al., 2005), whereas p190B is primarily regulated by subcellular localization (Matheson et al., 2006; Bustos et al., 2008). Our results are among only a few to include a direct comparison of p190A and p190B functions. Our finding of a role for the p190B-RhoGAP is consistent with the observation that p190B is localized to the terminal end buds of the developing mammary gland and is required for mammary ductal morphogenesis in vivo (Chakravarty et al., 2000, 2003). Moreover, others demonstrated that overexpression of p190B in mouse mammary gland promotes desmoplasia and tumor progression (Vargo-Gogola et al., 2006; Heckman-Stoddard et al., 2009; McHenry et al., 2010). However, whereas the mouse studies show the importance of p190B in mammary gland development and mammary tumor progression, these reports either did not access or were not able to find differences in RhoA activity but attributed their results to altered IGF signaling. Thus our results demonstrating the necessity of p190B in the spatial regulation of RhoA activity and subsequent ductal morphogenesis in compliant collagen gels in breast epithelial cells in vitro provide molecular insight for the results in vivo and suggest that p190B is an important regulator of RhoA during mammary gland development, in part by spatially controlling RhoA activation at cell–cell contacts.
Others have shown evidence for cross-talk between adherens junctions and matrix adhesions, such that events occurring at one site regulate events at the opposing site (Avizienyte et al., 2002; de Rooij et al., 2005; Wildenberg et al., 2006). On the basis of our results, we suggest a model in which the complex of p190B and p120-catenin at cell–cell contacts not only sequesters a large pool of inactive RhoA, but also modulates the amount of RhoA available to be activated at sites of cell–matrix interaction (see model in Figure 6). In support of this model, we found that RhoA activity at sites of cell–matrix interaction was significantly enhanced in rigid gels compared with compliant gels, whereas RhoA activity at sites of cell–cell contact remains unchanged in response to matrix compliance (Figure 6). Our data in rigid collagen gels suggest that the decrease in p120-catnein/p190B association frees RhoA to localize to the cell–matrix contact, where it is then activated (Figure 6). In addition, RhoA activity at cell–matrix adhesions significantly increases in compliant conditions when p190B is lost, further demonstrating that p190B is involved in the spatial regulation of RhoA. Taken together, these findings support the model of tensional homeostasis regulating cell phenotype (Paszek et al., 2005; Gehler et al., 2009), as the cell needs to generate internal contractility or tension through the regulation of RhoA activity and downstream actomyosin activity to oppose the tension or compliance of the local microenvironment, and this change in tension can alter cell phenotype. Our results also raise the possibility that other RhoA-regulatory molecules such as RhoGEFs may be spatially sequestered at the cell–matrix contact to locally activate Rho (Figure 6). Work from our lab determined that the RhoGEF GEF-H1 is activated in response to matrix stiffness, making it a possible candidate for RhoA activation at the cell–matrix contact in this model (Heck et al., 2012).
In summary, these results demonstrate a novel mechanism for regulation of RhoA activity by tensional homeostasis that is involved in the switch between epithelial ductal morphogenesis and mesenchymal invasive phenotypes in breast epithelial cells. Collagen stiffness is a major risk factor for breast carcinoma, and our data identifying a role for p190B and p120-catenin in the spatial regulation of RhoA in response to matrix rigidity suggest one mechanism by which breast carcinoma might be regulated. We propose that the complex of p120-catenin and p190B mediates cross-talk between cell–cell contacts and the cell matrix to govern the pool of RhoA that is able to localize at cell–cell contacts. At sites of cell–matrix adhesions, RhoA is activated, which results in intracellular contractility to balance cellular tension against the forces of the ECM. In contrast, in a compliant matrix, the ability of the cell to pull the matrix and around up allows the cells to undergo ductal morphogenesis, a process that requires the down-regulation of RhoA activity (Wozniak et al., 2003). Future work will be aimed at defining the upstream mechanisms by which collagen compliance regulates the complex of p120-catenin and p190B, as well as identifying other players in activation of RhoA at the matrix.
MATERIALS AND METHODS
Reagents
Collagen type I was obtained from BD Biosciences (Bedford, MA). Antibodies used include p190RhoGAP, p190RhoGAP-B, p190RhoGAP-A, p120RasGAP, and Rho (BD Biosciences), p120-catenin (Millipore, Billerica, MA), and vinculin and GFP (Sigma-Aldrich, St. Louis, MO). Horseradish peroxidase (HRP)–conjugated secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). Alexa secondary antibodies were purchased from Invitrogen (Eugene, OR). Cell culture media were purchased from Life Technologies (Grand Island, NY).
Cell culture, transfection, and real-time PCR
T47D cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained as described previously (Keely et al., 1995). For lentiviral production, HEK Phoenix-Ampho cells were obtained from National Gene Vectro Biorepository (Indianapolis, IN) and maintained as described. For p190RhoGAP-knockout studies, cells were stably transfected with human p190RhoGAP-A shRNA (sense sequence: #1, CGGTACATTAGAGATGCACAT, #2, CGGTTGGTTCATGGGTACATT), human p190RhoGAP-B shRNA (sense sequence: #1, CGAATACAGATCACAATATTAT, #2, ACGAATACAGATCACAATATTA, #3, CGGCTAATCTTCCATTTACATT, #4, CGGTCAGAAGCTTTGAAGTTAA) or shRNA control vectors pLK0.1 (for p190A shRNA constructs) and pSM2c or pGIPz (for p190B shRNA constructs; Open Biosystems, Huntsville, AL). For p120-catenin–knockout experiments hp120-catenin shRNA or pRetroSuper control vector (kind gifts from Albert Reynolds, Vanderbilt University, Nashville, TN) was stably transfected into T47D cells. For studies to determine localization of Rho, cells were either stably transfected with a GFP-Rho construct to visualize total Rho localization or infected with the unimolecular RhoA biosensor (RhoA-FLARE.sc; details at www.hahnlab.com; a kind gift from Klaus Hahn, University of North Carolina, Chapel Hill, NC) to determine spatial Rho activity.
To determine the isoform-specific knockdown of p190RhoGAP A and B, we performed quantitative real-time PCR. Total RNA was isolated from T47D cells transfected with five different p190A shRNA constructs, two different p190B shRNA constructs, control shRNA, and untransfected cells. Three million cells from each cell line were lysed with TRIzol reagent (Invitrogen, Carlsbad, CA), and RNA was purified using the RNEasy Kit (Qiagen, Valencia, CA). Total RNA quantity and purity were analyzed with an ND-1000 spectrophotometer (NanoDrop, Wilmington, DE). Using random primers, cDNA was generated from 500 μg of RNA template, and then isoform specific primers were used for amplification (p190A: forward, cgagggcgcaaggtttccat, and reverse, cctccgcgggtcttcgtctgt; p190B: forward, ggcggattccatttgacctc, and reverse, actatttctcgctgatgcctacca). The p190 expression was quantified using an Applied Biosystems 7900 Real Time PCR machine and SDS 2.1 software package (Applied Biosystems, Carlsbad, CA). We used 18S ribosomal primers as controls.
Cells were cultured in collagen type I gels as previously described (Keely et al., 1995). Gels containing T47D cells were poured into six-well plates and allowed to polymerize for 4 h at 37°C. Two milliliters of complete media was added, and gels were either detached from the sides of the dish (floating) or were left attached. The time at which the gels were rendered floating was considered day 0. Phase contrast microscopy was carried out using a Nikon TE300 inverted microscope (Nikon Instruments, Melville, NY) equipped with a CoolSNAP fx charge-coupled device camera (Photometrics, Tucson, AZ). Images were acquired using SlideBook, version 4.2 (Intelligent Imaging Innovations, Denver CO) and processed using Photoshop CS2 (Adobe Systems, San Jose, CA).
Western blotting, immunoprecipitations, and immunofluorescence
Protein expression was assessed through immunoblotting. Briefly, cells were lysed in denaturing Laemmli buffer, followed by protein separation using SDS–PAGE. After proteins were transferred onto polyvinylidene fluoride (PVDF) membrane, membranes were blocked using 5% milk plus 0.3% Tween-20 in Tris-buffered saline (TBS). Membranes were probed with 1:2000 anti-p190RhoGAPA, 1:1000 anti-p190RhoGAP-B, or 1:2000 anti–p120-catenin (BD Biosciences, San Jose, CA), 1:5000 anti-vinculin (Sigma-Aldrich), or 1:1000 anti-Rho (EMD Millipore, Billerica, MA), followed by incubation with 1:5000 HRP-conjugated secondary antibodies (Jackson ImmunoResearch). Membranes were visualized using ECL reagents (GE Biosciences, Pittsburgh, PA).
Immunoprecipitations of p190B or Rho were completed similar to those described previously (Wozniak and Keely, 2005). Briefly, collagen gels containing 10 million cells in RPMI/bovine serum albumin (BSA) were allowed to polymerize for 1 h, and then gels were released or left attached as described. Gels were incubated for an additional 3 h at 37°C. Cells in gels were lysed at 4°C for 10 min with 2× lysis buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2% NP-40, 2 mM NaF, 0.5% NaDOC, 1 mM pervanadate, and protease inhibitors). Lysates were centrifuged at 20,500 × g for 5 min to remove collagen and insoluble components, and the supernatants were incubated with either p190B or Rho antibody plus 30 μl GammaBind G-Sepharose (GE Biosciences) overnight at 4°C. Samples were washed extensively with lysis buffer, and bound proteins were eluted directly into Laemmli buffer. Samples were separated using SDS–PAGE and transferred onto PVDF membranes. Membranes were blocked with 3% BSA plus 0.3% Tween-20 in TBS and then incubated with Rho, p120-catenin, or p190B antibodies. After incubation with secondary antibodies, membranes were rinsed and then visualized using ECL reagents. For normalization of immunoprecipitations, densitometry was completed using ImageJ software (National Institutes of Health, Bethesda, MD), and then p120-catenin levels were normalized to levels of p190B, whereas in the Rho immunoprecipitations p190B levels were normalized to levels of Rho. All digital images for micrographs and immunoblots were processed and produced using Photoshop CS5 (Adobe Systems).
The localization of GFP-Rho, p190B, and p120-catenin in 3D collagen gels was assessed by immunofluorescence. Briefly, cells were cultured in floating or attached gels for 10 d, and then the gels were fixed in 4% paraformaldehyde, permeabilized in 0.2% Triton X-100, and incubated in blocking buffer (1% BSA + 1% donkey serum) before antibody incubation. All primary antibodies were used at 1:100 dilution and incubated for 1 h at room temp. Alexa secondary antibodies were used at 1:800 dilution with a 1-h room temperature incubation. Immunofluorescence and collagen matrix images were observed using multiphoton laser scanning microscopy and SHG on a custom-built multiphoton microscope platform (Wokosin et al., 2003) with an excitation source produced by a Spectra Physics Mai Tai DeepSee laser (Newport Corporation, Irvine, CA), tuned to a wavelength of 890 nm, mounted around a Nikon Eclipse TE300 inverted microscope. Images were focused onto a Nikon 60X Plan Apo water-immersion lens (numerical aperture 1.4), and SHG emission was observed at 445 nm and discriminated from fluorescence using a 445-nm, 20-nm narrow-bandpass emission filter (Semrock, Lake Forest, IL).
RhoA activity assays
As described previously (Keely et al., 2007), RhoA activity was measured using a RhoA G-LISA Kit (Cytoskeleton, Denver, CO) and normalized to total RhoA determined by Western blot analysis. T47D cells were cultured in floated or attached collagen gels at a density of one million cells per 1 ml of collagen gel. The gels were allowed to polymerize for 1 h and then released or left attached for an additional 2 h. The gels were then lysed on ice in 400 μl of the provided lysis reagent, and after centrifugation 60 μl of the supernatant was used in the plate assay following the G-LISA protocol, and 30 μl was loaded on an SDS–PAGE gel for Western blot analysis of total Rho levels. Active RhoA was normalized to Western blots for total RhoA determined by densitometry using ImageJ (Collins, 2007).
RhoA activity by FRET
The confocal data sets were collected on an Olympus FluoView FV1000 confocal microscope (Olympus, Center Valley, PA) and processed in ImageJ. The 16-bit tiff formats were read into ImageJ and the saturated pixels and those below a determined threshold excluded in all measurements. ROIs were recorded based upon anatomical features to include cell–cell or cell–matrix segments. From the ROI data, pixel-by-pixel ratio values of fretted yellow fluorescent protein (YFP) over cyan fluorescent protein (CFP) were calculated, and then a third, unfretted YFP channel was collected and the FRET ratio was normalized to these values. All fields of view and ROIs were summarized for each condition, and a Student's t test was used to look for individual significance in treatments and regional information.
GST pull-down assays
The p120-catenin isoforms 3A and 4A were PCR amplified from LZRS-GFP-mp120–3A and 4A (a kind gift from Albert Reynolds) with flanking EcoRI restriction sites. The digested PCR product was then cloned into the pGEX-4T1 bacterial expression vector. Deletion of the Rho-binding domain (Δ560–628 amino acids) from isoform 4A was generated by QuikChange Mutagenesis Kit (Stratagene, La Jolla, CA). After transformation into BL21 bacteria, protein expression was induced with isopropyl-β-d-thiogalactoside and bacteria allowed to grow at 37°C for 3–4 h. Pelleted bacteria were lysed (50 mM Tris, pH 8.0, 1 mM EDTA, pH 8.0, 5 mg/ml lysozyme, 1 mM dithiothreitol, 1% sarcosyl, and protease inhibitors) and centrifuged, and the supernatant was incubated with glutathione–Sepharose beads (GE Healthcare Bioscience) for 1 h at 4°C. The beads were then washed in lysis buffer, and the purified protein was used for pull-down assays.
T47D cells were cultured in 2D collagen-coated dishes with BSA in RPMI. For each pull-down, eight million cells were lysed in 2× lysis buffer (described earlier, except that detergent was decreased to 1%). After centrifugation the supernatants were incubated with GST–p120-catenin bound to glutathione–Sepharose beads overnight at 4°C. Samples were washed extensively with lysis buffer, followed by denaturation with Laemmli buffer and SDS–PAGE gel separation. Binding of p190B to the GST constructs was then determined by Western blotting (as described previously).
Statistical analysis
All statistical analysis was completed using Prism 4 (GraphPad Software, La Jolla CA). For direct comparisons of two conditions, a Student's t test was performed. For comparisons of multiple parameters an analysis of variance was completed, followed by a Bonferroni post hoc test. FRET image analysis was completed using the statistical program R (www.r-project.org/).
Supplementary Material
Acknowledgments
We thank Tracy Vargo-Gogola, Klaus Hahn, Albert Reynolds, and Panagiotis Z. Anastasiadis for constructs, Sean Carroll for use of the confocal microscope, Jimmy Fong and Aivar Grislis for assistance with FRET analysis, and Brian Burkle and Jessica Heck for critical evaluation of this study. This work was supported by National Institutes of Health Grants RO1 CA114462 (P.J.K.), RO1 CA142833 (P.J.K.), and R21 EB008811 (P.J.K. and K.W.E).
Abbreviations used:
- ECM
extracellular matrix
- FRET
Forster resonance energy transfer
- p190A
p190RhoGAP-A
- p190B
p190RhoGAP-B
- RBD
Rho-binding domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0386) on April 3, 2013.
REFERENCES
- Avizienyte E, Wyke AW, Jones RJ, McLean GW, Westhoff MA, Brunton VG, Frame MC. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat Cell Biol. 2002;4:632–638. doi: 10.1038/ncb829. [DOI] [PubMed] [Google Scholar]
- Boyd NF, et al. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- Boyd NF, Lockwood GA, Martin LJ, Knight JA, Byng JW, Yaffe MJ, Tritchler DL. Mammographic densities and breast cancer risk. Breast Dis. 1998;10:113–126. doi: 10.3233/bd-1998-103-412. [DOI] [PubMed] [Google Scholar]
- Burbelo PD, Drechsel D, Hall A. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J Biol Chem. 1995a;270:29071–29074. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- Burbelo PD, Finegold AA, Kozak CA, Yamada Y, Takami H. Cloning, genomic organization and chromosomal assignment of the mouse p190-B gene. Biochim Biophys Acta. 1998;1443:203–210. doi: 10.1016/s0167-4781(98)00207-3. [DOI] [PubMed] [Google Scholar]
- Burbelo PD, Miyamoto S, Utani A, Brill S, Yamada KM, Hall A, Yamada Y. p190-B, a new member of the Rho GAP family, and Rho are induced to cluster after integrin cross-linking. J Biol Chem. 1995b;270:30919–30926. doi: 10.1074/jbc.270.52.30919. [DOI] [PubMed] [Google Scholar]
- Bustos RI, Forget MA, Settleman JE, Hansen SH. Coordination of Rho and Rac GTPase function via p190B RhoGAP. Curr Biol. 2008;18:1606–1611. doi: 10.1016/j.cub.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnola PJ, Loew LM. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat Biotechnol. 2003;21:1356–1360. doi: 10.1038/nbt894. [DOI] [PubMed] [Google Scholar]
- Chakravarty G, Hadsell D, Buitrago W, Settleman J, Rosen JM. p190-B RhoGAP regulates mammary ductal morphogenesis. Mol Endocrinol. 2003;17:1054–1065. doi: 10.1210/me.2002-0428. [DOI] [PubMed] [Google Scholar]
- Chakravarty G, Roy D, Gonzales M, Gay J, Contreras A, Rosen JM. P190-B, a Rho-GTPase-activating protein, is differentially expressed in terminal end buds and breast cancer. Cell Growth Differ. 2000;11:343–354. [PubMed] [Google Scholar]
- Chamberlain CE, Kraynov VS, Hahn KM. Imaging spatiotemporal dynamics of Rac activation in vivo with FLAIR. Methods Enzymol. 2000;325:389–400. doi: 10.1016/s0076-6879(00)25460-8. [DOI] [PubMed] [Google Scholar]
- Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43(1 Suppl):25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol. 2005;171:153–164. doi: 10.1083/jcb.200506152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Schaffhauser B, Beug H, Foisner R. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J Cell Biol. 2000;148:173–188. doi: 10.1083/jcb.148.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, Eliceiri KW, Weaver VM, Calderwood DA, Keely PJ. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20:3224–3238. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G, Sheetz MP. Substrate rigidity and force define form through tyrosine phosphatase and kinase pathways. Trends Cell Biol. 2006;16:213–223. doi: 10.1016/j.tcb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Guo YP, Martin LJ, Hanna W, Banerjee D, Miller N, Fishell E, Khokha R, Boyd NF. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol Biomarkers Prev. 2001;10:243–248. [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Heck JN, Ponik SM, Garcia-Mendoza MG, Pehlke CA, Inman DR, Eliceiri KW, Keely PJ. Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol Biol Cell. 2012;23:2583–2592. doi: 10.1091/mbc.E11-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman-Stoddard BM, Vargo-Gogola T, McHenry PR, Jiang V, Herrick MP, Hilsenbeck SG, Settleman J, Rosen JM. Haploinsufficiency for p190B RhoGAP inhibits MMTV-Neu tumor progression. Breast Cancer Res. 2009;11:R61. doi: 10.1186/bcr2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmark J, Heldin NE, Nilsson M. N-cadherin-mediated adhesion and aberrant catenin expression in anaplastic thyroid-carcinoma cell lines. Int J Cancer. 1999;83:692–699. doi: 10.1002/(sici)1097-0215(19991126)83:5<692::aid-ijc21>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Jiang W, Sordella R, Chen GC, Hakre S, Roy AL, Settleman J. An FF domain-dependent protein interaction mediates a signaling pathway for growth factor-induced gene expression. Mol Cell. 2005;17:23–35. doi: 10.1016/j.molcel.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Kardash E, Reichman-Fried M, Maitre JL, Boldajipour B, Papusheva E, Messerschmidt EM, Heisenberg CP, Raz E. A role for Rho GTPases and cell-cell adhesion in single-cell motility in vivo. Nat Cell Biol. 2010;12:47–53. doi: 10.1038/ncb2003. [DOI] [PubMed] [Google Scholar]
- Keely PJ, Conklin MW, Gehler S, Ponik SM, Provenzano PP. Investigating integrin regulation and signaling events in three-dimensional systems. Methods Enzymol. 2007;426:27–45. doi: 10.1016/S0076-6879(07)26002-1. [DOI] [PubMed] [Google Scholar]
- Keely PJ, Fong AM, Zutter MM, Santoro SA. Alteration of collagen-dependent adhesion, motility, and morphogenesis by the expression of antisense alpha 2 integrin mRNA in mammary cells. J Cell Sci. 1995;108:595–607. doi: 10.1242/jcs.108.2.595. [DOI] [PubMed] [Google Scholar]
- Kurley SJ, Bierie B, Carnahan RH, Lobdell NA, Davis MA, Hofmann I, Moses HL, Muller WJ, Reynolds AB. p120-catenin is essential for terminal end bud function and mammary morphogenesis. Development. 2012;139:1754–1764. doi: 10.1242/dev.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Matsuda M. Localized RhoA activation as a requirement for the induction of membrane ruffling. Mol Biol Cell. 2005;16:4294–4303. doi: 10.1091/mbc.E04-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Plenum; 1999. [Google Scholar]
- Ludwig K, Parsons SJ. The tumor suppressor, p190RhoGAP, differentially initiates apoptosis and confers docetaxel sensitivity to breast cancer cells. Genes Cancer. 2011;2:20–30. doi: 10.1177/1947601911402680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malliri A, Collard JG. Role of Rho-family proteins in cell adhesion and cancer. Curr Opin Cell Biol. 2003;15:583–589. doi: 10.1016/s0955-0674(03)00098-x. [DOI] [PubMed] [Google Scholar]
- Matheson SF, Hu KQ, Brouns MR, Sordella R, VanderHeide JD, Settleman J. Distinct but overlapping functions for the closely related p190 RhoGAPs in neural development. Dev Neurosci. 2006;28:538–550. doi: 10.1159/000095116. [DOI] [PubMed] [Google Scholar]
- McGlade J, Brunkhorst B, Anderson D, Mbamalu G, Settleman J, Dedhar S, Rozakis-Adcock M, Chen LB, Pawson T. The N-terminal region of GAP regulates cytoskeletal structure and cell adhesion. EMBO J. 1993;12:3073–3081. doi: 10.1002/j.1460-2075.1993.tb05976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHenry, et al. P190B RhoGAP has pro-tumorigenic functions during MMTV-Neu mammary tumorigenesis and metastasis. Breast Cancer Res. 2010;12:R73. doi: 10.1186/bcr2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbant P, Hodgson L, Kraynov V, Toutchkine A, Hahn KM. Activation of endogenous Cdc42 visualized in living cells. Science. 2004;305:1615–1619. doi: 10.1126/science.1100367. [DOI] [PubMed] [Google Scholar]
- Numaguchi K, Eguchi S, Yamakawa T, Motley ED, Inagami T. Mechanotransduction of rat aortic vascular smooth muscle cells requires RhoA and intact actin filaments. Circ Res. 1999;85:5–11. doi: 10.1161/01.res.85.1.5. [DOI] [PubMed] [Google Scholar]
- Ohkubo T, Ozawa M. The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J Cell Sci. 2004;117:1675–1685. doi: 10.1242/jcs.01004. [DOI] [PubMed] [Google Scholar]
- Omelchenko T, Hall A. Myosin-IXA regulates collective epithelial cell migration by targeting RhoGAP activity to cell-cell junctions. Curr Biol. 2012;22:278–288. doi: 10.1016/j.cub.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, et al. Spatially distinct binding of Cdc42 to PAK1 and N-WASP in breast carcinoma cells. Mol Cell Biol. 2005;25:1680–1695. doi: 10.1128/MCB.25.5.1680-1695.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–342. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pavalko FM, Chen NX, Turner CH, Burr DB, Atkinson S, Hsieh YF, Qiu J, Duncan RL. Fluid shear-induced mechanical signaling in MC3T3-E1 osteoblasts requires cytoskeleton-integrin interactions. Am J Physiol. 1998;275:C1591–C1601. [PubMed] [Google Scholar]
- Pertz O. Spatio-temporal Rho GTPase signaling—where are we now. J Cell Sci. 2010;123:1841–1850. doi: 10.1242/jcs.064345. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Yan L, Ada-Nguema A, Conklin MW, Inman DR, Keely PJ. Nonlinear optical imaging of cellular processes in breast cancer. Microsc Microanal. 2008a;14:532–548. doi: 10.1017/S1431927608080884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008b;95:5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Self AJ, Kasmi F, Paterson HF, Hall A, Marshall CJ, Ellis C. Rho family GTPase activating proteins p190, BCR and RhoGAP show distinct specificities in vitro and in vivo. EMBO J. 1993;12:5151–5160. doi: 10.1002/j.1460-2075.1993.tb06210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder BA, Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL. Tensile mechanical properties of three-dimensional type I collagen extracellular matrices with varied microstructure. J Biomech Eng. 2002;124:214–222. doi: 10.1115/1.1449904. [DOI] [PubMed] [Google Scholar]
- Roof RW, Dukes BD, Chang JH, Parsons SJ. Phosphorylation of the p190 RhoGAP N-terminal domain by c-Src results in a loss of GTP binding activity. FEBS Lett. 2000;472:117–121. doi: 10.1016/s0014-5793(00)01439-3. [DOI] [PubMed] [Google Scholar]
- Roof RW, Haskell MD, Dukes BD, Sherman N, Kinter M, Parsons SJ. Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr 1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Mol Cell Biol. 1998;18:7052–7063. doi: 10.1128/mcb.18.12.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- Settleman J, Albright CF, Foster LC, Weinberg RA. Association between GTPase activators for Rho and Ras families. Nature. 1992a;359:153–154. doi: 10.1038/359153a0. [DOI] [PubMed] [Google Scholar]
- Settleman J, Narasimhan V, Foster LC, Weinberg RA. Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell. 1992b;69:539–549. doi: 10.1016/0092-8674(92)90454-k. [DOI] [PubMed] [Google Scholar]
- Su L, Pertz O, Mikawa M, Hahn K, Parsons SJ. p190RhoGAP negatively regulates Rho activity at the cleavage furrow of mitotic cells. Exp Cell Res. 2009;315:1347–1359. doi: 10.1016/j.yexcr.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Veeramani BPS, Yue DT. Robust approaches to quantitative ratiometric FRET imaging of CFP/YFP fluorophores under confocal microscopy. J Microsc. 2009;233:192–204. doi: 10.1111/j.1365-2818.2008.03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Olufemi L, Wang MT, Nie D. Role of Rho GTPases in breast cancer. Front Biosci. 2008;13:759–776. doi: 10.2741/2718. [DOI] [PubMed] [Google Scholar]
- Timpson P, et al. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 2011;71:747–757. doi: 10.1158/0008-5472.CAN-10-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NL, Nagle RB, Cress AE, Heimark RL. N-cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion with stromal cells. Am J Pathol. 1999;155:787–798. doi: 10.1016/S0002-9440(10)65177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005;33:6566–6578. doi: 10.1093/nar/gki965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargo-Gogola T, Heckman BM, Gunther EJ, Chodosh LA, Rosen JM. P190-B Rho GTPase-activating protein overexpression disrupts ductal morphogenesis and induces hyperplastic lesions in the developing mammary gland. Mol Endocrinol. 2006;20:1391–1405. doi: 10.1210/me.2005-0426. [DOI] [PubMed] [Google Scholar]
- White JG. An evaluation of confocal versus conventional imaging of biological structures by fluorescence light microscopy. J Cell Biol. 1987;105:41–48. doi: 10.1083/jcb.105.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Wokosin DL, Squirrell JM, Eliceiri KW, White JG. Optical workstation with concurrent, independent multiphoton imaging and experimental laser microbeam capabilities. Rev Sci Instrum. 2003;74:193–201. doi: 10.1063/1.1524716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, Keely PJ. Use of three-dimensional collagen gels to study mechanotransduction in T47D breast epithelial cells. Biol Proced Online. 2005;7:144–161. doi: 10.1251/bpo112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Anastasiadis PZ. p120 catenin is essential for mesenchymal cadherin-mediated regulation of cell motility and invasiveness. J Cell Biol. 2006;174:1087–1096. doi: 10.1083/jcb.200605022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Huveldt D, Kreinest P, Lohse CM, Cheville JC, Parker AS, Copland JA, Anastasiadis PZ. A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J Biol Chem. 2008;283:18344–18354. doi: 10.1074/jbc.M801192200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki H, Ohba Y, Kurokawa K, Itoh RE, Nakamura T, Mochizuki N, Nagashima K, Matsuda M. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J Cell Biol. 2003;162:223–232. doi: 10.1083/jcb.200212049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.