

ATP7A mediates copper absorption and feeds cuproenzymes in the trans-Golgi network. To regulate copper homeostasis, ATP7A cycles between the TGN and plasma membrane. The roles of clathrin, adaptor complexes, lipid rafts, and Rab22a are assessed in an attempt to decipher the regulatory proteins involved in ATP7A cycling.
Abstract
The transporter ATP7A mediates systemic copper absorption and provides cuproenzymes in the trans-Golgi network (TGN) with copper. To regulate metal homeostasis, ATP7A constitutively cycles between the TGN and plasma membrane (PM). ATP7A trafficking to the PM is elevated in response to increased copper load and is reversed when copper concentrations are lowered. Molecular mechanisms underlying this trafficking are poorly understood. We assess the role of clathrin, adaptor complexes, lipid rafts, and Rab22a in an attempt to decipher the regulatory proteins involved in ATP7A cycling. While RNA interference (RNAi)–mediated depletion of caveolin 1/2 or flotillin had no effect on ATP7A localization, clathrin heavy chain depletion or expression of AP180 dominant-negative mutant not only disrupted clathrin-regulated pathways, but also blocked PM-to-TGN internalization of ATP7A. Depletion of the μ subunits of either adaptor protein-2 (AP-2) or AP-1 using RNAi further provides evidence that both clathrin adaptors are important for trafficking of ATP7A from the PM to the TGN. Expression of the GTP-locked Rab22aQ64L mutant caused fragmentation of TGN membrane domains enriched for ATP7A. These appear to be a subdomain of the mammalian TGN, showing only partial overlap with the TGN marker golgin-97. Of importance, ATP7A remained in the Rab22aQ64L-generated structures after copper treatment and washout, suggesting that forward trafficking out of this compartment was blocked. This study provides evidence that multiple membrane-associated factors, including clathrin, AP-2, AP-1, and Rab22, are regulators of ATP7A trafficking.
INTRODUCTION
Copper is an essential trace element and an important cofactor for many enzymes with roles in neuronal function, embryonic development, pigmentation, and other processes (Uauy et al., 1998; Pena et al., 1999; Harris, 2000; Balamurugan and Schaffner, 2006; Schlief and Gitlin, 2006). As a redox-active metal, excess copper is toxic to cells due to the risk of generating damaging free radicals. Therefore tight regulation of copper balance is vital. Among the proteins known to be important in copper homeostasis is the copper-transporting ATPase ATP7A (Menkes protein, MNK), a member of the P-type ATPase family responsible for ATP-driven ion transport across membranes (Kuhlbrandt, 2004). ATP7A is ubiquitously expressed in the body, observed in the brain and several other tissues, such as kidney, lung, placenta, and mammary gland (Lutsenko et al., 2007), but not liver, which shows high expression of ATP7B. Defects in copper transport are linked to human disease, and mutations in the ATP7A gene are responsible for Menkes disease, occipital horn syndrome (Tümer, 2013), and X-linked distal hereditary motor neuropathy (Kennerson et al., 2010).
The ATP7A transporter meets the challenge of fluctuating copper levels by relocating to sites where copper transport is needed, and this trafficking seems to be crucial for the export of copper from the cell. At low basal levels of copper, ATP7A resides predominantly in the trans-Golgi network (TGN), with a small amount constitutively cycling between the TGN and the plasma membrane (PM; Petris et al., 1996). This location is compatible with ATP7A's role of pumping copper into the TGN lumen to supply cuproenzymes traversing the secretory pathway. When copper levels are raised, a majority of ATP7A relocates to the plasma membrane, or in some cell types to vesicles close to the plasma membrane, to export excess copper from the cell, returning to the TGN when copper levels are reduced (Petris et al., 1996; Nyasae et al., 2007). Further evidence suggests that increasing copper levels shifts ATP7A into a rapid recycling pool, preventing its return to the TGN and maintaining the transporter in the vicinity of the PM (Pase et al., 2003).
Specific motifs within proteins are essential for linking them to the trafficking machinery. Trafficking signals in the carboxyl-terminal cytosolic domain of ATP7A include a dileucine motif (LL positions 1487–1488), required for the internalization of ATP7A from the cell surface (Petris et al., 1998; Petris and Mercer, 1999; Francis et al., 1999); a class 1 PDZ target motif (DTAL positions 1497–1500), important for basolateral membrane targeting (Greenough et al., 2004); and phosphorylation of serine residues, required for trafficking to the PM (Veldhuis et al., 2009). The N-terminal copper-binding domains are also crucial, as it is clear that one of the six copper-binding domains is needed for trafficking to the PM (Goodyer et al., 1999; Strausak et al., 1999; Mercer et al., 2003). It is likely that a conformational shift triggered by copper binding exposes the target information within ATP7A to enable traffic. It was also proposed that glutathionylation and deglutathionylation of ATP7A might play a role in the transporter's trafficking (Singleton et al., 2010).
The importance of the dileucine motif for ATP7A internalization suggests a clathrin-mediated pathway for endocytosis, although attempts to block this pathway do not prevent ATP7A from internalizing (Cobbold et al., 2003; Lane et al., 2004). Clathrin is recruited to the membrane through interactions with the lipid-binding protein AP180 (Ford et al., 2001). Selection of cargo into clathrin-coated structures is mediated through recruitment by transient adaptors. Among the best characterized are the heterotetrameric adaptor proteins (APs) AP-1 and AP-2, consisting of μ1, β1, σ1, and γ for AP-1 and μ2, β2, σ2, and α for AP-2. Adaptors recognize signals in the cytoplasmic portion of membrane proteins that contain dileucine or tyrosine residues (Traub, 2009). Despite their similarity, AP-2 is associated with sorting cargo for clathrin-mediated endocytosis (CME) from the PM, whereas AP-1 is associated with sorting of cargoes shuttling between endosomes and the TGN (Robinson, 2004).
Additional regulatory components organize cargo transport between different compartments in the cell. Ras-related proteins in brain (Rabs) are GTPases that coordinate the complex processes of vesicular/tubular carrier formation, motility, tethering, and fusion. These small proteins function as molecular switches by cycling between a GTP-bound, active and a GDP-bound, inactive state, and a number of Rabs are involved in post-Golgi and endosomal trafficking (Zerial and McBride, 2001; Schwartz et al., 2007; Stenmark, 2009).
In this study we explore the trafficking mechanisms used by ATP7A to cycle within HeLa cells. We show that inhibition of clathrin-regulated pathways prevents ATP7A from being internalized at the PM before delivery to the TGN. Recruitment of ATP7A into the CME pathway appears to involve both AP-2 and AP-1 en route from the PM to the TGN. We also show that ATP7A does not require clathrin for the constitutive delivery on the outbound pathway from the TGN to the PM. However, this route appears to involve the GTPase Rab22, as expression of a constitutively active mutant of Rab22a prevents ATP7A trafficking to the PM in response to copper. Thus our findings implicate clathrin, AP-2, AP-1, and Rab22 as novel regulators of ATP7A trafficking and copper homeostasis.
RESULTS
ATP7A trafficking requires clathrin but not caveolin or flotillin
Multiple pathways operate to traffic membrane and soluble cargoes to and from the cell surface (Hansen and Nichols, 2009). To test whether clathrin- or lipid raft–mediated regulation was involved in ATP7A internalization, we depleted specific regulatory proteins using RNA interference (RNAi). We targeted the caveolin-, flotillin-, and clathrin-regulated steps in HeLa cells using small interfering RNA (siRNA) duplexes directed against caveolin 1 (cav1), caveolin 2 (cav2), flotillin 1 (flot1), or clathrin heavy chain (CLTC). The efficiency of protein depletion (knockdown) was confirmed by immunoblotting (Figure 1A). Note that the siRNA against cav1 causes a decrease in both cav1 and cav2 levels. This was reported previously (Bhatnagar et al., 2004) and is believed to be due to cav1 being required to stabilize cav2 (Razani et al., 2001).
FIGURE 1:

Clathrin is required for ATP7A trafficking. HeLa cells were treated with transfection reagent alone (mock), transfected with nonsilencing siRNA (-ve) or siRNA directed against cav1, cav2, flot1, or CLTC. (A) Immunoblot shows depletion of target proteins (indicated above the blot). A total of 10 μg of total protein was loaded per lane. Proteins are detected with the antibodies indicated on the left. (B) Cells were treated with BCS (a–e), CuCl2 (f–j), or CuCl2 followed by washout (k–o) at 37°C before fixation and IF staining with anti-ATP7A antibody (green) and DAPI nuclear stain (blue). Scale bar, 10 μm; where scale bar is not shown, scale is equivalent to that of n.
Using immunofluorescence (IF) and confocal microscopy we analyzed the localization of ATP7A in cells transfected with control or target siRNA (Figure 1B). In controls, under copper-limiting conditions after incubation with the copper chelator bathocuproinedisulfonic acid (BCS), ATP7A is observed predominantly in a perinuclear location (Figure 1B, a). This is consistent with reports of its basal presence in the TGN (Yamaguchi et al., 1996). Similarly, in cells subjected to knockdown for cav1, cav2, or flot1 (Figure 1B, b–d), ATP7A still resides predominantly in the TGN. However, when cells are transfected with siRNA against clathrin heavy chain, a large proportion of ATP7A is mislocalized to the cell surface (Figure 1B, e).
Although ATP7A shows predominant TGN localization under steady-state conditions, exposing cells to high levels of copper induces ATP7A to traffic out of the TGN to the cell surface (Petris et al., 1996; Cobbold et al., 2002). This relocalization is reversible upon copper washout, and ATP7A returns to the TGN. In cells subjected to cav1, cav2, or flot1 knockdown ATP7A moved to the cell surface when cells were incubated with excess copper ions (Figure 1B, g–i) and returned to the TGN after copper washout (Figure 1B, l–n), similar to control cells (Figure 1B, f and k). In contrast, in cells depleted of clathrin, ATP7A relocated to the PM in the presence of elevated copper (Figure 1B, j) but remained mislocalized to cell surface after copper washout (Figure 1B, o).
To confirm that mislocalization of ATP7A was a result of reduced clathrin levels, we colabeled clathrin-depleted cells with antibodies against clathrin heavy chain and ATP7A (Supplemental Figure S1A, b and d). Surface accumulation of fluorescence-labeled transferrin (Tf) confirms that CME is blocked in these cells (Supplemental Figure S1A, f). The results demonstrate that where Tf internalization is blocked by depletion of clathrin, ATP7A is mislocalized to the cell surface region (Supplemental Figure S1A, b, d, f, and h). This ATP7A cell surface pattern is similar in both BCS- and copper-treated cells (Supplemental Figure S1B, compare d and e), and after copper washout, ATP7A was unable to return to the TGN (Supplemental Figure S1B, f and i).
We further quantified ATP7A (and Tf) internalization in clathrin-depleted cells by assessing the mean area of ATP7A (or Tf) fluorescence labeling on a cell-by-cell basis (Supplemental Figure S2). In this assay inhibition of internalization will result in an increased fluorescence area as ATP7A (or Tf) is redistributed throughout the cell. The analysis was performed on 12 randomly imaged fields of view in triplicate, representing a sample of 3000–4000 cells per condition. Indeed, clathrin depletion results in a significant increase in the mean area of ATP7A fluorescence compared with ATP7A distribution in nontargeting siRNA controls. This result is similar to that observed for Tf, reconfirming that clathrin depletion results in an ATP7A-trafficking block.
Depletion of clathrin may affect the integrity of the TGN, causing ATP7A mislocalization. To verify that Golgi and TGN collapse was not an issue, we stained clathrin-depleted cells with the cis-Golgi marker GM130 or the TGN marker P230. Depleted cells were identified using cell surface accumulation of labeled Tf. Cells depleted of clathrin show normal Golgi and TGN architecture (Figure 2B). Thus clathrin depletion does not cause collapse of the Golgi/TGN, but it affects the trafficking of ATP7A as it moves between the cell surface and the TGN. The loss of ATP7A staining in the TGN of clathrin-depleted cells was not due to degradation because analogous levels of ATP7A were present in clathrin-depleted and nondepleted cells (Figure 2A). Despite >85% reduction in clathrin heavy chain levels in depleted cells, the levels of ATP7A were unaffected.
FIGURE 2:

Clathrin depletion does not affect ATP7A levels or Golgi/TGN architecture. (A) Lysates of siRNA-treated cells containing 10 μg of total protein per lane were separated by SDS–PAGE, followed by immunoblotting with the antibodies indicated on the left of the blot. ATP7A protein levels were quantified, normalized against actin levels, and expressed as percentage of mock control. The graph is representative of six independent experiments. (B) The integrity of the Golgi and the TGN were verified in CLTC-depleted cells by labeling with the cis-Golgi marker GM130 or the TGN marker P230 (green). Cells were incubated with Alexa Fluor 594-Tf (red) at 37°C before washing and fixation to identify clathrin-depleted cells. Scale bar, 10 μm.
ATP7A accumulates at the cell surface in clathrin-depleted cells
Our data suggest that ATP7A accumulates at the surface in cells depleted of clathrin. To verify this, we performed a cell surface biotinylation assay comparing surface levels of ATP7A in control and clathrin-depleted cells.
After siRNA treatment, cells were incubated with either BCS, to deplete copper in the culture media, or elevated copper, to stimulate trafficking of ATP7A to the cell surface. We then biotinylated the cell surface at 4°C, lysed the cells, and isolated the biotinylated cell surface proteins on NeutrAvidin beads. We analyzed the samples by SDS–PAGE and immunoblotting with anti-ATP7A antibodies. Figure 3 shows that in control cells treated with BCS, ATP7A was present at low levels on the cell surface (6.7 ± 3.9%), and this level was greatly increased when cells were incubated with copper (59.7 ± 6.7%). In contrast, in clathrin-depleted cells the amount of ATP7A at the cell surface is high for both BCS-treated (52.2 ± 1.7%) and copper-treated cells (54.1 ± 13.8% of total). Samples were also probed for actin as a negative control to demonstrate specificity for cell surface protein isolation (Figure 3A). Thus the cell surface biotinylation experiments show a significant relocation of ATP7A to the cell surface in clathrin-depleted cells, independent of copper. Of importance, this demonstrates that exocytic traffic of ATP7A from the TGN to the cell surface occurs in clathrin-depleted cells and therefore does not rely on clathrin.
FIGURE 3:
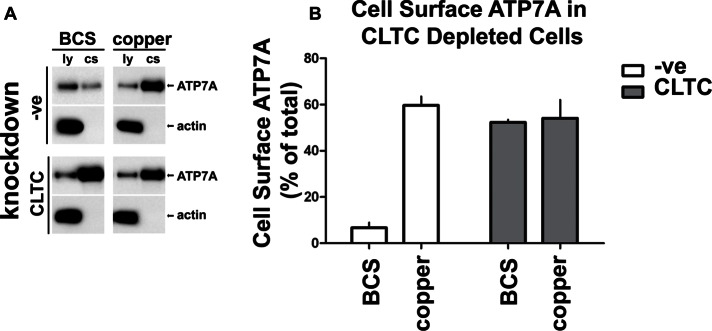
Depletion of clathrin causes ATP7A to accumulate at the cell surface. CLTC-deleted or control HeLa cells transfected with nonsilencing siRNA (-ve) were treated with BCS or CuCl2. Cell surface proteins were labeled with biotin at 4°C, followed by cell lysis and isolation of biotinylated cell surface proteins on NeutrAvidin beads. (A) Western blot of prebead lysate (ly) or eluted cell surface e1 protein samples (cs) probed with an antibody to ATP7A or actin. (B) Quantification of cell surface ATP7A, calculated as a percentage of total ATP7A in the cell lysate. The graph is representative of three independent experiments.
Internalization of ATP7A from the cell surface is dependent on clathrin adaptors AP180 and AP-2
We further investigated the role of clathrin in ATP7A internalization by inhibiting the recruitment of clathrin to membranes through the expression of a dominant-negative fragment of the tethering protein AP180. Overexpression of the carboxyl-terminus of AP180 (AP180-C), which lacks the lipid-binding domain, leads to the inhibition of both epidermal growth factor and Tf internalization, thus confirming its effect as a potent inhibitor of the CME pathway (Ford et al., 2001). We expressed AP180-C in HeLa cells and treated them with BCS, elevated copper, or elevated copper followed by washout. In BCS-treated cells, expression of AP180-C causes the redistribution of ATP7A to the PM (Figure 4, d and g), similar to that seen in copper-treated cells (Figure 4, e and h). When copper is washed out, ATP7A does not return to the TGN in cells expressing the AP180-C protein (Figure 4, f and i), in contrast to nontransfected cells.
FIGURE 4:

ATP7A internalization is inhibited in cells expressing AP180-C. Transiently transected HeLa cells expressing myc-tagged AP180-C were subjected to treatment with BCS, CuCl2, or CuCl2, followed by washout. AP180-C is detected with an antibody against the myc epitope (a–c, g–i red), and cells are counterstained for ATP7A (d–f, g–i green). Scale bar, 10 μm.
ATP7A contains a dileucine carboxyl-terminal sequence within the motif DKHSLL, implicated in the internalization of the transporter (Petris et al., 1998; Petris and Mercer, 1999; Francis et al., 1999). Sequences fitting [D/E]XXXL[L/I] motifs bind the clathrin adaptor AP-2, mainly through interactions with the σ2 subunit and contributions from the α subunit (Kelly et al., 2008). The depletion of the μ2 subunit of AP-2 by RNAi inhibits AP-2–mediated endocytosis (Fraile-Ramos et al., 2003; Motley et al., 2003), and we used this method to probe the role of the AP-2 pathway in ATP7A trafficking.
AP-2 μ2 subunit was efficiently depleted in HeLa cells (Figure 5A). In AP-2–depleted cells under BCS conditions, ATP7A was observed at the TGN (Figure 5B, d and g) and relocated to the cell surface when cells were treated with copper (Figure 5B, e and h). However, after copper washout, ATP7A fails to return to the TGN, as observed in the nondepleted cells (Figure 5B, f and i). These data further support that clathrin and the clathrin adaptors AP180 and AP-2 are essential for ATP7A traffic between the PM and the TGN. However, they do not appear essential for the movement of ATP7A from the TGN to the PM.
FIGURE 5:

Internalization of ATP7A is inhibited in cells depleted of AP-2. (A) Immunoblot comparing lysates from cells treated with nontargeting siRNA (-ve) or depleted of CLTC or AP-2 using siRNA against μ2 subunit of the AP-2 complex. A total of 10 μg of total protein was loaded per lane. Membranes were probed with the antibodies indicated on the left of the blot. (B) HeLa cells depleted of AP-2 were treated with BCS, CuCl2, or CuCl2, followed by washout. Cells showing knockdown of AP-2 are identified by accumulation of Alexa Fluor 594–Tf (a–c, g–i red) at the cell surface, and examples in the merge panels are marked by asterisk. Cells are counterstained for ATP7A (d–f, g–i green). Nondepleted cells showing transferrin internalization are used as an internal negative control; examples are marked with an arrow. Scale bar, 10 μm.
To confirm the involvement of the carboxyl-terminus in targeting ATP7A to the clathrin machinery, we used a CD8 chimera reporter system, a well-characterized method to study sorting signals in the carboxyl termini of transmembrane proteins (Nilsson et al., 1989; Kozik et al., 2010). We previously showed that substituting the CD8 cytoplasmic domain for the extreme carboxyl-terminal 24 amino acids of ATP7A containing the DKHSLL motif (residues 1477–1500, VVTSEPDKHSLLVGDFREDDDTAL*; resulting construct CD8-LL4) triggers the internalization of the normally cell surface located CD8 (Francis et al., 1999).
Control cells treated with nontargeting siRNA or cells depleted of clathrin heavy chain or AP-2 were transiently transfected with the CD8‑LL4 construct and allowed to internalize OKT8, an antibody against the CD8 extracellular domain (Supplemental Figure S3A, a–c), together with labeled Tf (Supplemental Figure S3A, d–f) before fixation. Cells depleted of clathrin or AP-2 were identified by accumulation of labeled Tf at the cell surface. Further confirmation of clathrin depletion was done by staining with an antibody against clathrin heavy chain (Supplemental Figure S3A, j and k). In depleted cells, CD8-LL4 was prevented from internalizing compared with the control, which internalized both OKT8 and Tf (Supplemental Figure S3A).
Using the approach described for Supplemental Figure S2, we quantified OKT8 internalization in CD8‑LL4–expressing cells depleted of clathrin or AP-2 or controls treated with nontargeting siRNA (Supplemental Figure S3B). From binary images we assessed the mean area of OKT8 (or Tf) fluorescence labeling on a cell-by-cell basis, with inhibition of internalization resulting in increased fluorescence area as OKT8 (or Tf) is redistributed throughout the cell. The quantification results confirm that CD8-LL4 internalization is significantly reduced in clathrin- or AP-2–depleted cells. Therefore the extreme C-terminus containing the DKHSLL motif is likely to be responsible for linking ATP7A to the AP-2 and clathrin machinery.
AP-1 is required for the TGN localization of ATP7A
AP-1 clathrin adaptor has also been shown to bind to [D/E]XXXL[L/I] motifs. AP-1 functions at the TGN and within the endosomal pathway (Bonifacino and Traub, 2003) and is involved in the recycling of M6PR to the TGN (Meyer et al., 2000). To test its putative role in the trafficking of ATP7A, we reduced the expression of the AP-1 complex by silencing its μ1A subunit. We were unable to obtain a suitable antibody to check the depletion of μ1A. However, because the γ adaptin subunit of AP-1 becomes cytosolic when μ1A is knocked down, we could identify depleted cells by diffuse rather than membrane-associated immunofluorescence of γ adaptin (Figure 6A, a–c; Meyer et al., 2000; Hirst et al., 2003, 2004; Byland et al., 2007). We also confirmed by Western blot that the depletion of μ1A does not affect the levels of the μ2 subunit of AP-2 (Figure 6C).
FIGURE 6:

AP-1 depletion blocks ATP7A trafficking to the TGN. (A) HeLa Cells depleted of AP-1 by transfection with siRNA targeting the μ1 subunit were treated with BCS, CuCl2, or CuCl2, followed by washout. Cells depleted of AP-1 are identified by loss of adaptin γ signal at the TGN (a–c, g–i red). Cells are counterstained for ATP7A (d–f, g–i green). Examples of depleted cells are marked in the merge panels with asterisk, and nondepleted cells are marked with an arrow. (B) AP-1–depleted cells, identified by loss of adaptin γ signal (red), are labeled for the trans-Golgi marker golgin‑97 (green) or the cis-Golgi marker GM130 (blue). Scale bar, 10 μm. (C) Immunoblot of lysates from control cells (-ve) or those depleted of AP-1 or AP-2 and blotted with antibodies indicated on the left of the blot. A total of 10 μg of protein was loaded per lane.
In AP-1–depleted cells treated with BCS, ATP7A staining is dispersed, in contrast to the TGN localization observed in nondepleted cells (compare Figure 6A, d and g). Copper treatment causes relocation of ATP7A to the cell surface, showing a similar phenotype to nondepleted cells (Figure 6A, e and h). However, upon copper washout, ATP7A again shows diffuse staining, similar to the BCS-treated cells (Figure 6A, f and i).
We were concerned that AP-1 depletion might compromise the integrity of the TGN and account for the dispersed ATP7A phenotype. To demonstrate that Golgi and TGN architecture is unaffected by the loss of AP-1, we stained depleted cells for the markers GM130 and golgin-97. Normal staining pattern for GM130 and golgin-97 was observed in control and AP-1–depleted cells (Figure 6B), suggesting that the dispersed ATP7A localization was a result of a trafficking block and not the collapse of the TGN.
ATP7A localization and trafficking are regulated by Rab22
Rab-mediated sorting events occur at the level of endosomes and the TGN. We assessed pathways mediating ATP7A trafficking by transiently expressing Rab constructs locked in either their GDP (dominant negative) or GTP (constitutively active) forms. Rab11 and Rab22 are implicated in the regulation of protein recycling and traffic to the TGN (for review see Schwartz et al., 2007; Stenmark, 2009). We first assessed the role of Rab11 in ATP7A trafficking. In cells under low copper conditions, expression of dominant-negative forms of Rab11a and b (S25N) had no effect on ATP7A localization to the TGN (Supplemental Figure S4, a and b). In addition, expression of the constitutively active form of Rab11b (Q70L) did not affect ATP7A localization to the TGN under low-copper conditions (Supplemental Figure S4c). When overexpressed, Rab11bQ70L localizes to puncta in some cells; after copper treatment, ATP7A did not enter the Rab11-positive structures (Supplemental Figure S4d). Our data suggest that ATP7A does not use the Rab11-regulated recycling pathway.
Rab22a has been proposed to regulate the recycling pathway from early endosomes to the PM (Weigert and Donaldson, 2005; Magadan et al., 2006; Barral et al., 2008). In addition, its close homologue Rab22b has been shown to facilitate the traffic between the TGN and the sorting endosome (Rodriguez-Gabin et al., 2001, 2009). To assess the role of Rab22a in ATP7A trafficking, we expressed the dominant-negative Rab22aS19N and constitutively active Rab22aQ64L mutants in cells.
Figure 7 shows experiments performed in cells treated with BCS. In cells expressing the dominant-negative Rab22aS19N mutant, ATP7A localized to the TGN in a pattern analogous to that observed in untransfected cells (Figure 7a). In contrast, in cells expressing the constitutively active Rab22aQ64L mutant, ATP7A was detected in a dispersed punctate pattern (Figure 7, b–f). The nature of the dispersed ATP7A-containing compartment was probed by colabeling the ATP7A puncta with markers for the cis-Golgi, TGN, and endosomal compartments. In Rab22aQ64L-expressing cells, the cis-Golgi marker GM130 also appeared dispersed, but did not colocalize with ATP7A (Figure 7c, inset in merge). The Tf receptor localization appeared unaltered, and Tf receptor did not colocalize with the ATP7A puncta (Figure 7d, inset in merge). The ATP7A-containing compartment was distinct from early endosomes, as shown by lack of colocalization with early endosome antigen 1 (EEA1; Figure 7e, inset in merge). However, this compartment may be at least partly made up of dispersed TGN, as indicated by a partial overlap of ATP7A and golgin‑97 staining (Figure 7f, inset in merge). This effect of Rab22aQ64L expression on the TGN was not previously described.
FIGURE 7:

Expression of the constitutively active mutant Rab22aQ64L fragments the TGN. HeLa cells transiently transfected with myc-tagged, dominant-negative Rab22aS19N (a) or the constitutively active Rab22aQ64L (b–f) were treated with BCS, fixed, and colabeled with an antibody against myc to detect the Rab22a mutants (a–f red) or with antibodies for the indicated markers (c–f pink). Cells are counterstained for ATP7A (a–f green). DAPI staining (blue) labels the nucleus. ATP7A appears in puncta in Rab22aQ64L-expressing cells. In merge, inset of f, the arrow indicates puncta showing no colocalization between ATP7A and golgin-97 in Rab22aQ64L-expressing cells, and the arrowhead indicates areas of colocalization. Scale bar, 10 μm.
When Rab22aQ64L-expressing cells were treated with copper, ATP7A remained in puncta and did not relocate to the cell surface (Figure 8a). The punctate structures did not colocalize with EEA1 (Figure 8b) and appeared mostly independent of golgin-97 (Figure 8c), suggesting that they might be a subcompartment of the TGN separate from golgin-97 TGN elements. ATP7A remained in a punctate pattern after copper washout (Figure 8d). Taken together, our data suggest that constitutively active Rab22aQ64L arrests ATP7A within a TGN compartment and prevents movement to the PM during copper stimulation.
FIGURE 8:

Expression of the Rab22aQ64L mutant inhibits ATP7A trafficking. HeLa cells transiently transfected with myc-tagged, constitutively active Rab22aQ64L were treated with CuCl2, fixed, and colabeled for ATP7A (green) and anti-myc to detect the Rab22a mutant (a), EEA1 (b), or golgin-97 (c) (all red). Cells expressing Rab22aQ64L (d) were treated with copper, followed by washout, and colabeled for ATP7A (green) and myc tag (red). Nuclei are labeled with DAPI (blue). Scale bar, 10 μm.
DISCUSSION
It is well established that ATP7A traffics between the TGN and the PM and/or sub-PM vesicles in response to changing intracellular copper levels (Petris et al., 1996; Nyasae et al., 2007). However, our understanding of how ATP7A recycling is regulated and the factors involved are not well understood.
Clathrin regulates the internalization of ATP7A from the cell surface
Previous studies showed that structural features in ATP7A are important for the copper transporter to internalize. Mutating a dileucine motif in the extreme carboxyl-terminus inhibits ATP7A internalization and strongly points toward the involvement of clathrin in ATP7A endocytosis (Francis et al., 1999; Petris and Mercer, 1999). Recently it was reported that the distal motor neuropathy causing mutation, P1386S, in TM domain 8 of ATP7A causes relocation of the C-terminus to the extracellular surface and accumulation of ATP7A at the PM (Yi et al., 2012). Furthermore, blocking CME by subjecting cells to hypertonic conditions (Pase et al., 2003; Lane et al., 2004) also prevents ATP7A internalization.
For the first time we directly assessed clathrin's involvement in ATP7A trafficking by depleting clathrin heavy chain levels using RNAi. Here we show that ATP7A becomes trapped at the cell surface when clathrin is knocked down or clathrin recruitment to the membrane is blocked by the overexpression of the AP180 carboxyl-terminus. Our results demonstrate a direct involvement of clathrin in ATP7A endocytic trafficking but, of interest, not in exocytosis of ATP7A from the TGN.
Despite these findings, other experimental approaches seem to indicate otherwise. Earlier studies by our lab and others (Cobbold et al., 2003; Lane et al., 2004) showed that dominant-negative mutants of dynamin (the large GTPase proposed to facilitate scission of clathrin-coated vesicles; Marks et al., 2001), which block CME (van der Bliek et al., 1993; Damke et al., 1994; Altschuler et al., 1998), do not inhibit the copper-regulated internalization of ATP7A (we reconfirmed these results for the present study, observing no effect of overexpression of dynamin 1 and 2 K44A mutants on ATP7A trafficking; Supplemental Figure S5). On further investigation we found that treating HeLa cells with the dynamin-inhibiting drug Dynasore during copper washout traps ATP7A at the plasma membrane (Supplemental Figure S6). These results suggest that a form of dynamin might be involved in the internalization of ATP7A and support a role for CME. According to the currently accepted, most common clathrin-mediated endocytosis model of PM proteins, these data appear contradictory. However, other reports indicate there are reasons that may explain them. Dynasore affects a number of dynamin isoforms, disabling not only dynamin 1, dynamin 2, and Drp1, the mitochondrial dynamin (Macia et al., 2006), but also potentially dynamin 3 (Lou et al., 2012). It is also possible that Dynasore is targeting another unidentified GTPase required at the early stages of invagination (Nankoe and Sever, 2006). This broad-range block of dynamin activity is sufficient to impair internalization of ATP7A from the PM, whereas blocking only either dynamin 1 or dynamin 2 is not. Identification of the specific target of Dynasore involved in ATP7A internalization via CME will need further investigation. In any case, the lack of effect of K44A mutants cannot be taken as full proof of clathrin-independent internalization.
There are other cases reported in which internalization is clathrin dependent but is not blocked by the expression of dominant-negative mutant dynamin. G protein–coupled receptors interact with β-arrestin, which acts as an adaptor for clathrin and AP-2 to promote internalization (Lefkowitz et al., 2006; Moore et al., 2007). The G protein–coupled receptors α2B-adrenergic receptor and the dopamine D2 receptor long isoform both require β-arrestin to internalize (Kim et al., 2004; Wang et al., 2004), and their endocytosis is not blocked by expression of dynamin mutants (Schramm and Limbird, 1999; Vickery and von Zastrow, 1999; Kim et al., 2004). We also investigated the caveolin and flotillin pathways as the most logical alternative candidates for regulating ATP7A endocytosis. Depletion of caveolin 1/2 or flotillin by RNAi appears to have no obvious effect on ATP7A cycling. It was reported that the stability of flotillin 1 and flotillin 2 proteins rely on each other, and the depletion of only one of them reduces the protein level of the other (Otto and Nichols, 2011); targeting flotillin 1 alone is sufficient to reduce clathrin-independent endocytosis (Glebov et al., 2006). These findings fit with our previous results showing that inhibitors of caveolae-mediated uptake do not affect ATP7A internalization (Cobbold et al., 2003). The results suggest that these pathways do not have a major involvement in ATP7A trafficking.
AP-2 is the clathrin adaptor required for ATP7A internalization from the PM
In cells depleted of AP-2, high copper levels stimulate ATP7A to traffic to the PM, but copper washout fails to trigger ATP7A's return to the TGN. Thus it is clear that when ATP7A reaches the PM, AP-2 is required for efficient internalization. Unlike the phenotype observed for clathrin depletion, we do not observe an obvious block of ATP7A at the PM in AP-2 depleted cells under low-copper conditions. We expected that if constitutive trafficking of ATP7A was a cycle between TGN and PM, then depleting AP-2 would show an ATP7A phenotype similar to clathrin knockdown. Given that the PM block of ATP7A was only observed after copper treatment, it suggests that, under low-copper basal conditions, ATP7A constitutively cycles mostly between the TGN and an intracellular location. The most likely reason ATP7A can be observed to accumulate strongly at the PM in clathrin-depleted cells is that clathrin knockdown affects not only internalization from the PM but also intracellular trafficking back to the TGN. Therefore ATP7A in vesicles, unable to return to the TGN, may be targeted to the PM and eventually accumulate there.
The AP-2 adaptor protein complex plays a role in the internalization of proteins such as HIV-Nef (Chaudhuri et al., 2007) through interactions with [D/E]XXXL[L/I] motifs. ATP7A contains a critical dileucine internalization signal within the sequence DKHSLL. Our results indicate that the region of the ATP7A carboxyl-terminus containing this motif mediates association of ATP7A with AP-2 and subsequently CME. Both clathrin and AP-2 depletion blocked internalization of a CD8 reporter containing the extreme 24 amino acids of the ATP7A carboxyl-terminus.
Other signals within the cytoplasmic domains of ATP7A may influence its internalization. ATP7A contains a PDZ binding motif (DTAL1500) at its extreme C-terminus (Greenough et al., 2004). Binding of G protein–coupled receptors to cellular PDZ-containing proteins recruits them to preexisting clathrin-coated pits but reduces their internalization rates (Puthenveedu and von Zastrow, 2006). It has been suggested that PDZ interactions tether the receptors to cortical actin, which delays the scission of the vesicle and results in a distinct pool of slow-release primary endosomal vesicles, as opposed to the quick release seen with Tf receptor. Recently a similar observation was reported for the glutamate transporter (EAAC1/EAAT3) (D'Amico et al., 2010). The authors propose that the PDZ motif in EAAC1 regulates transporter endocytosis by delaying its internalization through an AP-2–mediated pathway. Future studies will be needed to determine whether the PDZ binding domain of ATP7A influences its residence time on the cell surface.
ATP7A uses an AP-1 pathway to return to the TGN
AP-1 has been localized to clathrin-associated tubular structures in the TGN (Hinners and Tooze, 2003), and there is evidence that AP-1 functions in the endosome-to-TGN retrograde pathway (Meyer et al., 2000). Our data indicate that the constitutive and copper-induced trafficking of ATP7A back to the TGN involves an AP-1–mediated pathway.
ATP7A has been shown to traffic to melanosomes in a biogenesis of lysosome-related organelles complex-1 (BLOC-1)–regulated process (Setty et al., 2008). Hypopigmentation is observed in BLOC-1–deficient cells (Setty et al., 2007), and ATP7A most likely moves to the melanosome to supply the melanin biosynthetic cuproenzymes with copper at their site of function. The role of AP-1 in the transport of ATP7A to the melanosome has not been characterized. However, AP-1 has been implicated in the trafficking of other melanosome cargo along microtubules through interaction with the kinesin motor KIF13A (Delevoye et al., 2009). The authors speculate that BLOC-1 may cooperate with AP-1 in the delivery of melanosome cargo. This is supported by the observation that knockdown of AP-1 in a zebrafish model sensitized melanocytes to hypopigmentation in low-copper conditions (Ishizaki et al., 2010).
We observed a dependence on AP-1 for TGN localization of ATP7A under basal copper levels and for trafficking from the PM to the TGN after copper load and washout. AP-1 depletion does not appear to disrupt the TGN but, instead, selectively prevents the return of ATP7A to this compartment. In AP-1–depleted cells, ATP7A appears dispersed, and we hypothesize that it is predominantly found in small vesicles. This has not been directly observed, possibly because of the resolution limits of the light microscope. AP-1–mediated coating events are regulated by active forms of ARF, and we showed previously that inactivating cellular ARF disrupts the steady-state localization of ATP7A and inhibits its trafficking (Holloway et al., 2007). Our AP‑1 depletion data agree with our previous report and suggest that AP-1 is essential in the pathway used by ATP7A to return to the TGN.
Rab22a is involved in anterograde ATP7A traffic from the TGN
Our data suggest that Rab22 might regulate the trafficking of ATP7A at the TGN. In cells expressing the constitutively active mutant Rab22aQ64L, ATP7A can be observed in puncta in which ATP7A remains even after copper treatment or after copper treatment followed by washout. This compartment overlaps only partially with the TGN-resident golgin-97 and after copper treatment is independent of golgin-97, suggesting that ATP7A is able to access a region of the TGN separate from golgin-97 or that Rab22aQ64L arrests ATP7A trafficking in a post-TGN compartment. However, Rab22aQ64L clearly prevents the copper-regulated exit of ATP7A and its inclusion into anterograde intermediates that deliver this membrane protein to the PM.
Fragmentation of the Golgi was reported in Rab22aQ64L-expressing cells (Kauppi et al., 2002). Rab22a might regulate trafficking between Golgi and early endosomes, as Rab22a binds to EEA1. EEA1 has been shown to interact with syntaxin-6 (Simonsen et al., 1999), a soluble N-ethylmaleimide–sensitive factor attachment protein receptor involved in transport events at the TGN (Mallard et al., 2002; Nakamura et al., 2005). However, in our studies Rab22aQ64L did not alter the EEA1‑containing endosomes, and ATP7A remained distinct from the early endosomal compartments.
Rab22a has also been implicated in the clathrin-independent recycling of proteins: the major histocompatibility complexes MHCI and CD1a (Weigert et al., 2004; Barral et al., 2008) both use a Rab22a route for membrane protein recycling. Rab22a has also been associated with facilitating the exit of cargo from sorting endosomes (Magadan et al., 2006), endosome to TGN transport of the cation-independent mannose 6-phosphate receptor and cholera toxin (Mesa et al., 2005), and the sorting of nerve growth factor/pTrkA receptor into signaling endosomes (Wang et al., 2011). Our findings now suggest a new role for Rab22a in regulating the anterograde exit of ATP7A from the TGN to the PM.
In this study we clarify the importance of several regulators in the trafficking of ATP7A by identifying clathrin and the clathrin adaptors AP180, AP-2, and AP-1 and defining a novel role for Rab22a in the exit of ATP7A from the TGN. The results of this study are summarized in Table 1. We postulate that ATP7A traffics through the pathways shown in Figure 9. ATP7A constitutively cycles between the TGN and early endosome and/or small sub-PM vesicles, and trafficking out of the TGN into either of these destinations requires functional Rab22a. Elevated levels of copper direct ATP7A to the PM, and when copper levels are reduced, ATP7A requires clathrin and AP-2 for efficient endocytosis. In addition, AP-1 is required for the delivery of ATP7A to the TGN, and lack of functional AP-1 appears to arrest ATP7A in small vesicles. Additional regulators are likely to further modulate each step of this pathway and remain to be characterized.
TABLE 1:
Summary of treatment effect on endogenous ATP7A (or CD8-LL4) subcellular localization.
| Protein | Treatment | Control | Dynasore | Dynamin K44A | AP-1 siRNA | AP-2 siRNA | Clathrin siRNA | AP180-C | Rab22a Q64L |
|---|---|---|---|---|---|---|---|---|---|
| CD8-LL4 | No copper | Vesicles/perinuclear | – | – | – | PM | PM | – | – |
| ATP7A | No copper | TGN | – | TGN | Diffuse | TGN | PM | PM | Puncta |
| ↑copper | PM | – | PM | PM | PM | PM | PM | Puncta | |
| ↑copper/washout | TGN | PM | TGN | Diffuse | PM | PM | PM | Puncta |
–, not done.
FIGURE 9:

Regulators of ATP7A trafficking. Schematic representation of the proposed trafficking routes taken by endogenous ATP7A, either constitutively (left) or induced by copper level changes (right). Routes are marked by arrows, and blocks imposed by RNAi, mutant constructs, or drug treatment are shown (see also Table 1). Putative involvement of AP-2 in the constitutive pathway is shown as (?) (see the text). AP180-C shows a similar phenotype to clathrin knockdown and is not included in the scheme. Rab22aQ64L expression causes disruption of the TGN, which is not illustrated. Copper-induced forward pathway is shown as a direct trafficking step to the PM, although intermediary vesicles might be involved as in the constitutive route. V, vesicle.
MATERIALS AND METHODS
Antibodies and plasmids
Monoclonal antibodies against golgin-97 (CDF4) and transferrin receptor (H68.4) were from Invitrogen (Carlsbad, CA), actin (ACTN05 [C4]) from Abcam (Cambridge, MA), adaptin γ (110/3) and myc (9E10) from Sigma-Aldrich (St. Louis, MO), myc (9B11) from Cell Signaling Technology (Beverly, MA), and ATP7A (clone 34), caveolin 1, caveolin 2, flotillin 1, p230, and AP-2 μ2 from BD Biosciences (San Diego, CA); CD8 (OKT8) was produced from a mouse hybridoma cell line obtained from the American Type Culture Collection (Manassas, VA).
Polyclonal antibodies against EEA1 (C45B10) and clathrin heavy chain (P1663) were from Cell Signaling Technology; GM130 has been described (Nelson et al., 1998); ATP7A (Steveson et al., 2003) was a kind gift from Betty Eipper (University of Connecticut Health Center, Farmington, CT). All other antibodies were from BD Biosciences.
CD8 constructs containing the ATP7A C-terminus have been described previously (Francis et al., 1999; Cobbold et al., 2003). The plasmid for overexpression of the mutant protein Rab11aS25N (a kind gift from Richard Pagano, Mayo Clinic College of Medicine, Rochester, MN; Choudhury et al., 2002) was modified to incorporate a single green fluorescent protein (GFP) tag at the N-terminus (details provided on request) and checked by sequencing. Further Rab mutant constructs were generous gifts from the following: GFP-tagged Rab11bS25N and Rab11bQ70L, Beate Schlierf (Universität Erlangen-Nürnberg, Erlangen, Germany; Schlierf et al., 2000); myc-tagged Rab22aS19N and Rab22aQ64L, Vesa Olkkonen (National Institute for Health and Welfare, Helsinki, Finland; Kauppi et al., 2002); and myc-tagged AP180-C, Harvey McMahon (Laboratory of Molecular Biology, Cambridge University, Cambridge, United Kingdom; Ford et al., 2001). Plasmids were transfected into HeLa cells 12 h before experiments using ExGen 500 (Fermentas, Glen Burnie, MD) according to the manufacturer's instructions.
Confocal laser scanning microscopy
HeLa cells were cultured and immunofluorescence was performed as previously described (Holloway et al., 2007). Images were captured using a Zeiss LSM-510 META laser scanning confocal microscope with a 40×/1.3 numerical aperture (NA) or 63×/1.4 NA oil immersion objective (Carl Zeiss, Jena, Germany). 4′,6-Diamidino-2-phenylindole (DAPI) was excited using a 405-nm laser diode, and signal was collected through a bandpass (BP) 420- to 480-nm filter. Alexa Fluor 488 (or GFP) was excited with a 488-nm argon laser, and fluorescence was collected through a BP 505- to 530-nm emission filter; Alexa Fluor 594/555 was excited with a 543-nm laser, and fluorescence was collected through a BP 593- to 794-nm or BP 565–615 emission filter; and Alexa Fluor 633 was excited with a 633-nm laser, and emission was collected through a BP 655–710 nm. The confocal pinhole was set to Airy = 1, with each fluorochrome channel adjusted to the same z-slice optical thickness of ∼0.8 μm. Non–pixel-saturating conditions using low laser power were used. Postacquisition analyses were performed with the Zeiss LSM-510 Image Browser software. Photoshop Elements 8 (Adobe, San Jose, CA) image-editing software was used for further image processing.
Fluorescence image quantification
Line averaging of 2 was performed for each image. Photomultiplier gain was set to ensure that there was no saturation in any of the channels, and the same gain settings were maintained for each image. Three separate coverslips were imaged for each condition, and each coverslip was imaged in two randomly selected areas. Each image comprised a 2 × 2 tiled area equating to ∼40 μm2 (∼250–300 cells/image). This analysis was repeated for two separate experiments done on separate days. In experiments in which transfected cells expressing CD8-LL4 were analyzed an additional channel was generated that had a higher gain setting so that this could be used subsequently in the image analysis to select only transfected cells. Image analysis was performed in ImageJ (National Institutes of Health, Bethesda, MD) with a custom-written macro (details available on request), and analysis was performed in Excel 2010 (Microsoft, Redmond, WA) and Prism 5 (GraphPad Software, La Jolla, CA).
Trafficking assay
HeLa cells were treated with 200 μM CuCl2 in culture medium for 2 h at 37ºC and for the final 30 min in the presence of 50 μg/ml cycloheximide. For the copper washout, cells were treated with CuCl2 as previously described, washed once with culture medium, and incubated with culture medium containing 50 μg/ml cycloheximide and 200 μM copper chelating agent BCS for 4 h at 37ºC. The medium was replaced twice during the course of the washout. Addition of the protein synthesis inhibitor cycloheximide ensures that only internalized ATP7A is observed after the copper washout and not ATP7A synthesized during the time course of the experiment. Trafficking experiments using the cell-permeable dynamin-inhibiting drug Dynasore (Sigma-Aldrich) were carried out essentially as described (Kirchhausen et al., 2008), substituting fetal bovine serum (FBS) in the culture medium for 10% Nuserum (BD Biosciences) and omitting BCS during the washout. Fresh stock solution of Dynasore was used at a working concentration of 80 μM and included in the final 30 min of CuCl2 treatment and during washout. Medium was replaced three times during the washout period.
RNA interference
siRNA duplexes were purchased from either Ambion (Austin, TX) or Qiagen (Valencia, CA). Those targeting the mRNA of CLTC or the μ2 subunit of the AP-2 adaptor complex (AP2M1) were previously described (Murphy et al., 2008; Levecque et al., 2009). For AP-1 depletion, siRNA targeting the μ1 subunit (AP1M1) corresponding to the mRNA region GGUGUUUUCCGAGUACUUC was used. The following siRNA targeting regions were also used: GAGUUAGUGGAUUACUGC (cav1), GGAUUGAAUACUUGGACCC (cav2), and GGUUUACACUCGCCAUGGG (flot1).
HeLa cells were transfected with 10–20 nM duplex siRNA using HiPerfect transfection reagent (Qiagen) according to the manufacturer's instructions. Cells were transfected with siRNA on day 1, followed by a second transfection on day 2, 24 h apart. Cells were split on day 3 and seeded into fresh 24-well plates for Western blotting or into wells containing coverslips for immunofluorescence. Cells were lysed or processed for immunofluorescence on day 4, 72 h after the initial siRNA transfection. In negative controls, targeting siRNA was substituted for AllStars negative nontargeting siRNA (Qiagen). To monitor transferrin internalization cells were incubated with 50 μg/ml human Tf conjugated to Alexa Fluor 594/633 (Invitrogen) in the relevant media for 15 min at 37°C before washing, fixing, and processing. Protocols for Western blotting and quantification of band intensities are described elsewhere (Holloway et al., 2007).
Cell surface biotinylation assay
Confluent HeLa cells silenced for clathrin heavy chain or treated with nontargeting negative control siRNA were cultured in the presence of 200 μM BCS or 200 μM CuCl2 for 2 h in 35-mm dishes. Cell surface biotinylation was carried out at 4°C. Cells were cooled on ice and washed twice with cold DPBS, followed by incubation with Sulfo-NHS-SS-biotin (Pierce, Rockford, IL) at a concentration of 0.5 mg/ml in cold Dulbecco's phosphate-buffered saline (DPBS). The reaction was quenched in cold 10 mM glycine/DPBS, followed by 2× washes in cold DPBS. Cells were lysed in 0.5 ml of cold lysis buffer (150 mM NaCl, 1% NP-40, 50 mM Tris, pH 7.8, with protease inhibitors [EDTA free; Roche, Indianapolis, IN]), and 0.4-ml volumes of postnuclear supernatant were incubated with NeutrAvidin beads (Pierce) overnight at 4°C to pull out the cell surface proteins. Beads were washed 3× with cold lysis buffer and cell surface proteins eluted (e1) by heating at 80°C for 10 min in 0.05 ml of 2× reducing LDS loading dye (Invitrogen). The elution was removed, and a second elution was repeated (e2), as previously, to remove any remaining cell surface proteins attached to the beads. Postnuclear supernatant (0.01 ml loaded), postbead supernatant (unbound, 0.01 ml loaded), and e1 and e2 cell surface biotinylated proteins samples (0.02 ml loaded of each) were run on a 7% Tris acetate SDS–PAGE gels under reducing conditions (Invitrogen), followed by transfer to polyvinylidene fluoride membrane and Western blotting. The membrane was cut at around the 70-kDa mark to separate lower and upper parts of the blot, and detection of ATP7A or actin was carried out on the respective blots. ATP7A band intensities were measured as described (Holloway et al., 2007). The amount of biotinylated cell surface ATP7A was calculated as described (Nyasae et al., 2007). Briefly, surface ATP7A was expressed as a percentage of total ATP7A, that is, (e1 + e2)/(e1 + e2 + unbound×16) × 100. The unbound value was scaled up by 16 to reflect the total amount of unbound ATP7A in the postbead supernatant (taking into account the scale-up of e1 + e2). Figures are expressed as an average of three separate experiments.
Supplementary Material
Acknowledgments
We thank Betty Eipper, Richard Pagano, Beate Schlierf, Vesa Olkkonen, and Harvey McMahon for reagents and Keith Morris from the Microscopy Core Group for technical help. This work was supported by the Wellcome Trust (092071/Z/10/Z, 075491/Z/04).
Abbreviations used:
- AP
adaptor protein
- BCS
bathocuproinedisulfonic acid
- cav
caveolin
- CLTC
clathrin heavy chain
- CME
clathrin-mediated endocytosis
- EEA1
early endosome antigen 1
- flot
flotillin
- PM
plasma membrane
- Rab
Ras-related proteins in brain
- Tf
transferrin
- TGN
trans-Golgi network
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-08-0625) on April 17, 2013.
REFERENCES
- Altschuler Y, Barbas SM, Terlecky LJ, Tang K, Hardy S, Mostov KE, Schmid SL. Redundant and distinct functions for dynamin-1 and dynamin-2 isoforms. J Cell Biol. 1998;143:1871–1881. doi: 10.1083/jcb.143.7.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balamurugan K, Schaffner W. Copper homeostasis in eukaryotes: teetering on a tightrope. Biochim Biophys Acta. 2006;1763:737–746. doi: 10.1016/j.bbamcr.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Barral DC, Cavallari M, McCormick PJ, Garg S, Magee AI, Bonifacino JS, De Libero G, Brenner MB. CD1a and MHC class I follow a similar endocytic recycling pathway. Traffic. 2008;9:1446–1457. doi: 10.1111/j.1600-0854.2008.00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar A, Sheffler DJ, Kroeze WK, Compton-Toth B, Roth BL. Caveolin-1 interacts with 5-HT2A serotonin receptors and profoundly modulates the signaling of selected Galphaq-coupled protein receptors. J Biol Chem. 2004;279:34614–34623. doi: 10.1074/jbc.M404673200. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Byland R, Vance PJ, Hoxie JA, Marsh M. A conserved dileucine motif mediates clathrin and AP-2-dependent endocytosis of the HIV-1 envelope protein. Mol Biol Cell. 2007;18:414–425. doi: 10.1091/mbc.E06-06-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol. 2007;81:3877–3890. doi: 10.1128/JVI.02725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, Pagano RE. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J Clin Invest. 2002;109:1541–1550. doi: 10.1172/JCI15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold C, Coventry J, Ponnambalam S, Monaco AP. The Menkes disease ATPase (ATP7A) is internalized via a Rac1-regulated, clathrin- and caveolae-independent pathway. Hum Mol Genet. 2003;12:1523–1533. doi: 10.1093/hmg/ddg166. [DOI] [PubMed] [Google Scholar]
- Cobbold C, Ponnambalam S, Francis MJ, Monaco AP. Novel membrane traffic steps regulate the exocytosis of the Menkes disease ATPase. Hum Mol Genet. 2002;11:2855–2866. doi: 10.1093/hmg/11.23.2855. [DOI] [PubMed] [Google Scholar]
- Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delevoye C, et al. AP-1 and KIF13A coordinate endosomal sorting and positioning during melanosome biogenesis. J Cell Biol. 2009;187:247–264. doi: 10.1083/jcb.200907122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico A, Soragna A, Di Cairano E, Panzeri N, Anzai N, Vellea Sacchi F, Perego C. The surface density of the glutamate transporter EAAC1 is controlled by interactions with PDZK1 and AP2 adaptor complexes. Traffic. 2010;11:1455–1470. doi: 10.1111/j.1600-0854.2010.01110.x. [DOI] [PubMed] [Google Scholar]
- Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, McMahon HT. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 2001;291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- Fraile-Ramos A, Kohout TA, Waldhoer M, Marsh M. Endocytosis of the viral chemokine receptor US28 does not require beta-arrestins but is dependent on the clathrin-mediated pathway. Traffic. 2003;4:243–253. doi: 10.1034/j.1600-0854.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- Francis MJ, Jones EE, Levy ER, Martin RL, Ponnambalam S, Monaco AP. Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. J Cell Sci. 1999;112:1721–1732. doi: 10.1242/jcs.112.11.1721. [DOI] [PubMed] [Google Scholar]
- Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol. 2006;8:46–54. doi: 10.1038/ncb1342. [DOI] [PubMed] [Google Scholar]
- Goodyer ID, Jones EE, Monaco AP, Francis MJ. Characterization of the Menkes protein copper-binding domains and their role in copper-induced protein relocalization. Hum Mol Genet. 1999;8:1473–1478. doi: 10.1093/hmg/8.8.1473. [DOI] [PubMed] [Google Scholar]
- Greenough M, Pase L, Voskoboinik I, Petris MJ, O'Brien AW, Camakaris J. Signals regulating trafficking of Menkes (MNK; ATP7A) copper-translocating P-type ATPase in polarized MDCK cells. Am J Physiol Cell Physiol. 2004;287:C1463–C1471. doi: 10.1152/ajpcell.00179.2004. [DOI] [PubMed] [Google Scholar]
- Hansen CG, Nichols BJ. Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci. 2009;122:1713–1721. doi: 10.1242/jcs.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ED. Cellular copper transport and metabolism. Annu Rev Nutr. 2000;20:291–310. doi: 10.1146/annurev.nutr.20.1.291. [DOI] [PubMed] [Google Scholar]
- Hinners I, Tooze SA. Changing directions: clathrin-mediated transport between the Golgi and endosomes. J Cell Sci. 2003;116:763–771. doi: 10.1242/jcs.00270. [DOI] [PubMed] [Google Scholar]
- Hirst J, Miller SE, Taylor MJ, von Mollard GF, Robinson MS. EpsinR is an adaptor for the SNARE protein Vti1b. Mol Biol Cell. 2004;15:5593–5602. doi: 10.1091/mbc.E04-06-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Motley A, Harasaki K, Peak Chew SY, Robinson MS. EpsinR: an ENTH domain-containing protein that interacts with AP-1. Mol Biol Cell. 2003;14:625–641. doi: 10.1091/mbc.E02-09-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway ZG, Grabski R, Szul T, Styers ML, Coventry JA, Monaco AP, Sztul E. Activation of ADP-ribosylation factor regulates biogenesis of the ATP7A-containing trans-Golgi network compartment and its Cu-induced trafficking. Am J Physiol Cell Physiol. 2007;293:C1753–C1767. doi: 10.1152/ajpcell.00253.2007. [DOI] [PubMed] [Google Scholar]
- Ishizaki H, et al. Combined zebrafish-yeast chemical-genetic screens reveal gene-copper-nutrition interactions that modulate melanocyte pigmentation. Dis Model Mech. 2010;3:639–651. doi: 10.1242/dmm.005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi M, Simonsen A, Bremnes B, Vieira A, Callaghan J, Stenmark H, Olkkonen VM. The small GTPase Rab22 interacts with EEA1 and controls endosomal membrane trafficking. J Cell Sci. 2002;115:899–911. doi: 10.1242/jcs.115.5.899. [DOI] [PubMed] [Google Scholar]
- Kelly BT, McCoy AJ, Spate K, Miller SE, Evans PR, Honing S, Owen DJ. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature. 2008;456:976–979. doi: 10.1038/nature07422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerson ML, et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet. 2010;86:343–352. doi: 10.1016/j.ajhg.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim MY, Lee EJ, Ahn YS, Baik JH. Distinct regulation of internalization and mitogen-activated protein kinase activation by two isoforms of the dopamine D2 receptor. Mol Endocrinol. 2004;18:640–652. doi: 10.1210/me.2003-0066. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T, Macia E, Pelish HE. Use of Dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. In: William CJD, Balch E, Alan H, editors. In: Methods in Enzymology. San Diego, CA: Academic Press; 2008. pp. 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozik P, Francis RW, Seaman MN, Robinson MS. A screen for endocytic motifs. Traffic. 2010;11:843–855. doi: 10.1111/j.1600-0854.2010.01056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrandt W. Biology, structure and mechanism of P-type ATPases. Nat Rev Mol Cell Biol. 2004;5:282–295. doi: 10.1038/nrm1354. [DOI] [PubMed] [Google Scholar]
- Lane C, Petris MJ, Benmerah A, Greenough M, Camakaris J. Studies on endocytic mechanisms of the Menkes copper-translocating P-type ATPase (ATP7A; MNK). Endocytosis of the Menkes protein. Biometals. 2004;17:87–98. doi: 10.1023/a:1024413631537. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Levecque C, Velayos-Baeza A, Holloway ZG, Monaco AP. The dyslexia-associated protein KIAA0319 interacts with adaptor protein 2 and follows the classical clathrin-mediated endocytosis pathway. Am J Physiol Cell Physiol. 2009;297:C160–C168. doi: 10.1152/ajpcell.00630.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Fan F, Messa M, Raimondi A, Wu Y, Looger LL, Ferguson SM, De Camilli P. Reduced release probability prevents vesicle depletion and transmission failure at dynamin mutant synapses. Proc Natl Acad Sci USA. 2012;109:E515–E523. doi: 10.1073/pnas.1121626109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Magadan JG, Barbieri MA, Mesa R, Stahl PD, Mayorga LS. Rab22a regulates the sorting of transferrin to recycling endosomes. Mol Cell Biol. 2006;26:2595–2614. doi: 10.1128/MCB.26.7.2595-2614.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallard F, et al. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J Cell Biol. 2002;156:653–664. doi: 10.1083/jcb.200110081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks B, Stowell MH, Vallis Y, Mills IG, Gibson A, Hopkins CR, McMahon HT. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature. 2001;410:231–235. doi: 10.1038/35065645. [DOI] [PubMed] [Google Scholar]
- Mercer JF, Barnes N, Stevenson J, Strausak D, Llanos RM. Copper-induced trafficking of the cU-ATPases: a key mechanism for copper homeostasis. Biometals. 2003;16:175–184. doi: 10.1023/a:1020719016675. [DOI] [PubMed] [Google Scholar]
- Mesa R, Magadán J, Barbieri A, López C, Stahl PD, Mayorga LS. Overexpression of Rab22a hampers the transport between endosomes and the Golgi apparatus. Exp Cell Res. 2005;304:339–353. doi: 10.1016/j.yexcr.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Meyer C, Zizioli D, Lausmann S, Eskelinen EL, Hamann J, Saftig P, von Figura K, Schu P. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 2000;19:2193–2203. doi: 10.1093/emboj/19.10.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Vohra RS, Dunn S, Holloway ZG, Monaco AP, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Oxidised LDL internalisation by the LOX-1 scavenger receptor is dependent on a novel cytoplasmic motif and is regulated by dynamin-2. J Cell Sci. 2008;121:2136–2147. doi: 10.1242/jcs.020917. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Fukuda H, Kato A, Hirose S. MARCH-II Is a syntaxin-6-binding protein involved in endosomal trafficking. Mol Biol Cell. 2005;16:1696–1710. doi: 10.1091/mbc.E04-03-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankoe SR, Sever S. Dynasore puts a new spin on dynamin: a surprising dual role during vesicle formation. Trends Cell Biol. 2006;16:607–609. doi: 10.1016/j.tcb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Nelson DS, Alvarez C, Gao YS, Garcia-Mata R, Fialkowski E, Sztul E. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J Cell Biol. 1998;143:319–331. doi: 10.1083/jcb.143.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Jackson M, Peterson PA. Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell. 1989;58:707–718. doi: 10.1016/0092-8674(89)90105-0. [DOI] [PubMed] [Google Scholar]
- Nyasae L, Bustos R, Braiterman L, Eipper B, Hubbard A. Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: copper-dependent redistribution between two intracellular sites. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1181–G1194. doi: 10.1152/ajpgi.00472.2006. [DOI] [PubMed] [Google Scholar]
- Otto GP, Nichols BJ. The roles of flotillin microdomains—endocytosis and beyond. J Cell Sci. 2011;124:3933–3940. doi: 10.1242/jcs.092015. [DOI] [PubMed] [Google Scholar]
- Pase L, Voskoboinik I, Greenough M, Camakaris J. Copper stimulates trafficking of a distinct pool of the Menkes copper ATPase (ATP7A) to the plasma membrane and diverts it into a rapid recycling pool. Biochem J. 2003;378:1031–1037. doi: 10.1042/BJ20031181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena MM, Lee J, Thiele DJ. A delicate balance: homeostatic control of copper uptake and distribution. J Nutr. 1999;129:1251–1260. doi: 10.1093/jn/129.7.1251. [DOI] [PubMed] [Google Scholar]
- Petris MJ, Camakaris J, Greenough M, LaFontaine S, Mercer JF. A C-terminal di-leucine is required for localization of the Menkes protein in the trans-Golgi network. Hum Mol Genet. 1998;7:2063–2071. doi: 10.1093/hmg/7.13.2063. [DOI] [PubMed] [Google Scholar]
- Petris MJ, Mercer JF. The Menkes protein (ATP7A; MNK) cycles via the plasma membrane both in basal and elevated extracellular copper using a C-terminal di-leucine endocytic signal. Hum Mol Genet. 1999;8:2107–2115. doi: 10.1093/hmg/8.11.2107. [DOI] [PubMed] [Google Scholar]
- Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J. 1996;15:6084–6095. [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu MA, von Zastrow M. Cargo regulates clathrin-coated pit dynamics. Cell. 2006;127:113–124. doi: 10.1016/j.cell.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Razani B, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gabin A, Cammer M, Almazan G, Charron M, Larocca J. Role of rRAB22b, an oligodendrocyte protein, in regulation of transport of vesicles from trans Golgi to endocytic compartments. J Neurosci Res. 2001;66:1149–1160. doi: 10.1002/jnr.1253. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gabin AG, Yin X, Si Q, Larocca JN. Transport of mannose-6-phosphate receptors from the trans-Golgi network to endosomes requires Rab31. Exp Cell Res. 2009;315:2215–2230. doi: 10.1016/j.yexcr.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlief ML, Gitlin JD. Copper homeostasis in the CNS: a novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol Neurobiol. 2006;33:81–90. doi: 10.1385/MN:33:2:81. [DOI] [PubMed] [Google Scholar]
- Schlierf B, Fey GH, Hauber J, Hocke GM, Rosorius O. Rab11b is essential for recycling of transferrin to the plasma membrane. Exp Cell Res. 2000;259:257–265. doi: 10.1006/excr.2000.4947. [DOI] [PubMed] [Google Scholar]
- Schramm NL, Limbird LE. Stimulation of mitogen-activated protein kinase by G protein-coupled alpha(2)-adrenergic receptors does not require agonist-elicited endocytosis. J Biol Chem. 1999;274:24935–24940. doi: 10.1074/jbc.274.35.24935. [DOI] [PubMed] [Google Scholar]
- Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. Rab GTPases at a glance. J Cell Sci. 2007;120:3905–3910. doi: 10.1242/jcs.015909. [DOI] [PubMed] [Google Scholar]
- Setty SR, et al. BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol Biol Cell. 2007;18:768–780. doi: 10.1091/mbc.E06-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty SR, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS. Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature. 2008;454:1142–1146. doi: 10.1038/nature07163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Gaullier JM, D'Arrigo A, Stenmark H. The Rab5 effector EEA1 interacts directly with syntaxin-6. J Biol Chem. 1999;274:28857–28860. doi: 10.1074/jbc.274.41.28857. [DOI] [PubMed] [Google Scholar]
- Singleton WC, et al. Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B. J Biol Chem. 2010;285:27111–27121. doi: 10.1074/jbc.M110.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- Steveson TC, Ciccotosto GD, Ma XM, Mueller GP, Mains RE, Eipper BA. Menkes protein contributes to the function of peptidylglycine alpha-amidating monooxygenase. Endocrinology. 2003;144:188–200. doi: 10.1210/en.2002-220716. [DOI] [PubMed] [Google Scholar]
- Strausak D, La Fontaine S, Hill J, Firth SD, Lockhart PJ, Mercer JF. The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. J Biol Chem. 1999;274:11170–11177. doi: 10.1074/jbc.274.16.11170. [DOI] [PubMed] [Google Scholar]
- Traub LM. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- Tümer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013;34:417–429. doi: 10.1002/humu.22266. [DOI] [PubMed] [Google Scholar]
- Uauy R, Olivares M, Gonzalez M. Essentiality of copper in humans. Am J Clin Nutr. 1998;67:952S–959S. doi: 10.1093/ajcn/67.5.952S. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhuis NA, et al. Phosphorylation regulates copper-responsive trafficking of the Menkes copper transporting P-type ATPase. Int J Biochem Cell Biol. 2009;41:2403–2412. doi: 10.1016/j.biocel.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Vickery RG, von Zastrow M. Distinct dynamin-dependent and -independent mechanisms target structurally homologous dopamine receptors to different endocytic membranes. J Cell Biol. 1999;144:31–43. doi: 10.1083/jcb.144.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liang Z, Li G. Rab22 controls NGF signaling and neurite outgrowth in PC12 cells. Mol Biol Cell. 2011;22:3853–3860. doi: 10.1091/mbc.E11-03-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhao J, Brady AE, Feng J, Allen PB, Lefkowitz RJ, Greengard P, Limbird LE. Spinophilin blocks arrestin actions in vitro and in vivo at G protein-coupled receptors. Science. 2004;304:1940–1944. doi: 10.1126/science.1098274. [DOI] [PubMed] [Google Scholar]
- Weigert R, Donaldson JG. Fluorescent microscopy-based assays to study the role of Rab22a in clathrin-independent endocytosis. Methods Enzymol. 2005;403:243–253. doi: 10.1016/S0076-6879(05)03020-X. [DOI] [PubMed] [Google Scholar]
- Weigert R, Yeung AC, Li J, Donaldson JG. Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol Biol Cell. 2004;15:3758–3770. doi: 10.1091/mbc.E04-04-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Heiny ME, Suzuki M, Gitlin JD. Biochemical characterization and intracellular localization of the Menkes disease protein. Proc Natl Acad Sci USA. 1996;93:14030–14035. doi: 10.1073/pnas.93.24.14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L, Donsante A, Kennerson ML, Mercer JF, Garbern JY, Kaler SG. Altered intracellular localization and valosin-containing protein (p97 VCP) interaction underlie ATP7A-related distal motor neuropathy. Hum Mol Genet. 2012;21:1794–1807. doi: 10.1093/hmg/ddr612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.